
Abstract
Mitogen-activated protein kinases, which play a crucial role in signal transduction, are activated by phosphorylation in response to a variety of mitogenic signals. In the present study, the authors used Western blot analysis and immunohistochemistry to show that phosphorylated extracellular signal-regulated protein kinase (p-ERK) and c-Jun NH(2)-terminal kinase (p-JNK), but not p38 mitogen-activated protein kinase, significantly increased in both the neurons and astrocytes after traumatic brain injury in the rat hippocampus. Different immunoreactivities of p-ERK and p-JNK were observed in the pyramidal cell layers and dentate hilar cells immediately after traumatic brain injury. Immunoreactivity for p-JNK was uniformly induced but was only transiently induced throughout all pyramidal cell layers. However, strong immunoreactivity for p-ERK was observed in the dentate hilar cells and the damaged CA3 neurons, along with the appearance of pyknotic morphologic changes. In addition, immunoreactivity for p-ERK was seen in astrocytes surrounding dentate and CA3 pyramidal neurons 6 hours after traumatic brain injury. These findings suggest that ERK and JNK but not p38 cascades may be closely involved in signal transduction in the rat hippocampus after traumatic brain injury.
Mitogen-activated protein kinases (MAPKs), which play a crucial role in signal transduction, are activated by phosphorylation in response to a variety of mitogenic signals. The MAPK cascades are composed of extracellular signal-regulated protein kinase (ERK), c-Jun NH(2)-terminal kinase (JNK), and p38 pathways. The ERK is activated in response to growth factors (Boulton et al., 1991), oxidative stress (Aikawa et al., 1997), and intracellular calcium influx (Kurino et al., 1995). Phosphorylated ERK (p-ERK) plays an important role in the survival, proliferation, and differentiation of various cells (Seger et al., 1995). However, JNK and p38 are activated in response to the presence of inflammatory cytokines (Kyriakis et al., 1996), glutamate toxicity (Kawasaki et al., 1997), and hyperosmolality (Moriguchi et al., 1995). Phosphorylated JNK (p-JNK) and p38 (p-p38) cause alterations in the transcription factors that lead to neuronal apoptosis (Xia et al., 1995).
Recent studies have suggested that the activation of JNK and p38 pathways without the activation of an ERK pathway induce selective CA1 vulnerability to transient forebrain ischemia (Sugino et al., 2000; Hu et al., 2000). Meanwhile, selective hippocampal damage restricted to the dentate hilar neurons and the CA3 pyramidal neurons was observed after traumatic brain injury (TBI) in the rat (Cortez et al., 1989; Lowenstein et al., 1992). The mechanism of posttraumatic selective vulnerability of these neurons has not yet been fully elucidated. Until now, there have been no reports focusing on the expression and distribution of phosphorylated MAPKs after TBI. In the present study, we investigated, for the first time, the activation of the MAPK pathways in the rat hippocampus after experimental TBI. As a result, differential activation of the MAPK pathways was observed between TBI and transient forebrain ischemia in the rat hippocampus. These findings suggest that a distinct MAPK cascade may participate in the selective vulnerability of hippocampal CA3 neurons and dentate hilar neurons after TBI.
MATERIALS AND METHODS
Animal experimental procedures
Adult male Sprague-Dawley rats 9- to 10-weeks old and weighing 300 to 400 g were used. The rats were anesthetized with isoflurane in a 30:70 mixture of oxygen and nitrous oxide gas through a facemask. The rats were fixed in a stereotaxic flame. A 4.8-mm craniectomy was made over the right parietal cortex (3.8-mm posterior and 2.5-mm lateral to the bregma). A plastic Luer-Loc was placed over the craniectomy site with dental acrylic cement. The next day, the rats were anesthetized and implanted with a polyethylene catheter in the femoral artery. The rats were intubated with a 14-gauge angiocatheter and maintained on a mechanical ventilator after the infusion of pancronium bromide (tidal volume, 2.5–3.0 mL/kg; respiratory rate, 60 breaths per minute). The rectal temperature was measured with a rectal probe and maintained constant at a level of approximately 38°C using a heating pad. Arterial blood samples were analyzed intermittently. The rats were subjected to lateral fluid percussion injury at a moderate severity (3.5–4.0 ATM, 16-millisecond duration) using a fluid percussion device (model HPD-1700; Dragonfly R&D, Silver Spring, MD, U.S.A.) as previously described (Matsushita et al., 2000). The pressure pulse was measured extracranially with a pressure transducer (Model 211 B4; Kistler Instrument, Amherst, NY, U.S.A.) and recorded on a digital storage computer (MacLab; AD Instruments, NSW, Australia). After injury, the connection cap was removed and the scalp was sutured. The rats were placed on a heating pad to maintain a temperature of approximately 37°C until they could move independently. The Animal Care and Use Committee of the National Defense Medical College approved all animal procedures.
Tissue preparation
For Western blot analysis, the rats were killed by decapitation using intraperitoneal anesthesia with pentobarbital sodium at 5, 15, and 30 minutes and at 1, 6, 24, and 72 hours after injury (n = 4 per time point). The whole hippocampus was immediately excised while submerged in ice-cold artificial CSF (0.6 mmol/L NaH2PO4/2H2O, 3.35 mmol/L potassium chloride, 138.6 mmol/L sodium chloride, 9.9 mmol/L dextrose, 21 mmol/L sodium bicarbonate, 2.5 mmol/L calcium chloride, and 1 mmol/L magnesium chloride). The tissue specimens were aliquoted, frozen, and kept at −80°C. Sham-control animals were subjected to the same procedures except the actual insult, and were killed during the same time courses (n = 4 per time point). For immunohistochemical analysis, the rats were perfused transcardially with normal saline followed by 4% buffered paraformaldehyde at the same time points after injury (n = 3–5 per time point). The brains were removed and embedded in paraffin after fixation in 4% buffered paraformaldehyde, followed by 0.1 mmol/L phosphate-buffered saline (pH 7.4) for 24 hours at 4°C. Serial coronal sections (5-μm thick) were prepared. Existence of the morphologic changes, such as gliding contusional hemorrhage, were confirmed in all sections of TBI animals.
Western blot analysis
The ipsilateral hippocampus of the injured hemisphere was used for Western blot analysis. The frozen tissue specimens were homogenized with ice-cold homogenization buffer (20 mmol/L Tris-hydrochloride, pH 7.5; 1 mmol/L edetic acid, 5 mmol/L magnesium chloride, 1 mmol/L chlorophenothane, 20 μg/mL aprotinin, 1 mmol/L phenylmethylsulfonyl fluoride, and 2 mmol/L sodium orthovanadate). The protein content was determined using a bicinchoninic acid protein assay reagent kit (Pierce, Rockford, IL, U.S.A.). Equal amounts of protein (40 μg) were loaded in each lane on 10% polyacrylamide gel with a 4.75% stacking gel (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) with loading buffer (125 mmol/L Tris-hydrochloride, pH 6.8; 20% glycerol, 4% weight/volume sodium dodecyl sulfate, 10% mercaptoethanol, and 0.002% bromophenol blue). The protein was transferred to polyvinylidene difluoride membranes (Millipore, Tokyo, Japan) using an electrophoretic transfer system (TANK; Amersham Biosciences, Tokyo, Japan) at 60 V for 4 hours. The membranes were then incubated overnight at 4°C with primary antibody against polyclonal p-ERK 1/2 (1:500), p-JNK (1:500), or p-p38 (1:500) antibodies (New England Biolabs, Beverly, MA, U.S.A.). The membranes were incubated for 1 hour at room temperature with goat antirabbit immunoglobulin G alkaline phosphatase-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) diluted 1:3000 for p-ERK 1/2, p-JNK, and p-p38. The blots were developed with alkaline phosphatase substrate 5-bromo-4-chloro-3-indolyl phosphate along with nitroblue tetrazolium (AP Conjugate Substrate Kit; Bio-Rad, Hercules, CA, U.S.A.) in Tris buffer (pH 9.5). For an immunoblot analysis of the total amount of MAPKs, the membranes were incubated overnight at 4°C with primary antibody (Santa Cruz Biotechnology) diluted 1:1000 for ERK1, JNK1, and p38. The membranes were incubated for 1 hour at room temperature with goat antirabbit immunoglobulin G alkaline phosphatase-conjugated secondary antibody diluted 1:5000 for ERK1, JNK1, and p38. All other procedures were performed as described previously. The liver tissue specimens of rats undergoing ultraviolet treatment were used for positive staining to detect p-p38.
Immunohistochemistry
Immunohistochemistry was performed using the strep-t-avidin-biotin peroxidase complex method. A HISTOFINE SAB-PO Kit (Nichirei, Tokyo, Japan) was used for immunostaining by polyclonal antibodies against p-ERK1/2, p-SAPK/JNK, and p-p38 (New England Biolabs). After deparaffinization and hydration, the sections in 10-mmol/L sodium citrate (pH 6.0) were boiled for 10 minutes at 95°C. Endogenous peroxidase was blocked with 0.3% hydrogen peroxidase. The sections were incubated with phosphate-buffered saline containing 10% normal goat serum at room temperature to eliminate any nonspecific binding, and were incubated overnight at 4°C with polyclonal antibodies against p-ERK1/2 (1:100), phosphorylated stress-activated protein kinase/p-JNK (1:100), and p-p38 (1:250). The sections were incubated with biotinylated secondary antibody and horseradish peroxidase-linked streptavidin for 1 hour at room temperature. Peroxidase was observed with diamino benzidine. For an evaluation of morphologic changes, adjacent sections were counterstained with hematoxylin. For negative controls, normal rabbit immunoglobulin G was used instead of the primary antibodies. Rat brains were used for positive staining to detect p-p38 72 hours after transient forebrain ischemia.
Double immunostaining
The paraffin-embedded serial sections were double stained with the polyclonal antibody for glial fibrillary acidic protein (GFAP) and p-ERK1/2. Double staining was performed with the EnVision system (Dako, Kyoto, Japan) for GFAP and a histofine SAB-PO kit (Nichirei) for p-ERK1/2. After p-ERK1/2 immunostaining reaction with diamino benzidine, the sections were boiled at 95 to 100°C in citrate buffer (pH 6.0). The sections were incubated with prediluted rabbit anti-GFAP (Dako) for 1 hour at room temperature, followed by goat antirabbit immunoglobulin conjugation to alkaline phosphatase labeled-dextran polymer (Dako) for 30 minutes at room temperature. Color was developed with a fuchsin substrate chromogen system (Dako). Colocalization of GFAP (red) and p-ERK1/2 (brown) was identified either by the presence of both colors in the same cell or by immunostaining with a pinkish-brown mixed color.
Statistical analysis
All data are expressed as mean ± SD. The physiologic data were statistically analyzed by one-way analysis of variance. The densities of the immunoblotting band were quantified with a NIH (National Institutes of Health, Bethesda, MD, U.S.A.) image analyzer (version 1.61). Statistical significance for Western blots was determined using an analysis of variance followed by a post hoc Bonferroni-Dunn test. A P value of less than 0.05 was considered to be statistically significant.
RESULTS
Physiologic parameters were all within the normal ranges during surgical procedures and no significant differences were found between the TBI and sham-control rats after injury (Table 1).
Summary of physiologic parameters
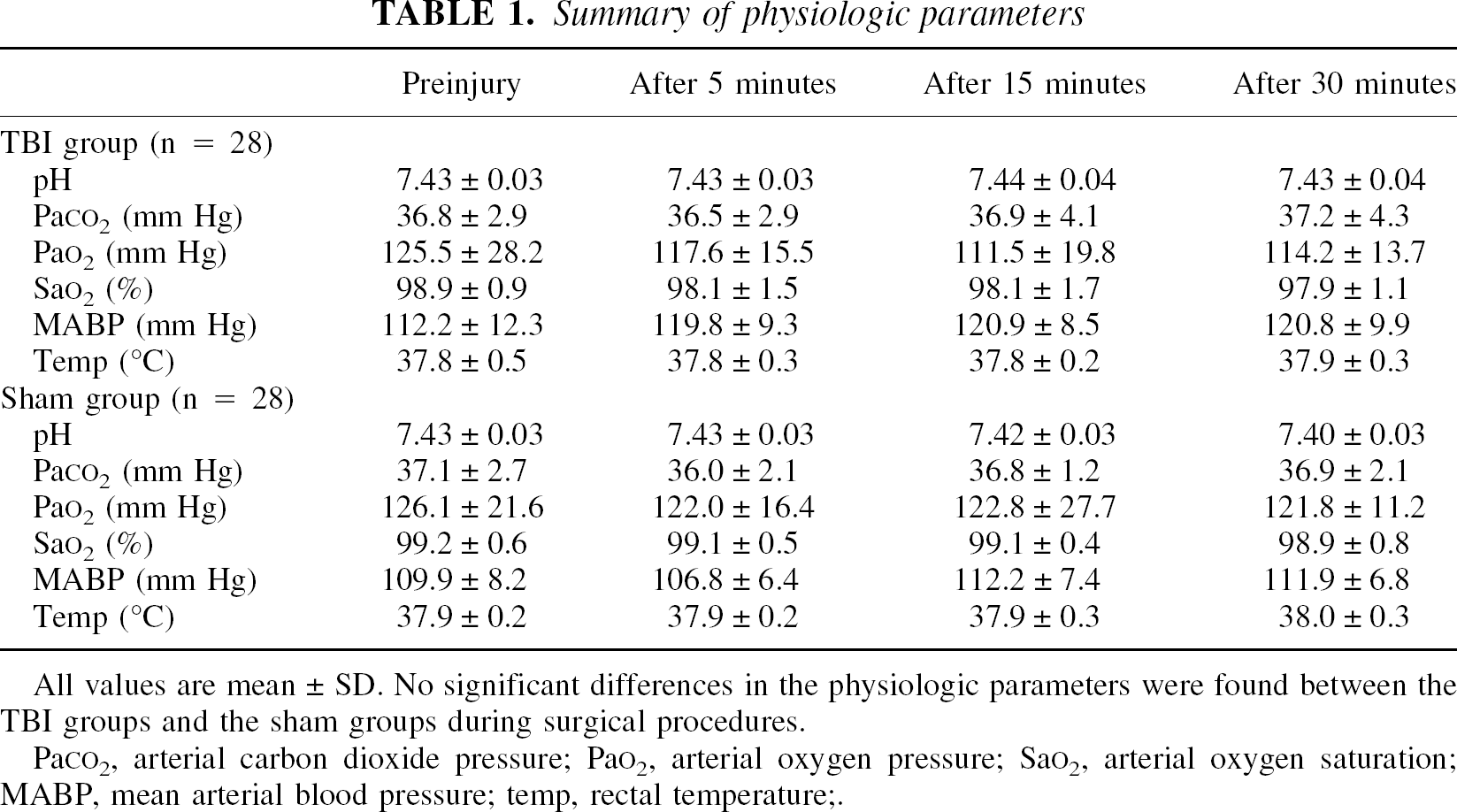
All values are mean ± SD. No significant differences in the physiologic parameters were found between the TBI groups and the sham groups during surgical procedures.
PaC
The p-ERK and p-JNK protein contents in the sham-control rats were not significantly different after injury. Our results showed that the p-ERK and p-JNK contents but not the p-p38 contents significantly increased in the ipsilateral hippocampus in response to TBI. A few differences in the immunoreactivities regarding the duration and magnification of p-ERK and p-JNK were observed after TBI. The immunoreactivity for p-JNK significantly increased 5 minutes after injury (157.2% ± 13.2%, P < 0.001) compared with sham-control rats, and thereafter gradually decreased to control levels 1 hour after injury (Figs. 1C and 1D). However, p-ERK immunoreactivity increased 5 minutes after injury (145.1% ± 26.6%, P < 0.001), reached a maximum level 15 minutes after injury (162.1% ± 18.9%, P < 0.001), and returned to control levels 24 hours after injury (Figs. 1A and 1B). Although we could detect p-p38 immunoreactivity in liver tissue specimens of the rats undergoing ultraviolet treatment, no discernible p-p38 protein was noted in the rat hippocampus after injury (Fig. 2A). In addition, no significant changes in the total amounts of ERK, JNK, and p38 were observed after injury compared with sham-control rats (Figs. 2B to 2D).
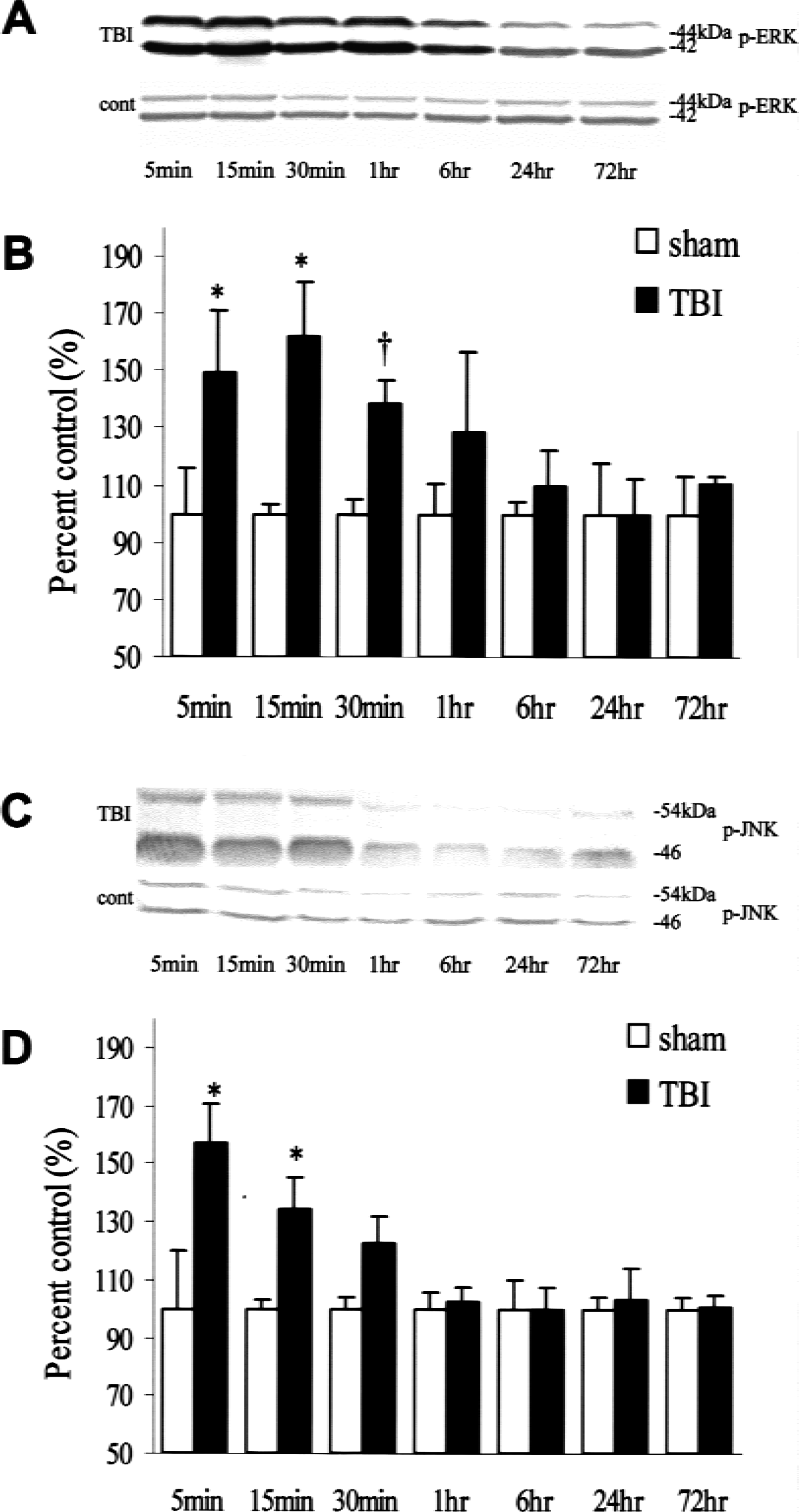
Time course of p-ERK and p-JNK expression in the hippocampus at the site of injury. Picture of a representative Western blot analysis showed the time course of p-ERK
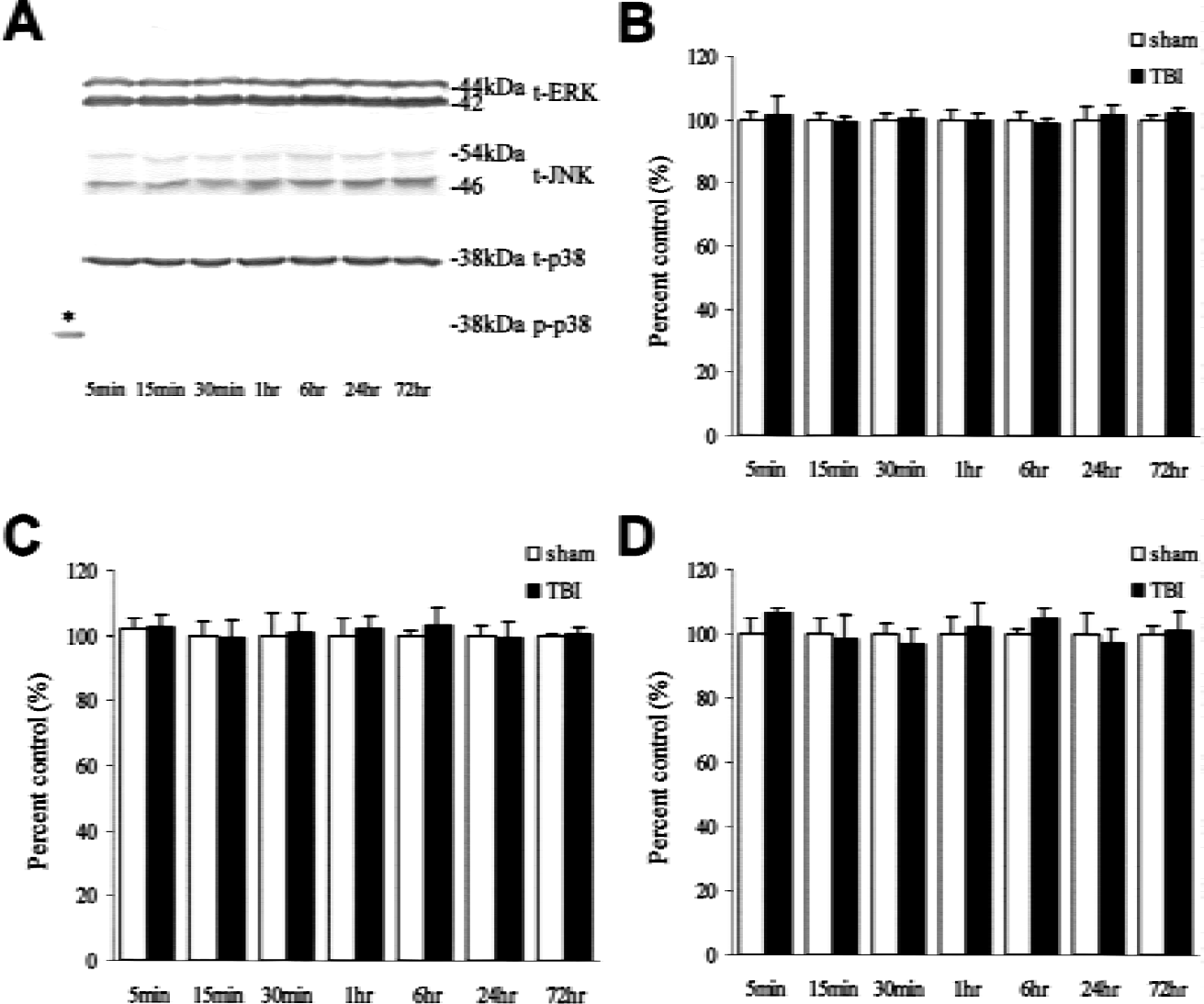
Time course of total amounts of ERK, JNK, and p38 expression in the hippocampus at the site of injury. Picture of a representative Western blot
Immunohistochemical results were consistent with those of Western blot analysis, suggesting that the immunoreactivity for p-p38 was not present after injury despite its detection in rat brains after 72 hours of transient forebrain ischemia (Figs. 4I and 4L). A few immunopositive-stained cells for p-ERK and p-JNK were detected in sham-control rats (Figs. 3A, 3B, 4A, 4D, 4G, and 4J). The immunoreactivity for p-JNK was induced and maintained uniformly in whole pyramidal neurons until 30 minutes after injury regardless of any regional selective vulnerability to TBI, and was primarily localized to the morphologically intact neurons (Figs. 4E, 4F, and 4K). In contrast, p-ERK immunoreactivity was immediately induced in the dentate gyrus, mossy fibers, and CA3 neurons (Figs. 3C to 3H, 4B, and 4C), but was not detected in the CA1 neurons after injury (Fig. 4H). Furthermore, the induction of the p-ERK expression was localized in the dentate hilar neurons (Figs. 3F and 3H) and the damaged CA3 neurons (Figs. 4B and 4C) with the appearance of a small, shrunken, and dark nucleus 15 to 30 minutes after injury.
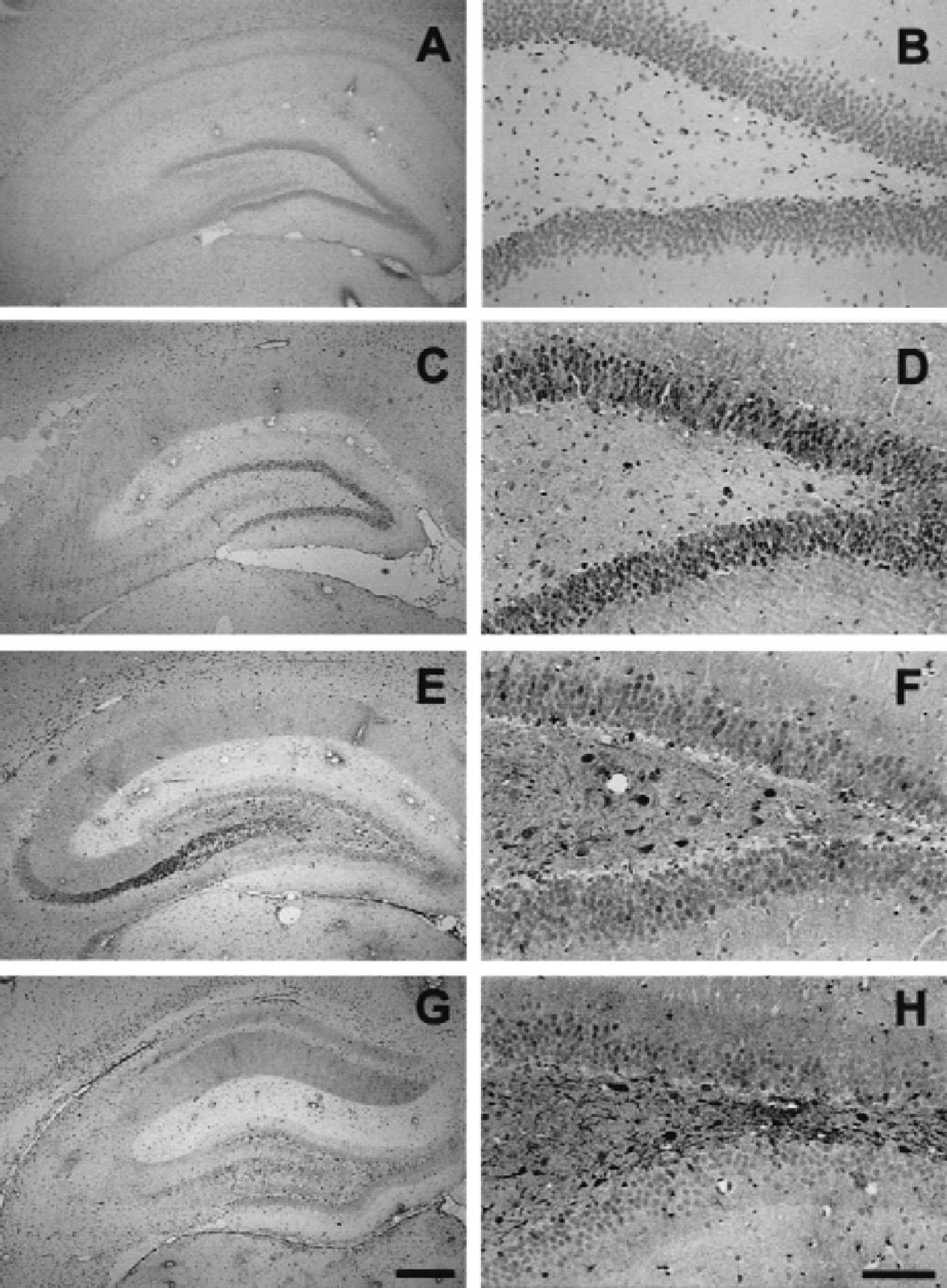
Photomicrographs showing the spatial distribution and time course of immunoreactivity for p-ERK in the ipsilateral hippocampus after traumatic brain injury. Sections were prepared from the sham controls (
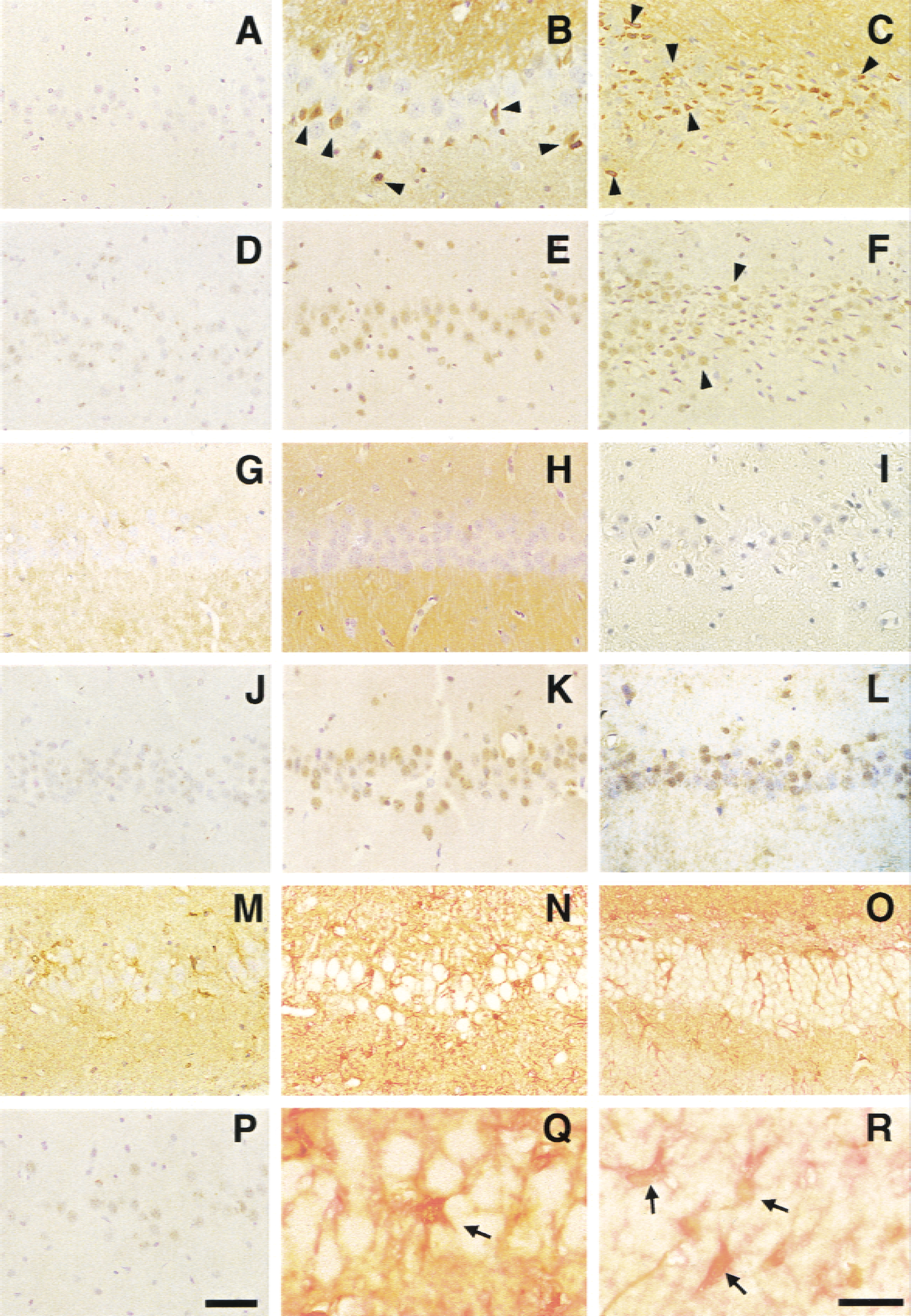
Photomicrographs showed the spatial distribution, cellular localization, and time course of immnoreactivity for p-ERK and p-JNK in the ipsilateral hippocampus after traumatic brain injury (TBI). Sections were prepared from the sham control
The current study showed that an induction of p-ERK was observed in astrocytes surrounding dentate neuronal cells (Figs. 4O and 4R) and pyramidal CA3 neurons (Figs. 4M, 4N, and 4Q) 6 hours after TBI, and thereafter was maintained until 72 hours after injury (data not shown).
DISCUSSION
Posttraumatic amnesia is a common feature during the acute phase after TBI, and seems to be related to hippocampal dysfunction (Scheff et al., 1997). Previous studies have suggested that a selective vulnerability to TBI was observed in hippocampal CA3 neurons (Cortez et al., 1989; Nawashiro et al., 1995; Hicks et al., 1996) and dentate hilar neurons (Lowenstein et al., 1992; Toth et al., 1997). However, the mechanism that would explain the posttraumatic selective vulnerability of these neurons has yet to be fully elucidated.
Recent studies have shown that TBI induces the expression of neurotrophin and neurotrophin-receptor messenger RNA in the rat hippocampus (Oyesiku et al., 1999; Truettner et al., 1999). Many neurotrophins, such as nerve growth factor and brain-derived neurotrophic factor, are thought to promote cell survival and differentiation by activating intracellular signal transduction through Ras-ERK cascades (Bonni et al., 1999).
Several studies of the participation of MAPK cascades in transient forebrain ischemia have revealed that the JNK and p38 MAPK pathways play important roles in CA1 neuronal cell death during ischemia (Sugino et al., 2000; Hu et al., 2000). In these articles, the immunoreactivity for p-JNK and p-p38 was induced in the CA1 and CA3 neurons 15 minutes after reperfusion, and thereafter gradually decreased in the CA1 neurons but remained at steady levels in the CA3 neurons until 72 hours after reperfusion. Meanwhile, the transient immunoreactivity for p-ERK was confirmed in the dentate gyrus and mossy fiber bundles. The authors hypothesized that the activation of ERK pathway might neuroprotective for the survival of granular cells of the dentate gyrus and CA3 neurons, which are resistant to ischemia, whereas the activation of the JNK and p38 cascades without a neuroprotective pathway in CA1 neurons might render these neurons vulnerable to ischemia. However, Namura et al. (2001) demonstrated that the inhibition of the ERK pathway using U0126, a MAP kinase/ERK kinase-specific inhibitor, resulted in reduced brain damage. This finding indicated that the ERK pathway contributed to brain injury after forebrain and focal cerebral ischemia.
The present results show that the immunoreactivity for p-p38 was not detected during after injury, and thereafter the immunoreactivity for p-JNK was induced transiently in whole pyramidal neurons regardless of any regional selective vulnerability to TBI. In contrast, the immunoreactivity for p-ERK was not detected in the CA1 neurons but was localized in the dentate hilar neurons and the damaged CA3 neurons with the appearance of a small, shrunken, and dark nucleus after 30 minutes of TBI. The distinct expression in both the magnitude and distribution of p-ERK and p-JNK reflects pathophysiologic differences between TBI and transient forebrain ischemia in the rat hippocampus. Therefore, we speculated that CA1 neurons might be resistant to traumatic stress because the expression of p-JNK was weak and transient, whereas p-ERK and p38 remained unchanged in the CA1 neurons after injury. In addition, the remarkable and prolonged expression of p-ERK in the dentate hilar neurons and the damaged CA3 neurons may promote cell damage in the posttraumatic vulnerable regions.
In the current study, an induction of p-ERK was observed in astrocytes surrounding dentate hilar neurons and CA3 pyramidal neurons 6 hours after TBI. There were close relations between inflammation, cytokine production, and astrogliosis (Lieberman et al., 1992). Reactive astrocytes induced the expression of a variety of molecules, such as neurotrophins and growth factor families (McKeon et al., 1997). Mandell et al. (2001) assessed that ERK phosphorylation triggered an astroglial reaction that led to reactive astrogliosis, which has both beneficial and detrimental consequences for the functional recovery of neurons. A recent study indicated that reactive astrocytes have a beneficial effect on both neuronal survival and the repair of the damaged blood-brain barrier (Bush et al., 1999). Therefore, the prolonged phosphorylation of ERK in astrocytes may play an important role in the pathophysiology of late-period TBI.
In this study, no significant changes in the total amounts of ERK, JNK, and p38 were observed after injury. These findings suggested that TBI immediately induced a transient phosphorylation of ERK and JNK, but this phenomenon may not be attributable to the alteration in the total amounts of ERK or JNK.
We are the first to demonstrate that the immunoreactivity of p-ERK and p-JNK significantly increase after TBI in the rat hippocampus. Our data suggest that the phosphorylation of ERK and JNK but not p38 were associated with the molecular sequelae of TBI, and that the discrepancy in the MAPK alterations reflected differences in selective vulnerability between the mechanical and ischemic events in the rat hippocampus. Therefore, the MAPK pathways may be important molecular targets for elucidating the mechanism of the hippocampal neuronal damage after TBI. However, the role of MAPK phosphorylation after TBI is still not fully understood. Further investigations using specific inhibitors will be necessary to elucidate the role of MAPKs phosphorylation, namely to determine whether activation of MAPK cascades is directly involved with neuronal damage or is simply an epiphenomenon reflecting intracellular signaling events after TBI.
Footnotes
Acknowledgments:
The authors thank Drs. Yoshitaro Matsushita, Kojiro Wada, and Takamoto Suzuki for their excellent technical assistance, and Drs. Hiroshi Katoh, Nobusuke Tsuzuki, and Shoichiro Ishihara for manuscript preparation.