
Abstract
Cerebral ischemia and intracerebral hemorrhage cause extensive damage to neurons, disrupt the extracellular matrix, and increase capillary permeability. Multiple substrates participate in the cellular damage, including free radicals and proteases. Matrix metalloproteinases and serine proteases are two classes of proteases that are normally present in brain in latent forms, but once activated, contribute to the injury process. These enzymes have a unique role in the remodeling of the extracellular matrix and in the modulation of the capillary permeability. Intracerebral injection of the matrix metalloproteinase, type IV collagenase, attacks the basal lamina around the capillary and opens the blood—brain barrier, Extracellular matrix-degrading proteases are induced by immediate early genes and cytokines, and regulated by growth factors. Activity of the matrix metalloproteinases is tightly controlled by activation mechanisms and tissue inhibitors of metalloproteinases. During ischemia and hemorrhage, multiple matrix metalloproteinases and serine proteases are produced along with their inhibitors. These proteolytic enzymes are involved in the delayed injury that accompanies the neuroinflammatory response. Synthetic inhibitors to metalloproteinases reduce proteolytic tissue damage, and may limit secondary neuroinflammation.
Keywords
Cells die when their membranes undergo irreversible changes. Ischemia and intracerebral hemorrhage initiate complex changes that result in release of glutamate, increase in intracellular calcium, and activation of intracellular proteolytic processes (Chan et al., 1984; Choi, 1992; Ginsberg and Pulsinelli, 1994). Free radicals and proteases are induced after a delay and cause irreversible damage to cell membranes. Shortly after ischemia begins, cells swell as the energy sources are depleted. A secondary, inflammatory response follows, which is fueled by the release of cytokines, chemokines, and immediate early genes. These mediators activate astrocytes and microglia, which release extracellular matrix-degrading proteases. Much has been learned about the manner in which free radicals participate in this process, but less is known about the role of the proteases. Recently, immunohistochemistry and molecular biology techniques have been used to study the role of the proteases in ischemic cell death. Normally, proteases are formed as latent enzymes that require activation. Because they are highly toxic, tight regulation takes place at multiple steps. Acid hydrolases, which are active at acid pH, are proteolytic enzymes that attack a wide spectrum of cellular components. Neutral proteases, such as matrix metalloproteinases (MMP) and plasminogen activators (PA), remodel the extracellular matrix (ECM) normally and attack substrates pathologically as part of the neuroinflammatory response (Matrisian, 1990; Andersson et al., 1992; Birkedal-Hansen et al., 1993).
Extracellular matrix-degrading enzymes act at neutral pH. Stimulation of brain cells with proinflammatory substrates induces MMP production. Neutral proteases remodel the ECM, making them important in normal development. Recently, they were found to be increased pathologically in several brain diseases, such as ischemic infarction (Rosenberg et al., 1996b). In brain, the MMP and serine proteases are secreted by microglia, astrocytes, and endothelial cells. Control of the toxic effects of MMP occurs at multiple steps, including gene induction, activation of latent species, and inhibition of enzymes. The major inhibitors involved in MMP regulation are the tissue inhibitors of metalloproteinases (TIMP) (Matrisian, 1990). This review focuses on the role of the neutral proteases in blood vessel pathology in the neuroinflammatory response, secondary to stroke and intracerebral hemorrhage. Several reviews on the role of MMP in other brain disorders are available (Romanic and Madri, 1994; Rosenberg, 1995; Yong et al., 1998).
BIOLOGY OF EXTRACELLULAR MATRIX AND MATRIX METALLOPROTEINASES
Extracellular matrix components surround brain cells and blood vessels, providing structure and regulating the neuronal microenvironment (Cserr, 1986). A multilayered basal lamina composed of type IV collagen, laminin, fibronectin, and heparan sulfate separates epithelial cells from brain cells (Yurchenco and Schittny, 1990). Glycosaminoglycans, including hyaluronic acid, heparan sulfate, and chondroitin sulfate, make up the remainder of the 15% to 20% of brain tissue that comprises the extracellular space (Margolis et al., 1986). Several new proteoglycans have been identified in the brain, including aggracan, versican, neurocan, and brevican, but their function is unclear (Sobel, 1998). Matrix metalloproteinases and the serine proteases are the primary ECM-degrading proteases that remodel the ECM by breaking down its various components (Reich et al., 1988). Immediate early genes, c-fos and c-jun, lipopolysaccharide (LPS), tumor necrosis factor-α (TNF-α), and interleukin-1β (IL-1β), along with other inflammatory mediators, stimulate MMP and serine protease production. Formed as latent enzymes, they require activation by other proteases and free radicals. Tissue inhibitors of metalloproteinases and PA inhibitors are also produced by the tissues to control the proteolytic process. The balance between the proteases and the inhibitors is critical for normal ECM turnover. Shifting the balance to the activated forms of the enzymes results in excessive proteolytic activity. On the contrary, when inhibitory forces predominate, ECM buildup can result in fibrosis. For example, an increase in MMP is seen in heart tissue from young rats with aldosterone-salt hypertension, whereas in aging normotensive rats, a reduction of MMP was seen, which may explain the fibrosis seen in the blood vessels (Robert et al., 1997). Fibrotic thickening of the basement membrane in the kidney tubular epithelium is related to an imbalance in the metalloproteinases (Carome et al., 1993). Cultured proximal tubular epithelial cells exposed to 1% oxygen for 24 hours have decreased MMP activity and increased TIMP, suggesting that hypoxia may be important in stimulating fibrosis (Orphanides et al., 1997). Thus, here is growing evidence that the remodeling of the ECM around blood vessels in kidney and heart is related to the balance between the proteases and the inhibitors, suggesting a similar situation exists in brain tissues.
Matrix metalloproteinases are a gene family with 14 members (Birkedal-Hansen et al., 1993; Yong et al., 1998). Several MMP have been identified in brain tissue, including gelatinases, stromelysin, and metalloelastase. For classification purposes they are divided into four main groups on the basis of their protein structure (Fig. 1). Matrilysin (MMP-7) is the smallest of the MMP, containing only the propeptide region and the catalytic domain, but it degrades most ECM components. Stromelysins (MMP-3, −10, and −11) have a hemopexin domain; they are active against many matrix macromolecules and activate other proteases. Gelatinase A (72-kDa type IV collagenase or MMP-2) and gelatinase B (92-kDa type IV collagenase or MMP-9) have a fibronectin binding domain. They specifically attack type IV collagen, laminin, and fibronectin, which are the major components of the basal lamina around cerebral blood vessels. Membrane-type metalloproteinases (MT-MMP), which activate MMP-2, have both transmembrane and furin binding domains.
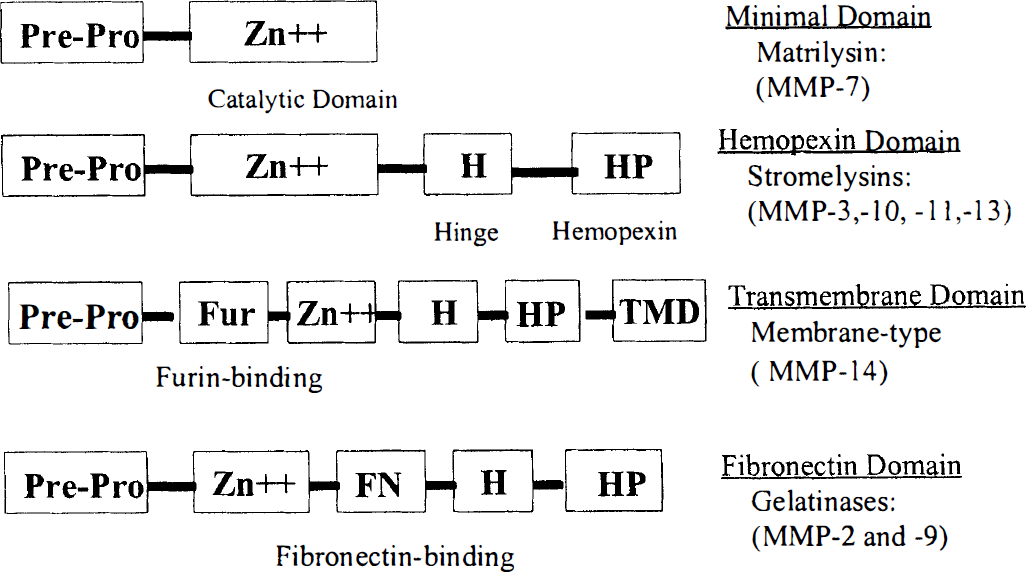
Classification of the matrix metalloproteinases (MMP) based on the different protein domains. Matrilysin has the simplest domain structure with a prepeptide and prodomain and the catalytic, zinc-containing domain. When the hemopexin domain is connected with a hinge protein, the stromelysins are formed. Membrane-type matrix metalloproteinases are formed by the addition of a furin-binding domain and transmembrane region. Finally, the gelatinases have the unique fibronectin-binding region, allowing them to bind to the basement membranes.
Astrocytes, microglia, and endothelial cells produce MMP in culture. Early studies showed that endothelial cells produce gelatinases when stimulated with phorbol esters (Herron et al., 1986a, 1986b). Brain tumor cells and fetal astrocytes also have the capacity to synthesize gelatinases when stimulated (Apodaca et al., 1990). Astrocytes constitutively produce gelatinase A, and synthesize gelatinase B in response to an inflammatory stimulus (Gottschall and Yu, 1995). Microglial cells in culture secrete gelatinase B when stimulated by LPS (Colton et al., 1993; Gottschall et al., 1995).
Although the gelatinases have been most intensively studied, several other MMP and the serine proteases, elastase and PA, may be involved in tissue disruption. Gelatinase A is a constitutively expressed enzyme that is normally present in the brain and CSF. On the contrary, gelatinase B is normally absent, but can be induced under proinflammatory conditions. Stromelysin and the related but smaller molecule, matrilysin, degrade multiple components of the ECM, including components of the basal lamina. Stromelysin may also contribute to the activation of the other proteases (Nagase, 1997). Membrane-bound metalloproteinase activates gelatinase A and is overexpressed in some brain tumors (Sato et al., 1994; Yamamoto et al., 1996).
The promoter region of the gelatinase B gene contains activator protein-1 (AP-1) and nuclear factor-κB sites that respond to a variety of inflammatory stimuli (Fig. 2). Immediate early gene products, including c-FOS and c-JUN, form a heterodimer that stimulates gelatinase B production by binding to the AP-1 site. Lipopoly saccharide acts through both AP-1 and nuclear factor-κB sites. Steroids block gelatinase B production by inhibiting the binding of the FOS/JUN heterodimer to the AP-1 site (Jonat et al., 1990). Regulation of the gelatinase A promoter region involves silencer and AP-2 sites. Regulation also occurs at the posttranscriptional level. Inhibition of protein synthesis blocks translation of the message.
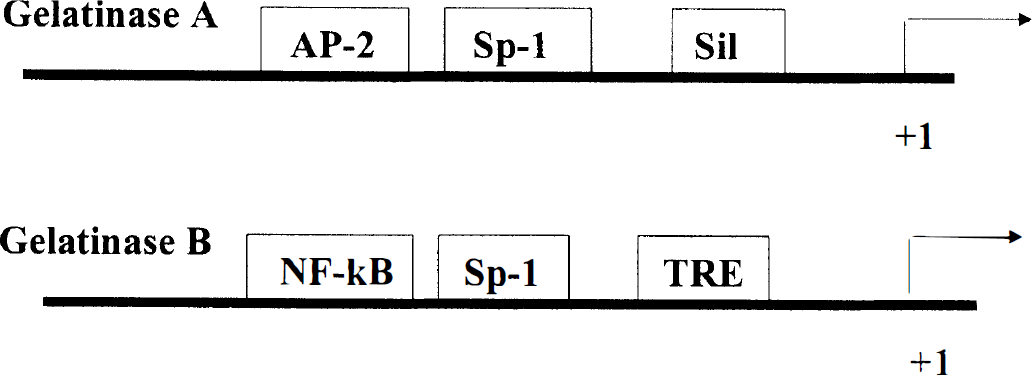
Promoter region of the gelatinase genes. The constitutively expressed gelatinase A (72-kDa type IV collagenase) has an AP-2 site, a Sp-1 site, and a silencer (sil) region. It is most likely involved in the remodeling of the extracellular matrix. The gelatinase B (92-kDa type IV collagenase) has the proinflammatory elements, nuclear factor-κB, Sp-1, and TRE (AP-1 site). The inflammatory elements respond to the FOS/JUN heterodimer, tumor necrosis factor-α (TNF-α), and lipopolysaccharide, and are blocked by the steroid—steroid receptor complex.
Activation mechanisms for the gelatinases in vivo are unknown (Nagase et al., 1996). Some information has been obtained from studies in cell cultures. In other tissues the main activator of MMP-2 is MT1-MMP, which is localized to the cell surface (Sato et al., 1994). Activation involves attachment of TIMP-2 to MT1-MMP. Other proteases appear to be important in the activation of MMP-9. Possible activators include plasmin or stromelysin (Nagase et al., 1996). Recently, we found that LPS-injected rat brains produced the activated 84-kDa form of MMP-9, along with the latent 92-kDa form (Mun-Bryce and Rosenberg, 1998). Activated gelatinases are rarely observed in brain tissues of animals assayed for gelatinase after injury. This could be caused by either the rapid breakdown of the highly toxic activated species or their location in spatially confined regions. Another possibility is that they are present in higher concentrations in localized regions, but are diluted during the tissue sampling process. A recent report described the ability of plasmin to activate MMP-2 and MMP-9; cell-to-cell contact was necessary for the activation process to continue (Mazzieri et al., 1997).
Tissue inhibitors of metalloproteinases balance the activity of MMP (Matrisian, 1990; Stetler-Stevenson, 1996). Four TIMP have been described. Each has a predilection for specific MMP, but have the ability to inhibit any of the MMP. They are small proteins, ranging from 21 kDa to 28 kDa in molecular mass, and they have many homologous regions in the gene. Synthetic inhibitors have been developed for the treatment of metastatic cancer and arthritis. Most are hydroxamate-based inhibitors, which nonspecifically bind to the zinc site of MMP (Gordon et al., 1993; Stetler-Stevenson et al., 1996).
Tissue plasminogen activator (tPA) and metalloproteinases interact in matrix remodeling and disruption. Mice lacking the tPA gene show less damage in the hippocampus after intracerebral injection of the excitotoxin, kainate, than wild-type mice, and they have smaller lesions after an ischemic insult (Tsirka et al., 1995; Wang et al., 1998). Plasmin generated by the action of tPA degrades laminin in hippocampal cells injured by kainate injection (Chen and Strickland, 1997). Inhibitors of plasminogen reduce inflammation in experimental allergic encephalomyelitis (Brosnan et al., 1980). Whether these agents would block the inflammatory response in ischemic brain remains to be studied.
MATRIX METALLOPROTEINASES IN INTRACEREBRAL HEMORRHAGE
Intracranial bleeding is either a primary or secondary event in many neurologic conditions. In primary intracerebral hemorrhage (ICH) the mass forms rapidly from the rupture of a blood vessel or from trauma. Secondary or delayed bleeding, on the other hand, occurs with infection, stroke, trauma, and brain tumors. Ischemic infarcts can undergo hemorrhagic transformation. Several mechanisms have been proposed to explain intracranial bleeding. Early neuropathologists postulated that the hemorrhagic mass resulted from either the rupture of an aneurysm on a hypertensive blood vessel or breakdown of the ischemic tissue in an enzymatic reaction (Zulch, 1971). However, a combined autopsy and radiographic study of postmortem injected brains failed to find evidence of Charcot-Bourchard aneurysms in hypertensive patients (Challa et al., 1992). Another mechanism for bleeding is reperfusion after an embolic stroke (Fisher and Adams, 1951). Evidence to support or refute these theories is difficult to obtain because tissues are destroyed by the bleeding, obscuring the origin of the blood.
Computed tomography (CT) and magnetic resonance imaging (MRI) scanning have lead to a reevaluation of the clinical causes of ICH. An early study before the use of CT showed that when radiolabeled red blood cells were injected into patients with suspected ICH before death, they were absent from the hemorrhagic mass after death, suggesting that progressive bleeding was uncommon (Herbstein and Schaumberg, 1974). However, serial CT scans in patients with a stroke have shown that hemorrhagic mass lesions can enlarge (Hornig et al., 1986; Chen et al., 1989; Okada et al., 1989; Broderick et al., 1990; Bogousslavsky et al., 1991). In patients with an ICH detected within 3 hours of onset, 30% had enlargement of the mass in serial CT scans over 24 hours (Brott et al., 1997). Serial CT and MRI scans in patients with stroke show a rate of hemorrhagic transformation of about 40% (Hornig et al., 1986, 1993). The hemorrhagic transformation may occur without symptoms, but in some instances it is life-threatening. Another cause of intracranial bleeding in stroke is treatment with thrombolytic agents (Wolpert et al., 1993). Recombinant tPA given intravenously within 3 hours of an ischemic stroke increased the risk of significant intracerebral bleeding from less than 1% to more than 6% (Anonymous, 1995).
Intracerebral hemorrhage has been studied in animals by the intracerebral injection of autologous blood. Another method is the intracarotid injection of emboli, which simulates the clinical conditions thought to be associated with bleeding (Lyden et al., 1987). Balloons inserted into the caudate and inflated to 50 µL have also been used to study the mass effect created by an ICH (Sinar et al., 1987). Autologous blood or a balloon simulates the rapid formation of a clot, such as occurs in a traumatic hematoma or the rupture of a vessel with release of a large amount of blood.
Intracerebral injection of bacterial collagenase causes a dose-dependent, highly reproducible hemorrhagic mass (Rosenberg et al., 1990b). Ultrastructural studies have shown that the basal lamina around the cerebral blood vessels, which contains type IV collagen, is disrupted by the bacterial collagenase (Rosenberg et al., 1993). Spectrophotometric measurement of hemoglobin in the brain tissue after a collagenase-induced hemorrhage has been used to show the extent of the bleeding (Choudhri et al., 1997).
As an ICH develops, neuronal tissue is compressed by the accumulating blood and edema. The collagenase-induced hematoma reaches maximal size by 4 hours, but the cerebral edema progresses at the site of the bleed and in distant regions for 24 to 48 hours (Fig. 3). Magnetic resonance imaging can be used to study the growth of the hemorrhage and to measure the effect of drugs on hemorrhage size (Elger et al., 1994; Brown et al., 1995; Del Bigio et al., 1996). Behavioral changes resolve by 2 weeks after a collagenase-induced hemorrhage, making this a model for testing agents that enhance recovery (Chesney et al., 1995; Del Bigio et al., 1996). Little is known about the effect of the mass lesion on the surrounding cells, which are compressed by the expanding mass and exposed to the toxic blood products. Thrombin is released by the hematoma and may be involved in the tissue damage (Lee et al., 1997)
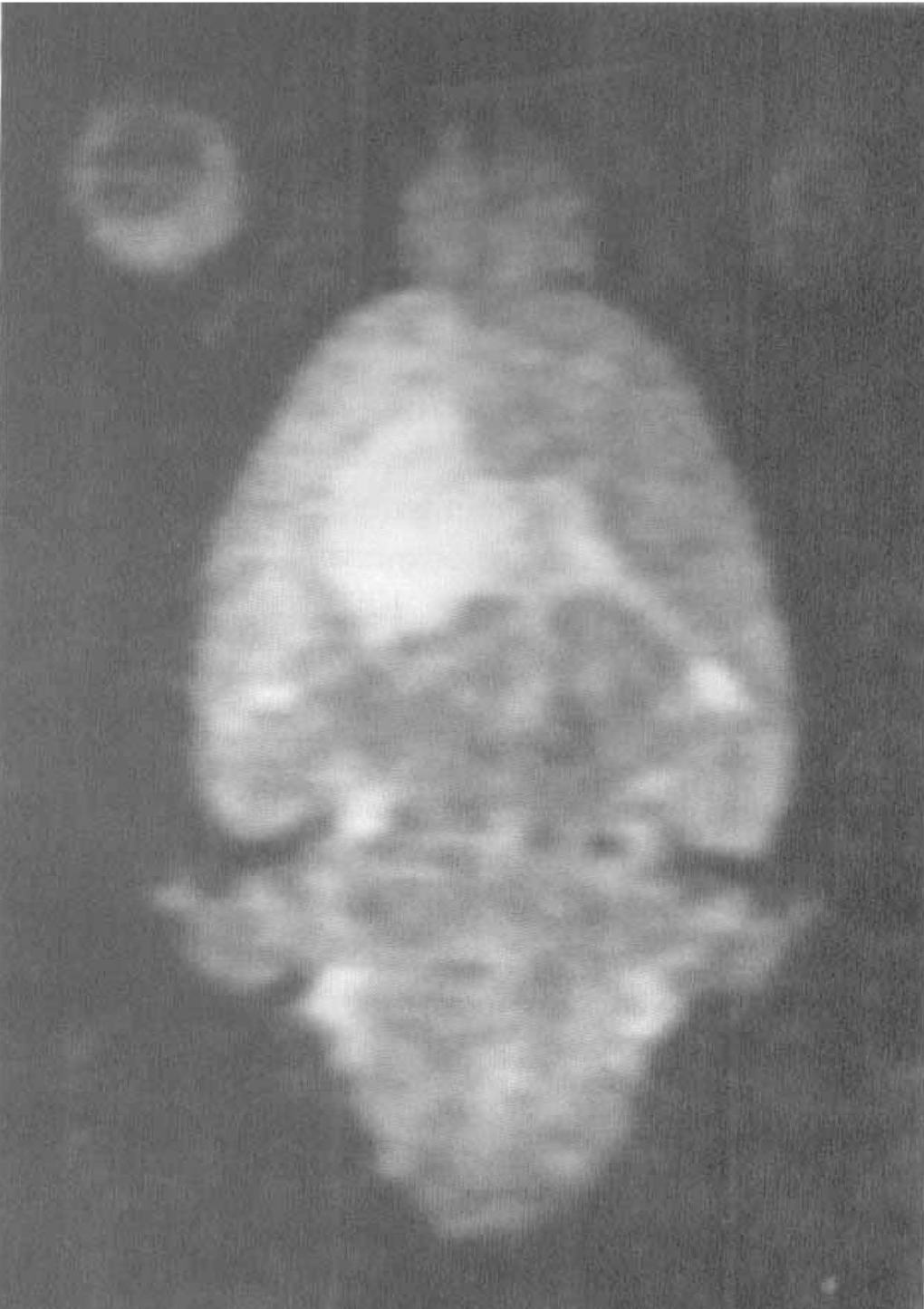
Magnetic resonance imaging scan (T2-weighted) from a rat injected with 0.5 U of bacterial collagenase 48 hours before imaging. The orientation is shown by the eyes in the front of the skull, and the cerebellum in the rear. The hemorrhagic lesion is seen as a hyperintense (white) region in the caudate. Overlying cortex shows the secondary edema (whiter area next to the bleed). There is a shift of the midline structures caused by the mass.
Patterns of edema fluid flow have been studied by autoradiography after a collagenase-induced hemorrhage (Rosenberg et al., 1990a). Radiolabeled tracers injected at the time of hematoma formation are found around the mass and in the opposite hemisphere (Fig. 4). White matter fiber pathways are followed into both posterior regions, primarily over the hippocampi. In the rat a hippocampal commissure connects the two brain hemispheres, forming a conduit that allows edema to move from the side of the hemorrhagic mass into the opposite hemisphere. As humans lack the hippocampal commissure, edema fluid remains in the injured hemisphere. The blood flow to the posterior regions remains normal in spite of the edema (Rosenberg et al., 1992). However, proton magnetic resonance spectroscopy has shown that lactate is formed in the edematous posterior regions and pH remains normal (Mun-Bryce et al., 1993).
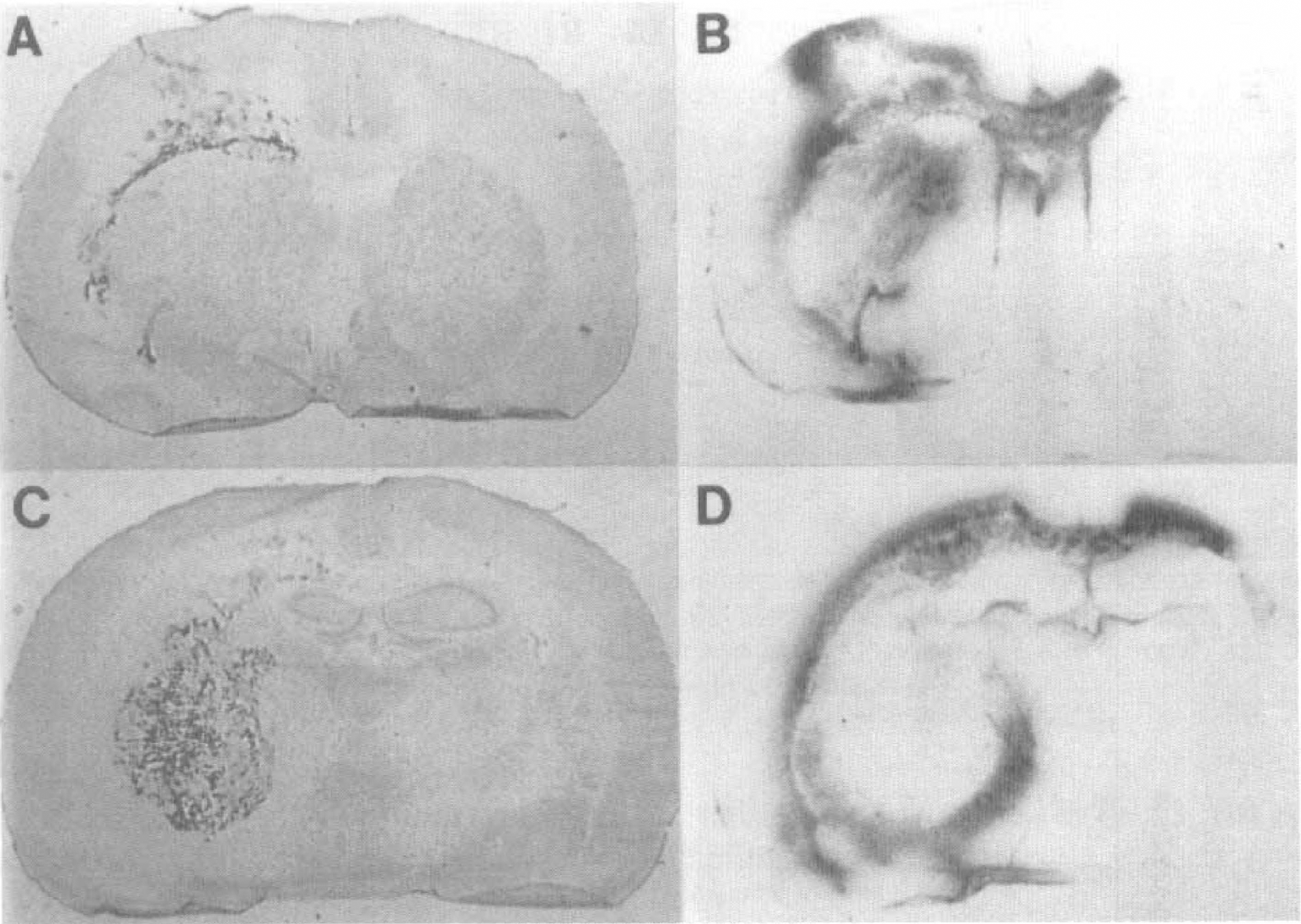
Autoradiograms and histology of collagenase-induced hemorrhage in rat brain.
Proteolytic enzymes are elevated secondary to the bacterial collagenase injury. Brain tissue containing bacterial collagenase lesions was analyzed by gelatin-substrate zymography. Zymograms are electrophoresis gels with gelatin embedded in the gels (Rosenberg and Navratil, 1994). Gelatinases hydrolyze the gelatin in the gels. When the gels are stained for protein, a clear band is seen in the region of the proteolytic activity. By 24 hours the gelatinase activity caused by the bacterial collagenase was decreasing, but new bands began to appear from endogenously produced gelatinases. Delayed production of gelatinase B occurred after several hours (Fig. 5A). At the hemorrhage site gelatinase B was markedly increased with both the latent 92-kDa and the active 84-kDa species observed. Gelatinase B was absent on the uninjured side. Plasminogen activators were also measured (Fig. 5B). By 24 hours after the injury tPA was reduced, and urokinase-type plasminogen activator (uPA) was increased. Levels of 72-, 84-, and 92-kDa gelatinases were significantly higher at the injury site compared with the opposite side. Likewise the differences in tPA and uPA between the two sides were significantly different (Fig. 5C and D). When an inhibitor of MMP was given to rats with bacterial collagenase lesions, a reduction in the secondary production of edema was seen (Rosenberg and Navratil, 1997). A uPA was detected in the injured tissue with the highest amounts seen after several days.
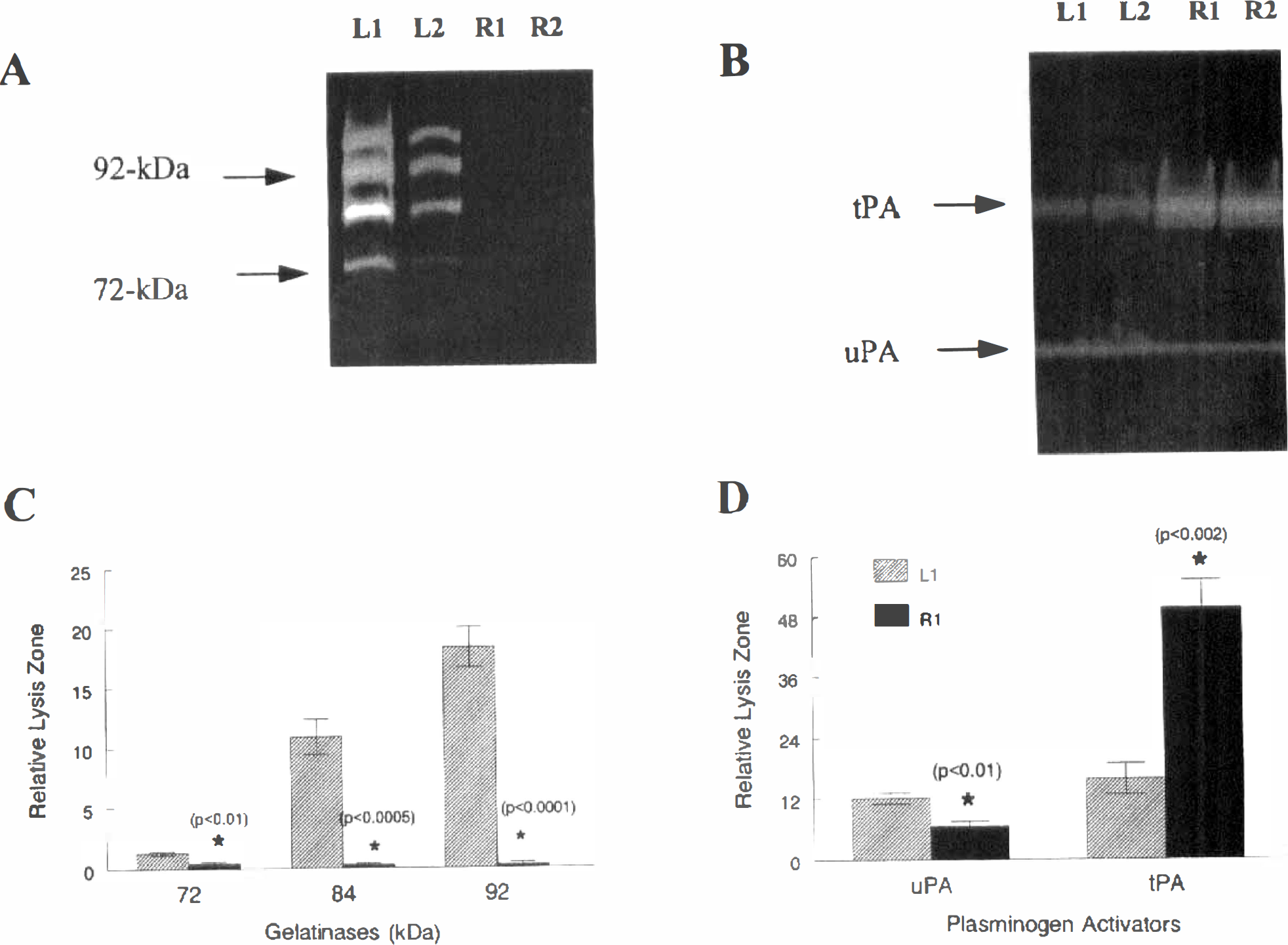
Effect of bacterial collagenase injection on the new production of matrix metalloproteinases. Rats were injected with bacterial collagenase (0.5 U) into the caudate.
A modification of the bacterial collagenase model that added an injection of heparin was recently described (Del Bigio et al., 1996). Neutrophils are associated with the edema, as shown by white matter hyperintensity in T2-weighted images, 12 hours after collagenase-heparin injection. Leukocytes respond to inflammatory cytokines by migrating to sites of damage or infection. These immune cells produce MMP and oxidative radicals (Weiss, 1989). In addition, resident cells of the CNS, including microglia and astrocytes, are also activated by inflammatory mediators, leading to the production of MMP (Sawada et al., 1989; Colton et al., 1993; Lee et al., 1993; Gottschall and Yu, 1995). Secretion of MMP by various cell types can lead to amplification of tissue damage (Birkedal-Hansen, 1993).
MATRIX METALLOPROTEINASES IN CEREBRAL ISCHEMIA
In an ischemic lesion MMP are produced within hours after the onset of permanent occlusion of a blood vessel (Rosenberg et al., 1996b). Gelatinase B levels begin to increase, reaching statistically increased levels by 12 hours, and remain elevated at 24 hours and 5 days (Fig. 6A). Gelatinase A, on the other hand, is normal until 5 days after the infarction, when it begins to rise along with uPA (Fig. 6B). Matrix metalloproteinases have been studied by immunohistochemistry in humans with strokes (Anthony et al., 1997). Gelatinase B was seen in neutrophils by 1 week after the infarct. Later than 1 week macrophages that expressed matrilysin and gelatinase A were observed. Human brain tissue after a stroke shows gelatinase A after several months, whereas gelatinase B is elevated within days after the infarct (Clark et al., 1997).
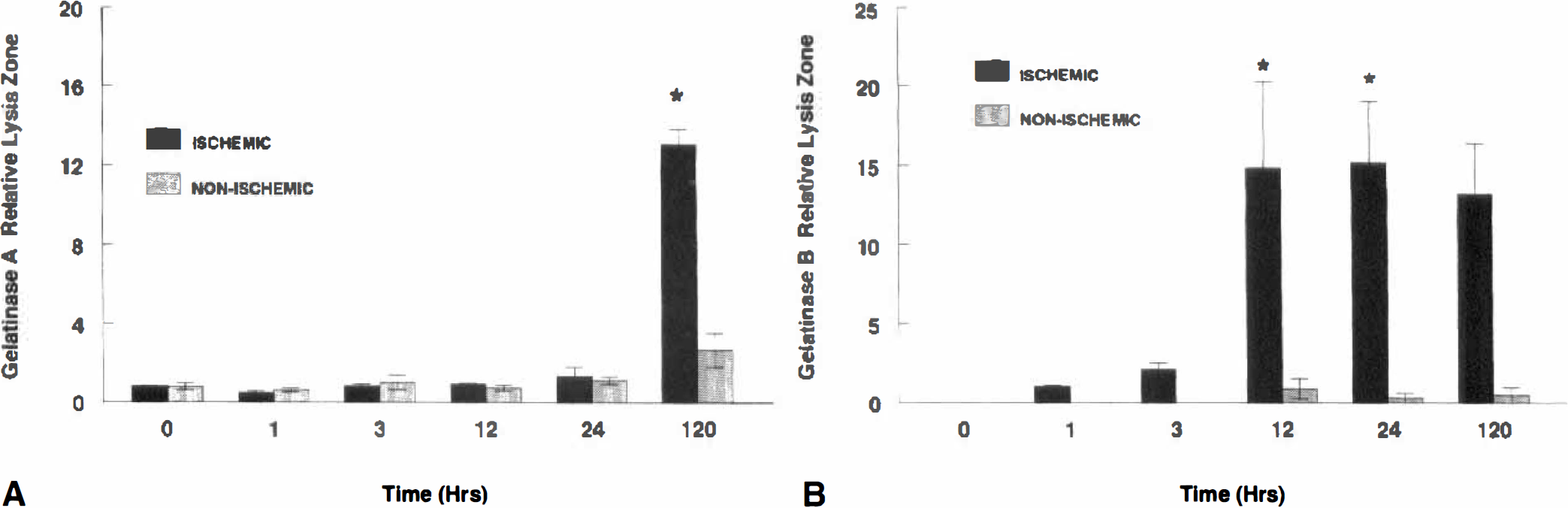
Production of gelatinases in spontaneously hypertensive rats (SHR) at different times after the permanent occlusion of the middle cerebral artery.
Reperfusion adds a new level of complexity to the ischemic damage. Reintroduction of blood with neutrophils and oxygen into the ischemic tissue creates complex changes. When the duration of the ischemic insult is short, restoring blood flow may be beneficial and the tissue damage reversible. Delay in reperfusion, however, leads to reperfusion injury. In rats subjected to 2 hours of middle cerebral artery occlusion, an initial opening of the blood—brain barrier (BBB) was seen after 3 hours of reperfusion (Yang and Betz, 1994). The initial opening occurs after 3 hours during the hyperemic phase. At 48 hours, a second opening of the BBB is seen (Belayev et al., 1996).
Reperfusion increases cerebral edema (Yang and Betz, 1994). The increase in brain water seen by 24 hours is before the time of maximal increase in capillary permeability, suggesting it is because of cytotoxic edema. By the time the capillary leakage is fully developed at 48 hours, the edema has reached maximal levels. Separating the factors affecting vasogenic and cytotoxic edema is difficult. The maturational effect seen in the delayed opening of the BBB may be caused by toxic factors formed by endogenous brain cells, which is amplified by invading leukocytes.
Reperfusion increases the risk of intracerebral bleeding. Multiple factors influence bleeding, including the length of occlusion, the size of the lesion, and the age of the subject. The risk of ICH after thrombolytic treatment has been studied in humans. Older patients showing larger ischemic lesions on CT scan had a higher risk of an ICH when treated with thrombolytic agents (Anonymous, 1995). Only the time between insult and treatment, however, had statistical predictability. Many issues remain unresolved regarding the risks of bleeding. Although it is highly likely that proteolytic attack on the cerebral blood vessel contributes to the tendency toward cerebral edema and hemorrhage, the causative agent is uncertain. If the proteases are brought to the injury site by infiltrating leukocytes, then treatments aimed at leukocyte trafficking would be best. However, if endogenous protease production is found, then agents that interfere with protease production or activation may be needed. Possibly both approaches will be necessary. Finally, the role of free radicals, particularly the production of peroxynitrite, will need to be considered, as it may also add to the capillary injury.
BLOOD—BRAIN BARRIER PROTEOLYSIS
Several proteases have been shown to increase BBB permeability (Robert and Godeau, 1974). Besides MMP, intracerebral injection of elastase opens the BBB, causing perivascular hemorrhages at the injection site (Armao et al., 1997). Another enzyme that may disrupt the BBB is plasmin, which causes a delayed opening of the BBB, leading to fibroticlike lesions at 24 hours after injection. Heparatinase and cathepsin G had the least effects on blood vessel permeability (Armao et al., 1997). Elastase is produced by stimulating microglial cells (Nakajima et al., 1992). However, the in vivo identification of elastases in brain tissue has not been reported, making its physiologic role in brain pathology uncertain. Neutrophil elastase breaks down TIMP and activates MMP, which could enhance the proteolytic response (Janoff, 1985).
Tumor necrosis factor-α, which causes an opening of the BBB, is linked to the expression of gelatinase B (Fig. 7). However, 24 hours elapsed between the injection of TNF-α and the increase in capillary permeability (Rosenberg et al., 1995). Tumor necrosis factor-α prolongs the stimulation by c-JUN of the AP-1 site (Brenner et al., 1989). Intracerebral injection of LPS also leads to opening of the BBB. By 8 hours after LPS injection, increased levels of gelatinase B were found by zymography. An activated species having a molecular weight of 84 kDa was detected by zymography, suggesting that the inflammatory response resulted in the production and activation of gelatinase B. Opening of the BBB was reduced by a metalloproteinase inhibitor, providing further support that the increase in MMP was linked to the BBB injury (Mun-Bryce and Rosenberg, 1998)
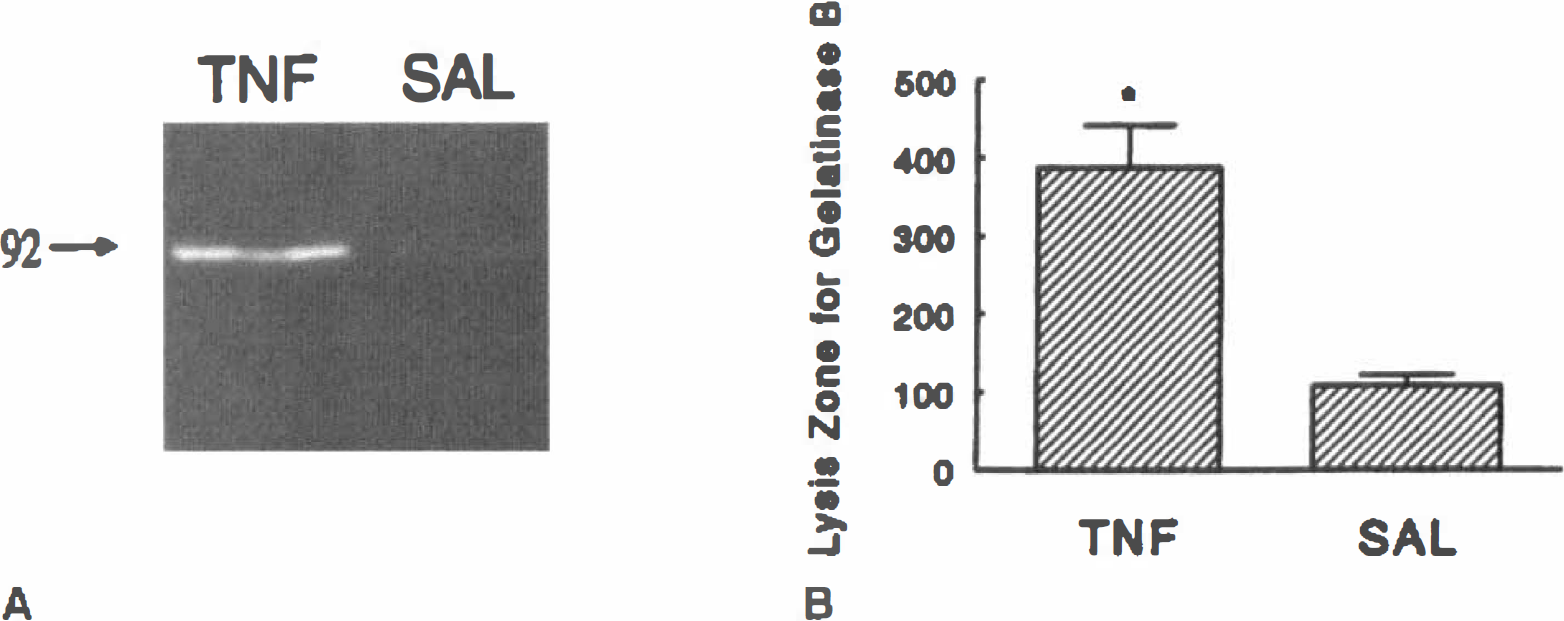
The effect of TNF-α on the induction of MMP is shown. Rats were injected intracerebrally with either TNF-α or saline (SAL).
Our studies on the mechanisms of BBB injury by proteases suggest that the ECM surrounding the endothelial cells is important in the inflammatory damage (Fig. 8). Normally, astrocytes constitutively release gelatinase A into the extracellular space. At times of growth and remodeling, MT-MMP are activated, converting latent gelatinase A into an active form. Excessive proteolytic activity results in unregulated cleavage of type IV collagen and other components of the basal lamina around the cerebral blood vessels. During neuroinflammation, this could be the signal for the endothelial cell to increase its permeability in response to injury and pathogenic threat. Microglial cells become activated. Release of cytokines, such as TNF-α and IL-1β, amplify the injury cascade (Feuerstein et al., 1994; Martiney et al., 1998). Immediate early genes, c-fos and c-jun, are produced and form the heterodimer that activates the AP-1 sites of the gelatinase B gene. Stromelysin is also activated early in this process. Stromelysin, plasmin, and, possibly, free radicals may activate the proinflammatory gelatinase B. Further attack on the basal lamina occurs both from gelatinases released by the endothelial cells and from the astrocytes and microglia. Chemokines and other neutrophil attractants amplify the reaction by recruiting the inflammatory cells to the injury where they release free radicals and proteases. Elastase from neutrophils further disrupts the integrity of the capillary, leading to necrosis and irreversible damage.
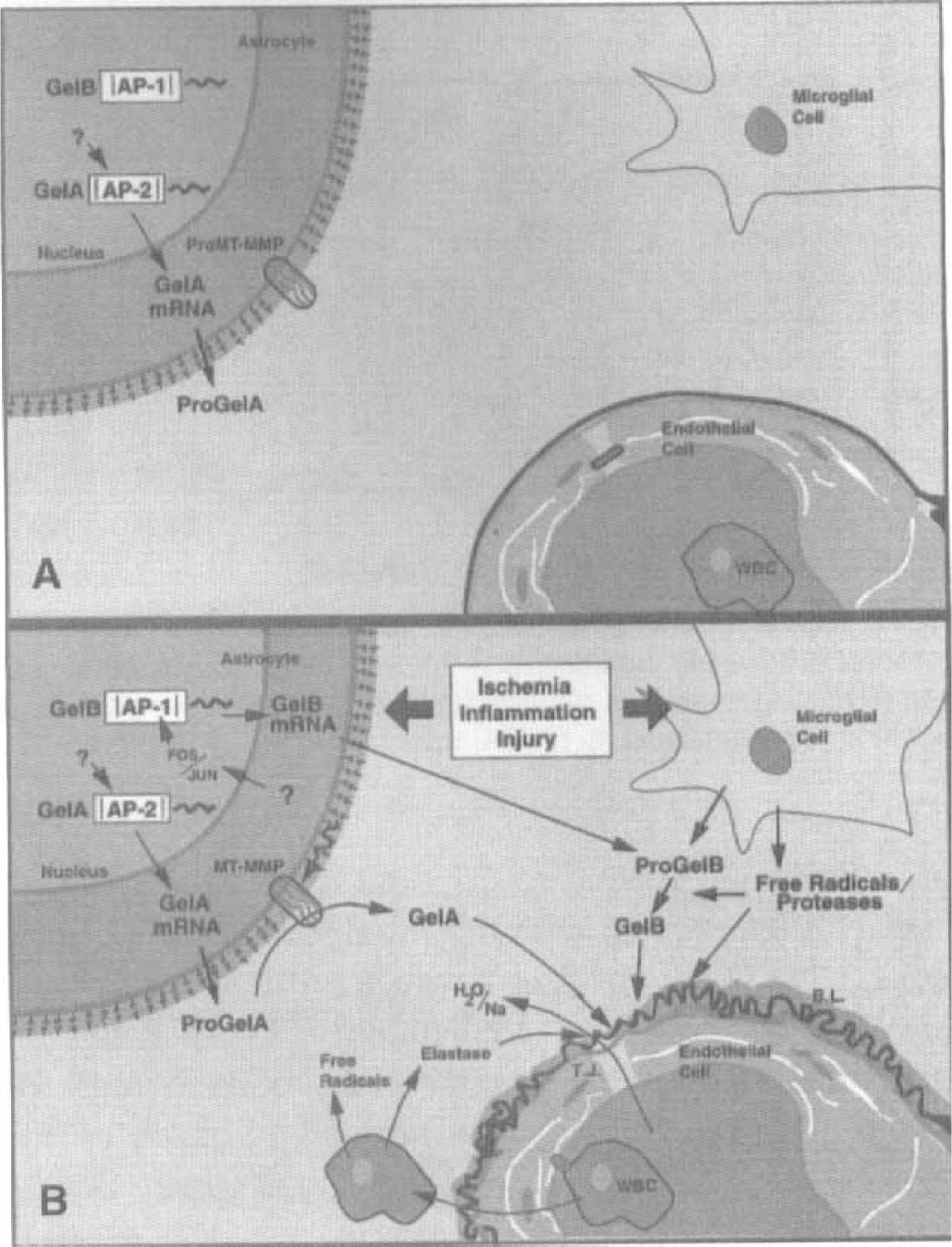
Hypothetical mechanism for the proteolytic opening of the BBB.
Tissue disruption by proteases is an unwanted effect of neuroinflammation. With opening of the BBB components of serum, such as fibrin, thrombin, and complement, cross the endothelial cell into brain tissue. Fibrin leads to activation of PA and conversion of plasminogen into plasmin. When plasmin and complement are combined in the extracellular space, bystander demyelination takes place, which could be the mechanism for vascular demyelination (Cammer et al., 1978). Remodeling of the ECM after the injury leads to fibrosis around the cerebral vessels. Deposited fibrin could trigger fibrinolysis, creating a cycle of blood vessel injury, fibrosis, and fibrinolysis, and ultimately leading to bystander damage to surrounding cells. Although still speculative, such a mechanism could explain the regions of demyelination seen around blood vessels with chronic fibrosis.
We have shown that MMP are increased in cerebral ischemia and hemorrhage. These matrix-degrading enzymes contribute to the acute tissue damage, along with other proteases and free radicals, and to the opening of the BBB. Overproduction of gelatinase B occurs during the early and proinflammatory phases of stroke, whereas gelatinase A is found in the later reparative phases when the blood vessels begin to regrow. Most likely free radicals and other still to be identified proteases, including plasmin, stromelysins, and elastases, will also contribute to the tissue lysis. The challenge will be to unravel the beneficial from the deleterious aspects of the proteolytic changes. Once the time course of the expression of the various proteases and their inhibitors after stroke and hemorrhage is understood, treatments with agents that regulate their action can be planned. Future studies should be directed to understanding the role of the gelatinases and other enzymes, such as stromelysins and elastases, in reperfusion injury and hemorrhage. Finding sites for potential intervention could reduce secondary proteolytic damage and enhance recovery.