
Abstract
Matrix metalloproteinases (MMPs) may contribute to the pathophysiology of cerebral ischemia by degrading matrix components in the neurovascular unit. In this study, the authors document a pathway by which MMPs interfere with cell—matrix interactions and trigger caspase-mediated cytotoxicity in brain endothelial cells. Hypoxia—reoxygenation induced endothelial cytotoxicity. Cytoprotection with zDEVD-fmk confirmed that cell death was partly caspase mediated. The temporal profile of caspase-3 activation was matched by elevations in MMP-2 and MMP-9. MMP inhibitors significantly decreased caspase-3 activation and reduced endothelial cell death. Degradation of matrix fibronectin confirmed the presence of extracellular proteolysis. Increasing integrin-linked kinase signaling with the β1 integrin-activating antibody (8A2) ameliorated endothelial cytotoxicity. The results suggest that MMP-9 and MMP-2 contribute to caspase-mediated brain endothelial cell death after hypoxia—reoxygenation by disrupting cell—matrix interactions and homeostatic integrin signaling.
Matrix metalloproteinases (MMPs) constitute a family of extracellular proteases that are involved in the remodeling of extracellular matrix (Hartung and Kieseier, 2000; Yong et al., 1998). Although normal MMP activity is required in development and wound healing, a pathologic role for MMPs has also been suggested in acute and chronic neurodegenerative disorders such as stroke, spinal cord injury, Alzheimer's disease, and multiple sclerosis (Campbell and Pagenstecher, 1999; Lukes et al., 1999; Rosenberg 2002; Yong et al., 2001). Dysregulated MMP activity may damage brain by degrading matrix substrates and interrupting cell—cell or cell—matrix homeostatic interactions necessary for cell survival (Chen and Strickland, 1997; Mun-Bryce and Rosenberg, 1998; Tsirka et al., 1997). In particular, two members of the MMP family, MMP-2 and MMP-9, can degrade critical neurovascular substrates such as collagen IV, laminin, and fibronectin, all of which are major components of vascular basal lamina. Neurovascular matrix degradation then leads to blood—brain barrier perturbations, edema, inflammatory infiltration and parenchymal injury (Asahi et al., 2001a; Mun-Bryce and Rosenberg, 1998; Rosenberg 2002; Yong et al., 2001). More recently, it has been shown that MMP-9 may also directly trigger anoikislike neuronal cell death by interrupting cell—matrix survival signaling (Gu et al., 2002).
In a nonhuman primate model of focal cerebral ischemia, regional neuronal injury was statistically correlated with local loss of microvascular integrity (Tagaya et al., 2001), suggesting an important link between vascular and parenchymal compartments in tissue homeostasis. Presumably, stroke disrupts these homeostatic interactions thus triggering cascades of cell death in brain (del Zoppo 2003; Lo et al., 2002, 2003). Although neuronal and glial cell death ultimately mediates brain tissue injury, proximal interactions with endothelial dysfunction play a critical role. However, the molecular mechanisms of cerebral endothelial cell injury after stroke remain to be fully elucidated. Here, we examined the role of MMPs, hypothesizing that upregulation of MMP-2 and MMP-9 after hypoxia—reoxygenation triggers caspase-3–mediated cytotoxicity by interrupting cell—matrix interactions and perturbing homeostatic integrin signaling in human cerebral endothelial cells.
MATERIALS AND METHODS
Cell culture
Human brain microvascular endothelial cells (Cell Systems Corporation, Kirkland, WA, U.S.A.) were grown and exposed to hypoxia—reoxygenation as previously described (Lee and Lo, 2003). For matrix experiments, plates or dishes were treated with 10 μg/mL human plasma fibronectin (Sigma, St. Louis, MO, U.S.A.) and incubated for 2 hours at 37°C before seeding. For inhibitor experiments, BB-94 (British Biotech, London, United Kingdom), GM6001 (Chemicon, Temecula, CA, U.S.A.), human TIMP-1 (Chemicon), and zDEVD-fmk (Enzyme Systems, Aurora, OH, U.S.A.) were added 30 minutes before hypoxia. 8A2 antibody for β1 integrin activation and mouse IgG (Sigma) for control were used.
Gelatin zymography
Culture medium was centrifuged at 10,000g for 5 minutes at 4°C to remove cells and debris. The cleared medium was concentrated using Microcon (Millipore, Bedford, MA, U.S.A.) with a 10-kd pore diameter cutoff, then each sample was mixed with equal amounts of sodium dodecyl sulfate (SDS) sample buffer (Novex, Carlsbad, CA, U.S.A.) and electrophoresed on 10% SDS-polyacrylamide gels (Novex) containing 1 mg/mL gelatin as the protease substrate. Following electrophoresis, gels were placed in 2.7% TritonX-100 for 1 hour to remove SDS, and then incubated for 20 hours at 37°C in developing buffer (50 mmol/L Tris base, 40 mmol/L HCl, 200 mmol/L NaCl, 5 mmol/L CaCl2, and 0.2% Briji 35; Novex). After incubation, gels were stained in 30% methanol, 10% acetic acid, and 0.5% w/v Coomassie Brilliant Blue for 1 hour followed by destaining. To confirm pro and processed active forms of MMP-2 or MMP-9, conditioned medium were treated for 2 hours at 37°C with p-aminophenylmercuric acetate (APMA), which converts zymogen to active enzyme (data not shown). Mixed human MMP-2 and MMP-9 standards (Chemicon) were used as positive controls. Relative gelatinolytic activity (MMP-2 and MMP-9) was quantified and expressed as a ratio of the loaded positive controls via measurement of optical density using the NIH image analysis software.
Western blot analysis
Cultures were rinsed twice with ice-cold phosphate-buffered saline (PBS) and the cells were collected into cell lysis buffer (20 mmol/L Tris, pH 7.5; 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton X-100, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L β-glycerophosphate, 1 mmol/L Na3VO4, 1 μg/mL leupeptin, and 1 mmol/L PMSF). Cell lysates were then homogenized and centrifuged at 10,000g for 15 minutes at 4°C and protein concentration was determined with the Bradford assay (Bio-Rad, Hercules, CA, U.S.A.). Samples were boiled in SDS sample buffer (Novex) with 10 mmol/L DTT and proteins (30 μg/lane) were separated on 4% to 20% SDS-polyacrylamide gels. The separated proteins were transferred to polyvinylidene difluoride membranes (Novex), treated with blocking buffer (Tris-buffered saline containing 0.2% I-block [Tropix, Bedford, MA, U.S.A.] and 0.1% Tween 20). Membranes were probed overnight at 4°C with anticleaved caspase-3 (Cell Signaling Technology, Beverly, MA, U.S.A.) and antifibronectin (Sigma) antibody. Anti—rabbit immunoglobulin G conjugated with horseradish peroxidase was used as the secondary antibody and immune complexes were visualized by enhanced chemiluminescence (Amersham, Piscataway, NJ, U.S.A.).
Lactate dehydrogenase release assay
Hypoxia—reoxygenation induced cytotoxicity was quantified by a standard measurement of lactate dehydrogenase (LDH) release using the LDH assay kit (Roche, Indianapolis, IN, U.S.A.).
DNA fragmentation assay
Genomic DNA isolation was performed using the apoptotic DNA ladder detection kit (Chemicon). Lysed cells with TE buffer were incubated with solution containing RNase A at 37°C for 10 minutes and Proteinase K at 55°C for 30 minutes. DNA was precipitated at -20°C for 2 hours with 3 mol/L ammonium acetate and the DNA ladder was visualized under ultraviolet light with ethidium bromide staining.
Fibronectin fragment analysis
Fibronectin (1 μg) from human plasma (Sigma) was incubated with purified human MMP-2 or MMP-9 (Chemicon) in 50 mmol/L Tris-HCl (pH 7.5), 0.1 mol/L NaCl, and 10 mmol/L CaCl2 buffer at 37°C for 6 hours. For enzyme activation, 10 μmol/L APMA was added in the buffer before reaction. The MMP inhibitor BB-94 was prepared at 1 mmol/L in dimethyl-sulfoxide (DMSO) and used at 1 μmol/L. The reactions were stopped with SDS sample buffer (Novex) with 10 mmol/L DTT and SDS polyacrylamide gel electrophoresis was performed using precast 4% to 20% gels followed by Western blotting.
Integrin-linked kinase (ILK) assay
Equivalent amounts of protein (200 μg) from cell lysates were incubated for 4 hours at 4°C in the presence of anti-ILK antibody (2 μg/mL; Upstate, Lake Placid, NY, U.S.A.). The protein/antibody complex was then precipitated with protein A and centrifuged. The complex was then washed three times with lysis buffer, and two times with kinase buffer containing 50 mmol/L HEPES (pH 7.0), 10 mmol/L MnCl2, 10 mmol/L MgCl2, 2 mmol/L sodium fluoride, and 1 mmol/L Na3VO4. Myelin basic protein (2 μg; Upstate) and ATP were added in kinase buffer, incubated for 20 minutes at 30°C. The reaction was stopped by adding sample buffer and proteins were separated on a 16% SDS polyacrylamide gels followed by Western blot analysis with anti—phospho-MBP antibody (Upstate).
Statistical analysis
Quantitative data were analyzed using analysis of variance followed by Tukey honestly significant difference tests between individual groups. Data were expressed as mean ± SD. A value of P < 0.05 was considered significant.
RESULTS
Hypoxia—reoxygenation induces caspase-3-mediated cytotoxicity in human cerebral endothelial cells
Cell death was quantified by measurement of LDH, a stable cytoplasmic enzyme released into the cell culture medium. Although this assay does not distinguish between necrotic or apoptotic cell death, it is a generally accepted cumulative index of cytotoxicity in cell culture systems where eventually cell lysis occurs independent of the precise death pathways involved. Cell death increased upon reoxygenation for periods of more than 12 hours after 4 hours hypoxia (Fig. 1A). The presence of DNA laddering suggested the role of a caspase-mediated apoptoticlike mechanism (Fig. 1B). Concomitantly, caspase-3 cleavage and activation occurred over the reoxygenation period (Fig. 1C). To confirm the involvement of caspase-3 in this model of hypoxia—reoxygenation-induced endothelial cell death, we tested the effects of caspase-3 inhibition. Pretreatment with zDEVD-fmk 30 minutes before the start of hypoxia significantly ameliorated cytotoxicity in a dose-dependent manner (Fig. 1D).
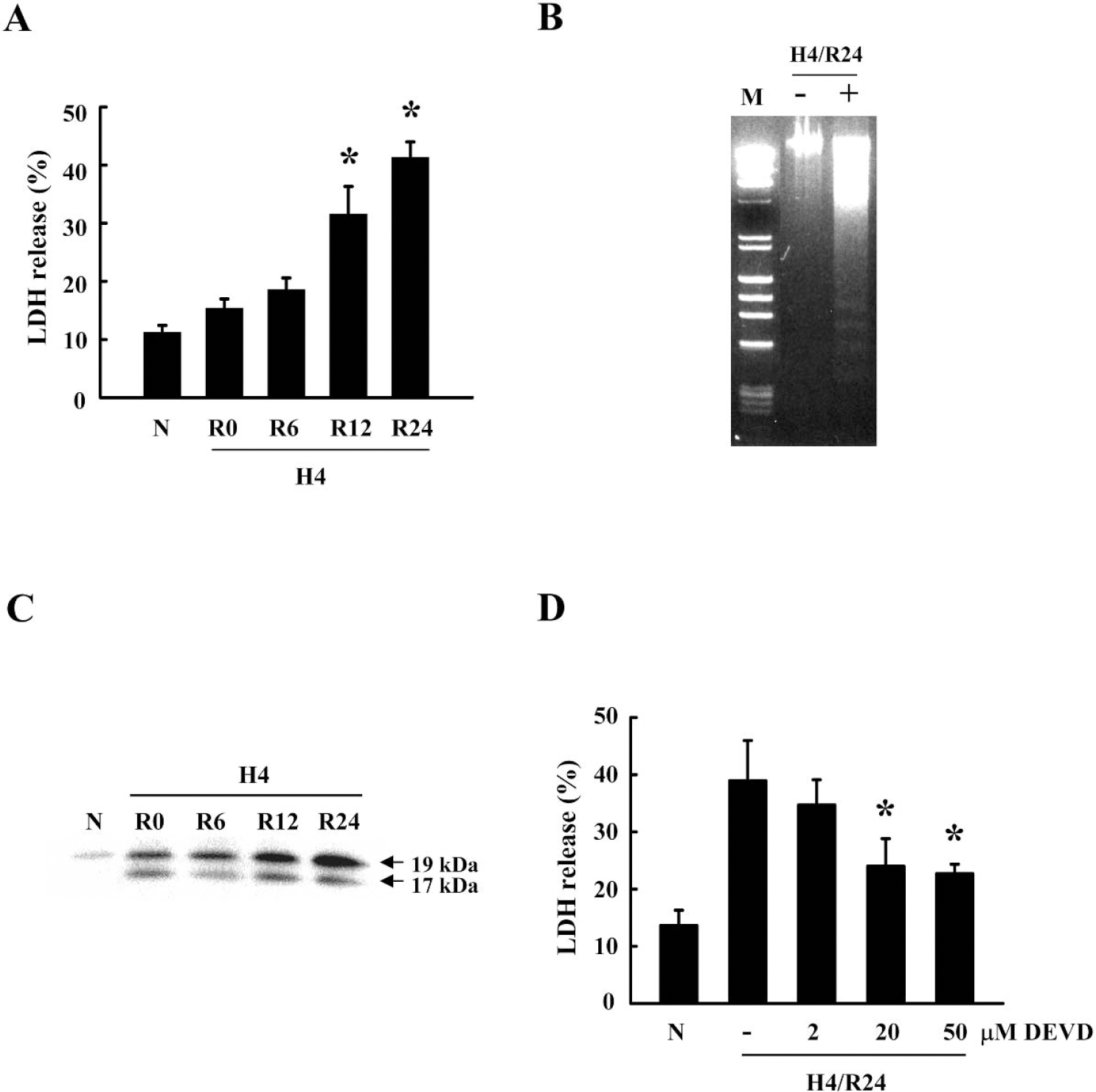
Involvement of caspase-3 in brain endothelial cell death after hypoxia—reoxygenation. (
Endothelial cell death is mediated by MMPs after hypoxia—reoxygenation
Gelatin zymography showed that baseline levels of MMP-2 but not MMP-9 were detectable in conditioned media under normal conditions (Fig. 2A, lane 2). Hypoxia—reoxygenation increased both MMP-2 and MMP-9 production. Total MMP-2 levels including both pro and processed active forms continuously increased during reoxygenation for up to 24 hours (Fig. 2A and Fig. 2B). In contrast, only processed active forms of MMP-9 were elevated, reaching a level of twofold over baseline levels after 24 hours (Fig. 2A and Fig. 2C).
Since the promoter regions of many MMP genes are known to contain nuclear factor kB (NF-kB) transcription factor sites, we tested the effects of the relatively selective NF-kB inhibitor SN50 in our system. Gelatin zymography showed that pretreatment with SN50 significantly reduced MMP-9 in a dose-dependent manner (Fig. 2D). No changes were observed for MMP-2 (data not shown).
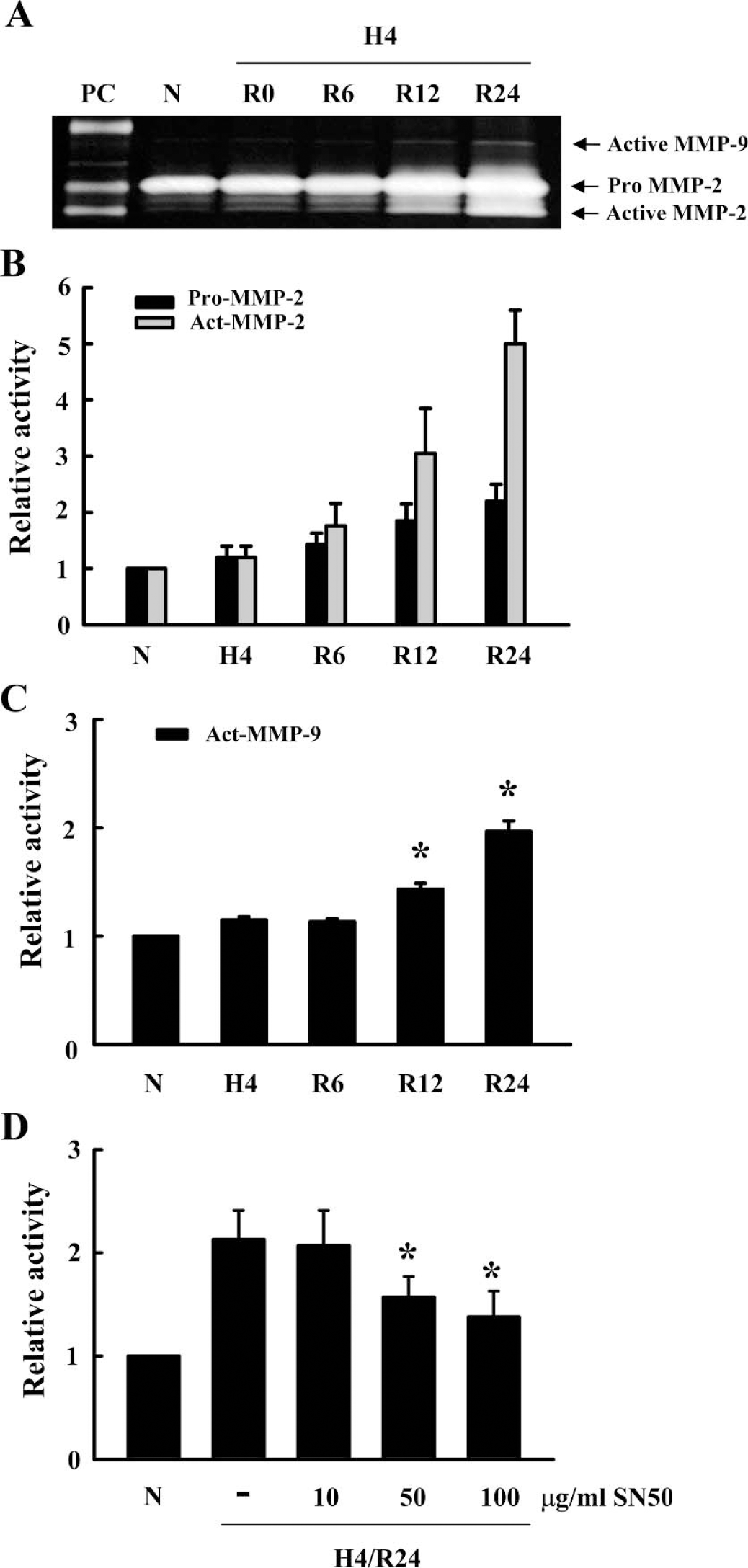
Upregulation of endothelial MMP-2 and MMP-9 after hypoxia—reoxygenation. (
To show that MMPs were involved in brain endothelial cytotoxicity, we tested two broad-spectrum metalloproteinase inhibitors, BB-94 (1 μmol/L) and GM6001 (5 μmol/L), as well as the endogenous tissue inhibitor of metalloproteinase recombinant TIMP-1 (2 μg/mL). All inhibitors significantly reduced caspase-3 activation (Fig. 3A) and cell death (Fig. 3B) after hypoxia—reoxygenation.
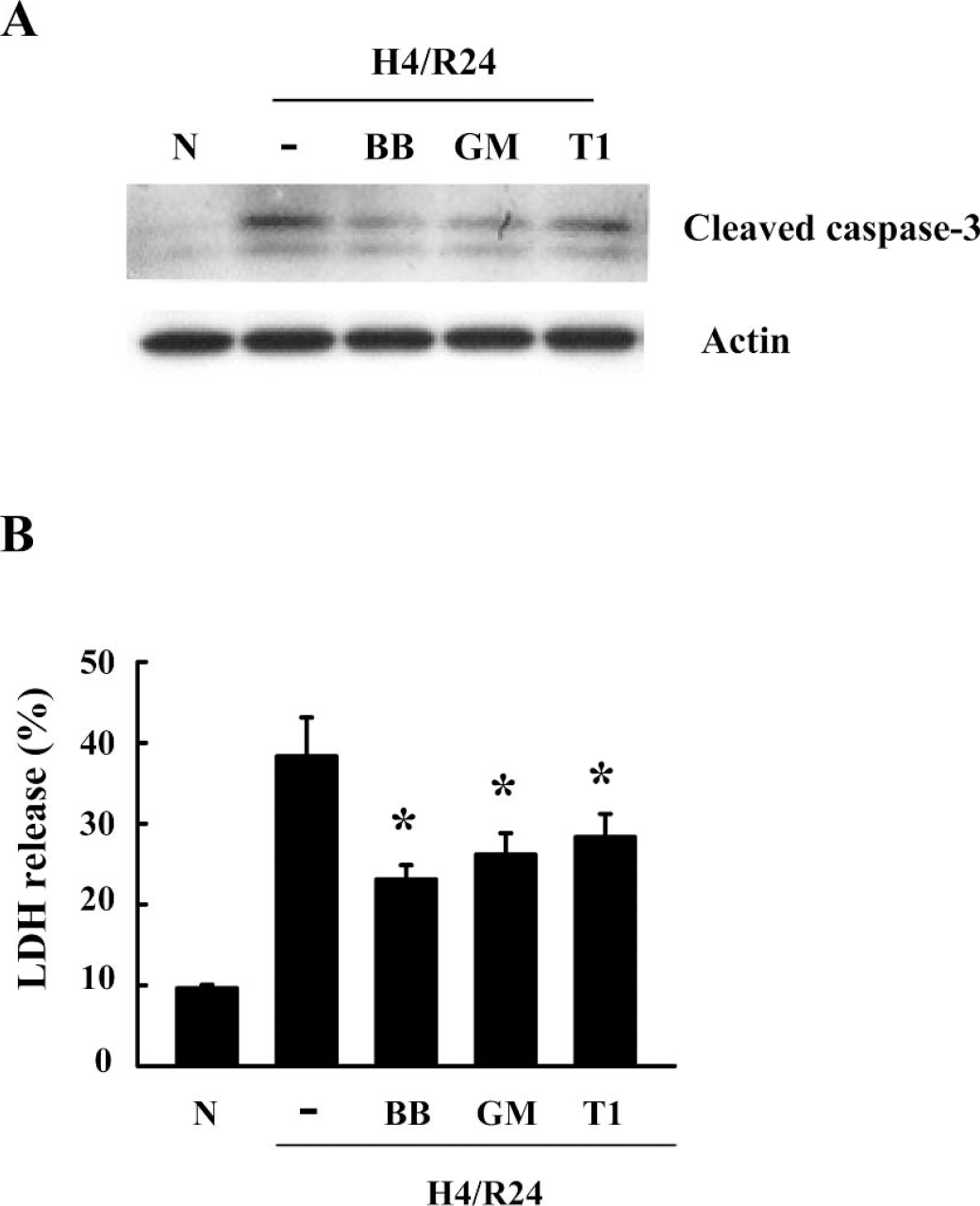
Effects of MMP inhibitors on caspase-3–mediated endothelial cell death. Cells were pretreated with 1 μmol/L BB-94 (BB), 5 μmol/L GM6001 (GM), and 2 μg/mL TIMP1 (T1) for 30 minutes before hypoxia. All MMP inhibitors (
MMPs degrade extracellular matrix and disrupt integrin signaling
Endothelial cells are dependent on adhesion to extracellular matrix for their continued survival. Thus, disruption of appropriate cell—matrix interactions can disrupt homeostasis and initiate cell death. In a cell-free system, exogenously added recombinant MMP-2 (Fig. 4A, left lanes) and MMP-9 (Fig. 4A, right lanes) degraded the matrix protein fibronectin. The metalloproteinase inhibitor BB-94 reduced fibronectin degradation caused by exogenous MMPs, thus confirming the specificity of the response. Next, we tested whether similar events occurred in our brain endothelial cell cultures grown on fibronectin-coated plates. After cells were damaged by 4 hours hypoxia and 24 hours reoxygenation, fibronectin degradation was detected (Fig. 4B). The metalloproteinase inhibitor BB-94 ameliorated fibronectin degradation (Fig. 4B).
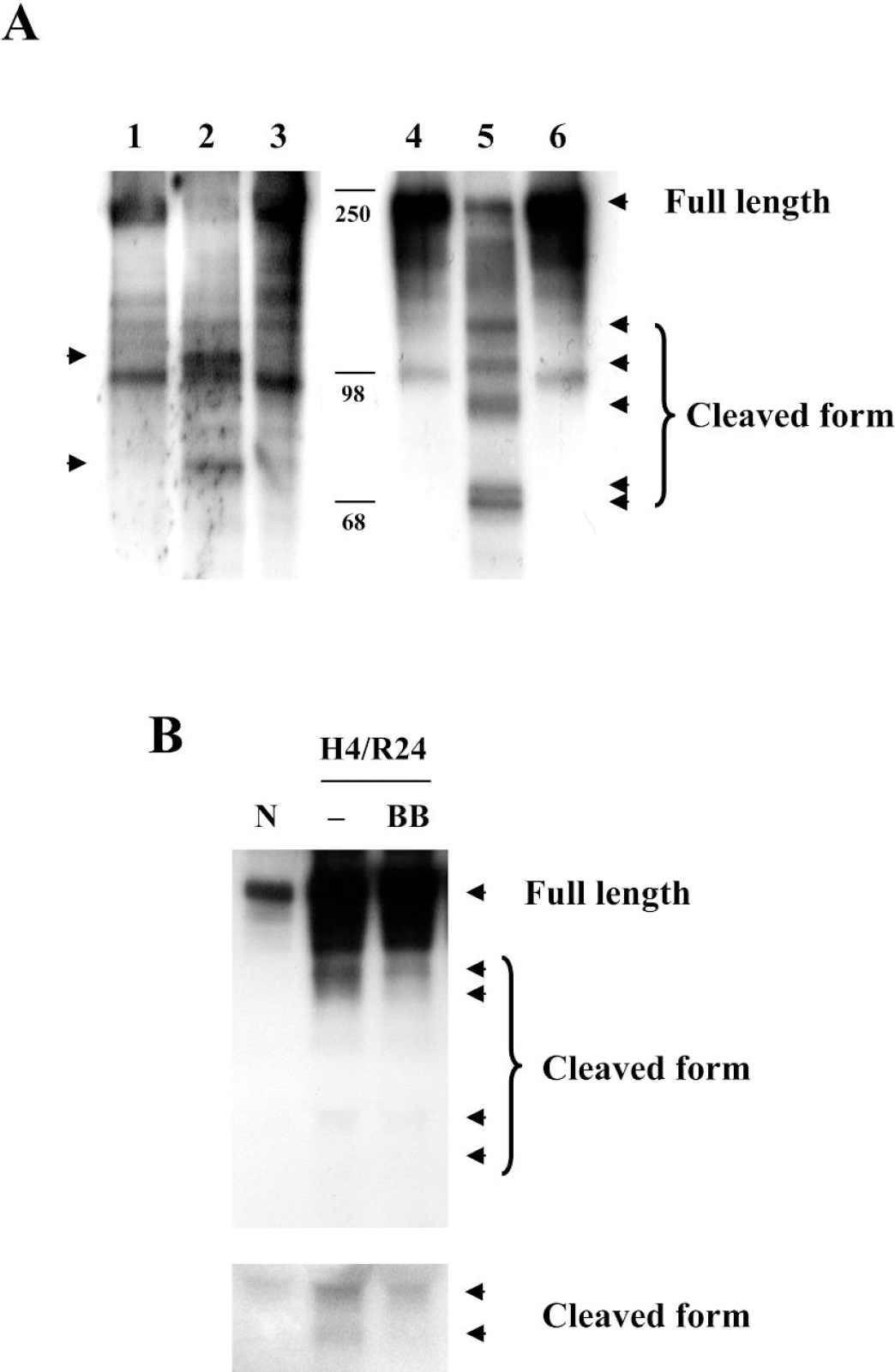
MMP-mediated degradation of matrix fibronectin. (
Integrin-mediated signaling is dependent on homeostatic interactions with extracellular matrix components. Hence, matrix-degrading enzymes such as MMPs may induce anoikislike cell death in our model by disrupting cell—matrix interactions. A major pathway involves matrix fibronectin-mediated integrin signaling. In our brain endothelial model, the β1 integrin-activating antibody 8A2 upregulated integrin-linked kinase (Fig. 5A). Pretreatment with 8A2 (10 μg/mL) at 30 minutes before hypoxia reduced caspase-3 cleavage and activation after hypoxia—reoxygenation (Fig. 5B). Concomitantly, rescue of integrin signaling with 8A2 also significantly ameliorated endothelial cytotoxicity (Fig. 5C).
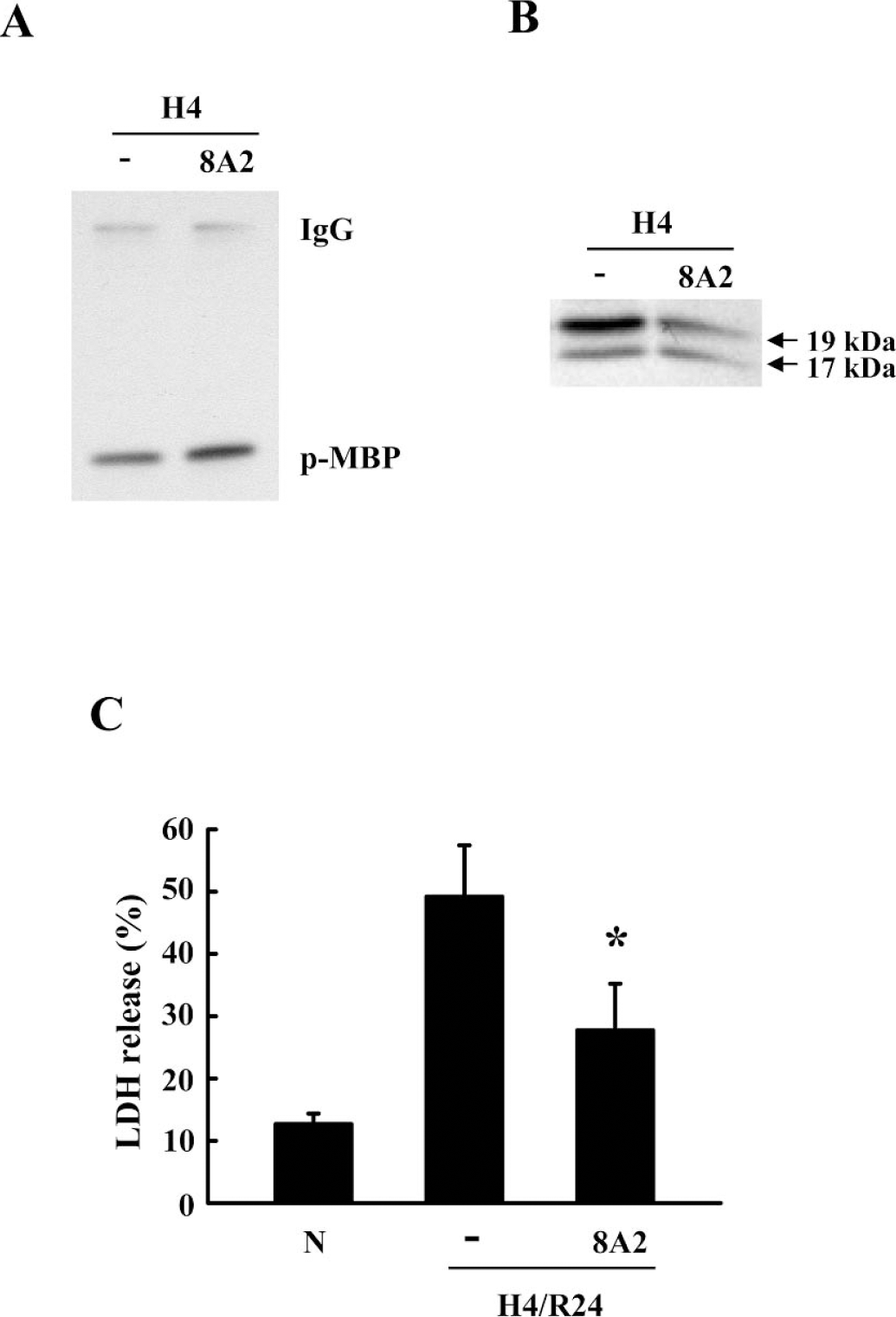
Protective effects of β1 integrin activation. (
DISCUSSION
A singular focus on neuron death pathways alone has not yielded successful stroke therapies (Fisher and Ratan, 2003; Gladstone et al., 2002). An emerging concept in stroke research now proposes that all cells of the neurovascular unit (typically comprising endothelium, astrocytes, and neurons) provide the functional substrate that mediates an integrative tissue response to ischemia and infarction (Lo et al., 2003). This modular concept emphasizes cell—cell and cell—matrix interactions, and further suggests that microvascular reactions to ischemia form a proximal trigger for parenchymal injury (del Zoppo and Mabuchi, 2003; Lo et al., 2003). Therefore, investigations into cerebral endothelial responses in stroke are urgently needed. Here, we used a human brain endothelial cell model to demonstrate that extracellular proteolysis due to dysregulated MMPs degrade cell—matrix interactions, and disruption of homeostatic integrin signaling then promotes caspase-mediated cell death (Fig. 6).
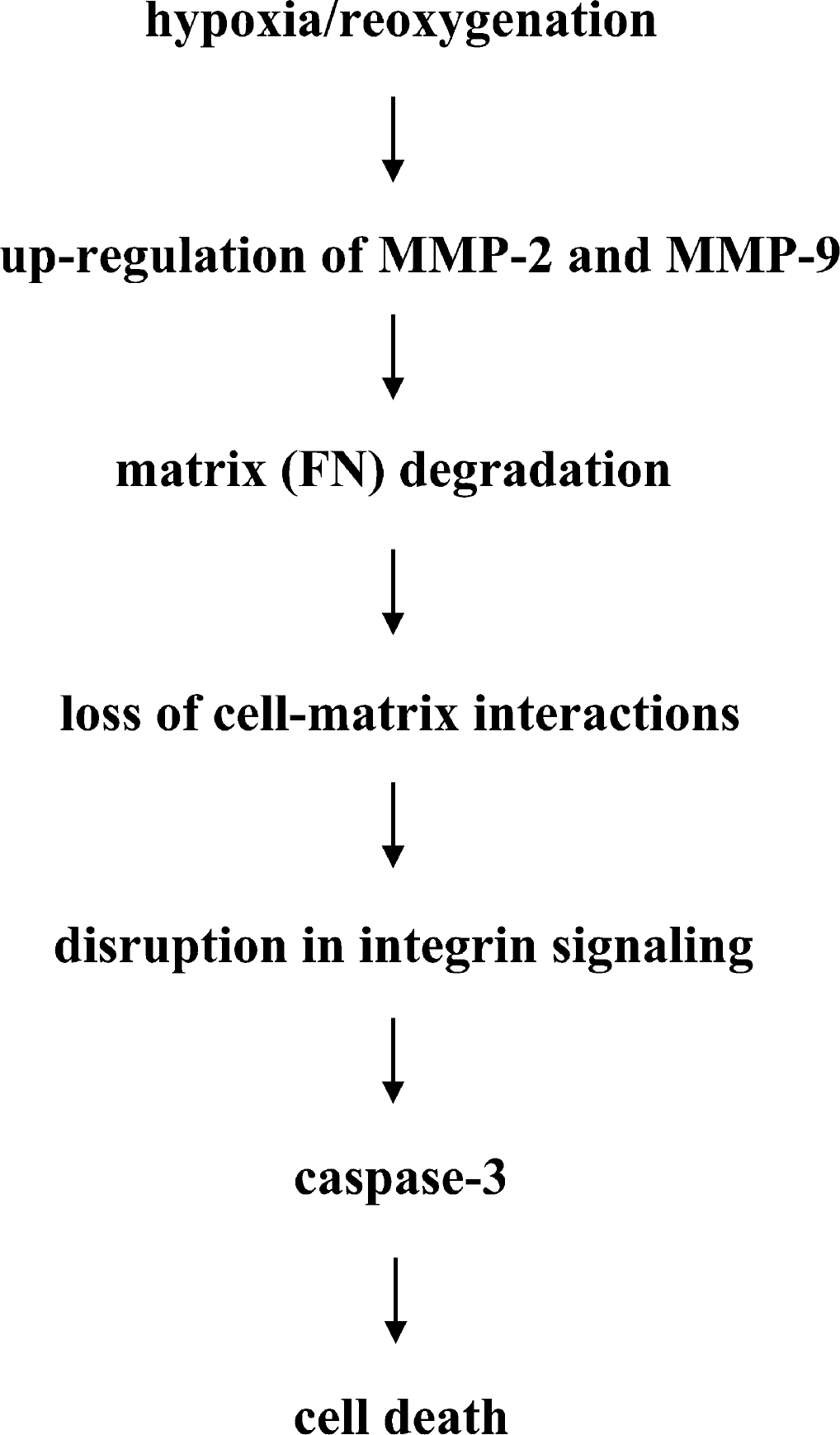
Schematic diagram shows extracellular proteolysis due to dysregulated MMPs that degrade cell—matrix interactions. Subsequent disruption in integrin signaling promotes caspase-mediated cell death in our model.
MMPs, especially the gelatinases MMP-2 and MMP-9, are upregulated in animal models of cerebral ischemia and hemorrhage (Lo et al., 2002; Rogenberg, 2002; Yong et al., 2001), and MMP-9 gene knockout protects against blood—brain barrier leakage (Asahi et al., 2001a; Wang et al., 2000). These findings are consistent with the idea that dysregulated MMPs degrade basal lamina substrates (e.g., collagen, laminin, fibronectin), impair microvascular function, and lead to parenchymal edema and injury. Although vascular matrix degradation and edema are usually considered to be the major pathways for MMP-mediated pathology, direct actions on cells may also occur. For example, MMP inhibition and MMP-9 gene knockout reduced hippocampal neuronal injury after transient global cerebral ischemia, even though blood—brain barrier leakage does not occur in this model (Lee et al., 2004). Extracellular proteolysis promotes anoikislike death in epithelial cells (Frisch and Francis, 1994). It is increasingly recognized that similar anoikislike pathways may operate in neurons as well (Lesay et al., 2001). Loss of neuron—matrix interactions promotes neurotoxicity by downregulating cell-survival integrin-signaling pathways (Gary et al., 2003; Gary and Mattson, 2001). In mouse hippocampus, kainate excitotoxicity is induced by tPA-mediated degradation of neuron—laminin interactions (Indyk et al., 2003; Tsirka et al., 1997). In addition to the plasminogen system, MMPs comprise the other major extracellular protease system in mammalian brain (Lo et al., 2002). MMP-9–mediated disruption of cell—matrix interactions promotes neuronal death (Gu et al., 2002). Here, we showed that similar mechanisms may be involved, insofar as endogenous apoptotic machinery has been previously demonstrated to be present in endothelial cells (Aoudjit and Vuori, 2001; Lin and Maiese, 2001; Meguro et al., 2001; Xu et al., 2001). After hypoxia—reoxygenation, MMP-2 and MMP-9 were upregulated in human brain endothelial cells, and subsequent degradation of extracellular matrix fibronectin appeared to trigger a form of caspase-mediated cytotoxicity. In this study, both cell-free and cell culture systems showed that MMP-2 and MMP-9 can degrade matrix fibronectin.
Fibronectin signaling is required to stave off apoptosis in many cell types (Di Matola et al., 2000; Jeong et al., 2001). A recent study showed that fibronectin knockout mice suffered increased neuronal apoptosis and infarction after cerebral ischemia (Sakai et al., 2001). Although the precise survival signals conveyed by matrix fibronectin remain to be fully dissected, interactions with various integrins are involved. Loss of constitutive integrin signaling recruits caspase-8 and triggers apoptosis (Stupack et al., 2001). The intermediary pathway may involve ILK, which activates the prosurvival signal Akt (Gary et al., 2003). Indeed, ILK protects a venous endothelial cell line against oxidized low-density-lipoprotein—induced cytotoxicity (Zhang et al., 2001). Here, we showed that directly activating β1 integrin with 8A2 antibody (Seki et al., 1996) increased ILK signaling, decreased caspase-3 levels, and ameliorated cell death.
The present finding linking MMPs to endothelial cytotoxicity is important because clinical data suggest that patients with elevated MMP-9 levels are prone to cerebral hemorrhage and injury after thrombolytic therapy (Horstmann et al., 2003; Montaner et al., 2003). Nevertheless, there may be a few caveats worth considering. First, although gel zymograms showed elevations in active MMP-2 and MMP-9 protein, we did not measure mRNA and so cannot define the mechanisms of MMP upregulation. However, hypoxia—reoxygenation induces transcriptional responses in gelatinases (Ben-Yosef et al., 2002; Kolev et al., 2003). NF-kB mediates MMP-9 regulation (Horstmann et al., 2003; Wang et al., 2003), and we showed that the NF-kB inhibitor SN50 downregulated MMP-9 in our model. Secondly, we cannot distinguish between MMP-2 and MMP-9 effects. Active forms of both gelatinases are increased in our model, and the MMP inhibitors used here (BB-94, GM6001 and recombinant TIMP1) can inhibit both proteases. Future studies using specific RNAi strategies may be required to clarify this point. However, it should be noted that MMP-9–knockout mice are protected against focal ischemia (Asahi et al., 2000, 2001a), whereas MMP-2 knockouts are not (Asahi et al., 2001b), suggesting a dominance for MMP-9 in this system. A third question relates to quantitation. Exactly how much of brain endothelial cell death seen here is accounted for by MMPs and caspase? MMP inhibitors reduced caspase activation in our model, but neither MMP inhibitors nor caspase inhibitors completely prevented cytotoxicity. Only about 30% to 40% of cell death was ameliorated. These data suggest that although MMP-mediated phenomena comprise an important portion of the lethal events after brain endothelial injury, other parallel pathways operate as well, further underscoring the need for combination therapies in stroke. For example, cross-talk between calpain and caspase proteolytic systems have been implicated in neuronal cell death (Chen et al., 2002; Rami, 2003; Neumar et al., 2003; Wang, 2000), and the use of calpain inhibitors may have significant cytoprotective properties here. In addition to the outside-in integrin signals delineated here, activation of extrinsic death pathways via Fas and tumor necrosis factor-α receptors may also amplify intrinsic mitochondrial death mechanisms (Eldadah and Faden, 2000; Ferrer and Planas, 2003; Plesnila et al., 2001; Wallach et al., 1999). Once death cascades are initiated, other caspase-independent signals may contribute, including the recently defined apoptosis-inducing factor (AIF) and poly-ADP-ribose polymerase (PARP) pathways (Ferrer and Planas, 2003; Susin et al., 1999; Yu et al., 2002, 2003). A fourth potential limitation concerns our cell culture model. Although the human brain endothelial system has been used before, the focus here on cell—matrix interactions may be hampered by the two-dimensional nature of the model. It is impossible to recapitulate all aspects of three-dimensional extracellular matrix and brain cell structure in cell culture. In vivo, more complex and redundant pathways may operate.
Finally, although our simplified schema in Fig. 6 suggests a linear pathway for brain endothelial injury, we aware that multiple mechanisms become activated after stroke. Endothelial cells undergoing apoptosis become more prone to interact with platelets and leukocytes (Bombeli et al., 1999; Schwartz et al., 1999), which further perturbs cerebral perfusion. MMPs may also cleave and liberate proinflammatory signals such as tumor necrosis factor-α and ICAM-1 (Rosenberg, 2002; Yong et al., 2001), hence promoting secondary tissue injury. Here, we showed that MMP inhibition decreased levels of caspase-3, but caspases can in turn cleave and activate MMPs (Levkau et al., 2002), so that positive feedback loops may occur. Furthermore, kinase signaling pathways are notorious for having complex cross-talk so that much more than ILK will be involved, including prosurvival or prodeath MAP kinase cascades (Griendling et al., 2000). Ultimately, however, these data show a central role for MMP-mediated caspase cell death in brain endothelium, which may comprise an important upstream trigger in stroke.
In summary, we document a role for MMPs in caspase-mediated brain endothelial cell death after hypoxia—reoxygenation. Extracellular proteolysis degrades cell—matrix interactions and disrupts integrin signaling. Further investigations into how MMP's disrupt cell—cell and cell—matrix homeostasis within all cells of the neurovascular unit are warranted. A deeper understanding of how endothelial injury and death in brain affect neurovascular function may reveal new therapeutic opportunities not only for stroke, but perhaps also for other neurodegenerative conditions where vascular dysfunction might be involved.
Footnotes
Acknowledgments
The authors thank Ken Arai and Xiaoying Wang for many helpful discussions, and are extremely grateful to John Harlan for kindly providing the 8A2 integrin-activating antibody.