
Abstract
Valproic acid (VPA), a histone deacetylase (HDAC) inhibitor, is known to protect against cerebral ischemia. The effects of VPA on blood–brain barrier (BBB) disruption were investigated in rats subjected to transient middle cerebral artery occlusion (MCAO). Postischemic VPA treatment remarkably attenuated MCAO-induced BBB disruption and brain edema. Meanwhile, VPA significantly reduced MCAO-induced elevation of matrix metalloproteinase-9 (MMP-9), degradation of tight junction proteins, and nuclear translocation of nuclear factor-κB (NF-κB). Sodium butyrate, another HDAC inhibitor, mimicked these effects of VPA. Our findings suggest that BBB protection by VPA involves HDAC inhibition-mediated suppression of NF-κB activation, MMP-9 induction, and tight junction degradation.
Keywords
Introduction
Focal cerebral ischemia induces blood–brain barrier (BBB) disruption. The loss of BBB integrity allows intravascular proteins and fluid to penetrate into the cerebral parenchymal extracellular space, thereby incurring vasogenic edema formation and further brain damage. Matrix metalloproteinases (MMPs) mediate BBB disruption and vasogenic edema after cerebral ischemia by degrading the extracellular matrix, basal lamina proteins, and tight junctions around the BBB (Rosell and Lo, 2008). Among MMPs, MMP-2 and MMP-9 have been most intensively studied for their involvement in BBB disruption after stroke (Yang et al, 2007).
Valproic acid (VPA), a histone deacetylase (HDAC) inhibitor, is commonly used for the treatment of seizures and bipolar disorder. VPA has a wide range of neuroprotective properties in cellular and animal models of neurodegenerative diseases (Chuang et al, 2009). In a rat focal cerebral ischemic model subjected to middle cerebral artery occlusion (MCAO), VPA decreases brain infarct volume and neurologic deficits, and exhibits antiinflammatory properties (Kim et al, 2007). The present study was conducted to examine whether postischemic VPA treatment ameliorates transient MCAO-induced BBB disruption and brain edema and, if so, to assess whether these neuroprotective effects are associated with inhibition of HDAC activity and MMP elevation in the brain.
Materials and methods
Middle Cerebral Artery Occlusion and Drug Administration
All animal experiments were performed according to protocols approved by the National Institutes of Health Animal Care and Use Committee. Male Sprague-Dawley rats (230 to 250 g; Charles River Laboratories, Wilmington, MA, USA) were subjected to 60 minutes of MCAO followed by reperfusion for 24, 48, or 72 hours, as previously described (Kim et al, 2007). Sham-operated rats underwent neck surgery without arterial occlusion. VPA (200 or 300 mg/kg; Sigma, St Louis, MO, USA) or sodium butyrate (SB; 300 mg/kg; Sigma) was intraperitoneally injected every 12 hours after first injection, as indicated.
Evans Blue Extravasation
BBB integrity was evaluated by Evans blue extravasation. Evans blue (2% in saline, 4 mL/kg; Sigma) was given intravenously at the onset of reperfusion. Rats were transcardially perfused with saline to remove the intravascular dye at indicated times. Each hemisphere was weighted, homogenized in 2 mL 50% trichloroacetic acid, and then centrifuged (4,000 × g for 40 minutes). The supernatant was measured for absorbance at 620 nm by spectrophotometry. The results were expressed as μg/g tissue calculated against a standard curve.
IgG Extravasation
BBB disruption was also assessed by using a one-step immunohistochemical detection of IgG (Zechariah et al, 2010). Rats were transcardially perfused with saline followed by 4% paraformaldehyde. After blocking of endogenous peroxidase and immersion in 5% normal goat serum, sections (30 μm) were incubated overnight at 4°C with biotinylated goat anti-rat IgG (1:200; Vector Laboratories, Burlingame, CA, USA), and stained with an avidin peroxidase kit and diaminobenzidine (Vector Laboratories). IgG extravasation was densitometrically analyzed by subtracting optical densities in the contralateral from the ischemic hemisphere.
Brain Water Content
Brain water content was examined to assess brain edema. The hemispheres were weighed to obtain the wet and dry weights before and after drying at 100°C for 48 hours, and the percentage of water content was calculated as (wet weight–dry weight) × 100/wet weight.
Gelatin Zymography
The activities of MMP-2 and MMP-9 were measured by gelatin zymography (Yang et al, 2007), using human recombinant pro- and active MMP-2/9 (Calbiochem, Gibbstown, NJ, USA) as standards. Briefly, brain tissues were homogenized in a lysis buffer (50 mmol/L Tris-HCl, pH 7.6, 150 mmol/L NaCl, 5 mmol/L CaCl2, 0.05% BRIJ-35, 0.02% NaN3, 1% Triton X-100), and centrifuged. Aliquots containing equal amounts of protein from each sample were incubated for 1 hour with gelatin-Sepharose 4B (GE Healthcare, Little Chalfont, UK) with constant shaking. The pellets were washed with a working buffer (lysis buffer without Triton X-100) and resuspended in 100 μL of elution buffer (working buffer with 10% dimethylsulfoxide) for 30 minutes and then centrifuged. The samples were loaded on 10% Zymogram Gelatin Gels (Invitrogen, Carlsbad, CA, USA). After electrophoresis, the gels were incubated in Renaturing Buffer (Invitrogen) for 1 hour at room temperature and then Developing Buffer (Invitrogen) overnight at 37°C. The gels were stained for 1 hour in 1% Coomassie blue (Invitrogen) and then washed with water to obtain the clearest background for photography.
Western Blotting
Western blotting, performed as described (Kim et al, 2007), was used to detect protein levels of MMP-9 (Millipore, Billerica, MA, USA), acetylated histone-H3 at K9 and K14 (Millipore), and the tight junction proteins claudin-5 (Invitrogen), zonula occludens-1 (ZO-1; Santa Cruz, Santa Cruz, CA, USA), occludin (Invitrogen), and p65 (Millipore).
Statistical Analysis
Data are expressed as means ± s.e.m. Comparisons between two groups and multiple groups were evaluated by the Student's t-test and one-way analysis of variance, respectively. Differences were considered statistically significant at P < 0.05.
Results
Valproic Acid Protects Against Transient Middle Cerebral Artery Occlusion-Induced Evans Blue Extravasation and Brain Edema
A marked increase in Evans blue content was noted in the ipsilateral hemisphere compared with the contralateral side in MCAO rats 24 and 48 hours after reperfusion, with no significant difference between these two time points. Evans blue extravasation was significantly exacerbated at 72 hours compared with that at 24 and 48 hours. VPA (200 and 300 mg/kg, injected immediately after MCAO) markedly and dose dependently reduced Evans blue extravasation in the ipsilateral hemisphere 24 hours after reperfusion (Figures 1A and 1B). VPA (200 mg/kg) also significantly attenuated Evans blue extravasation measured at 48 and 72 hours. In addition, VPA (300 mg/kg) administrated at the onset of reperfusion significantly decreased Evans blue extravasation, but this effect was not as robust as that seen when given immediately after MCAO. Moreover, VPA (200 mg/kg, injected immediately after MCAO) markedly ameliorated MCAO-induced IgG extravasation 24 hours after reperfusion (Figure 1C), thus confirming BBB protection by VPA. Concomitant with BBB disruption, brain water content was notably increased in the ipsilateral hemisphere at 24 hours, which was dose dependently reduced by VPA given at the onset of MCAO (Figure 1D). In contrast, MCAO induced a much smaller increase in Evans blue extravasation and brain water content in the contralateral hemisphere. For subsequent experiments, a lower dose (200 mg/kg) of VPA was injected immediately after the onset of MCAO to achieve the maximal protection against BBB disruption measured at 24 hours after reperfusion.
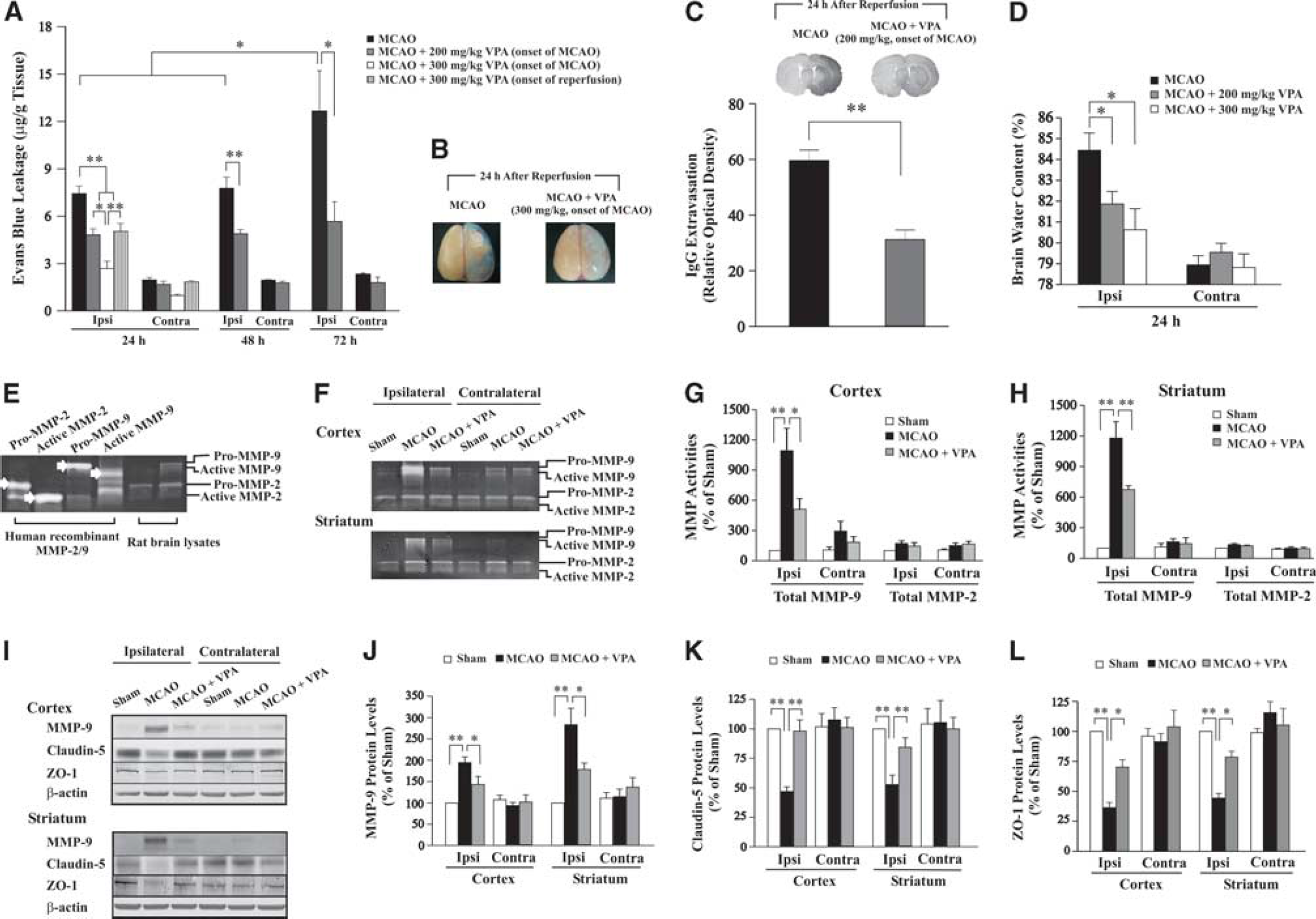
Valproic acid (VPA) attenuated transient middle cerebral artery occlusion (MCAO)-induced blood–brain barrier (BBB) disruption, matrix metalloproteinase-9 (MMP-9) elevation, and tight junction degradation. (
Valproic Acid Attenuates Transient Middle Cerebral Artery Occlusion-Induced Matrix Metalloproteinase-9 Elevation and Tight Junction Degradation
Pro- and active forms of MMP-2/9 were identified by reference to their respective standards in zymography (Figure 1E). Total MMP-9 activities in ipsilateral cortex and striatum of MCAO rats were robustly increased, with only a slight enhancement in the contralateral brain areas (Figures 1F–1H). Postischemic VPA treatment significantly suppressed the induction of total MMP-9 activities in both areas, whereas total MMP-2 activities were unaffected by MCAO. Western blotting showed that MMP-9 protein levels were remarkably induced in ipsilateral cortex and striatum (Figures 1I and 1J), similar to the results obtained with zymography. Conversely, protein levels of claudin-5 and ZO-1, tight junction proteins involved in BBB integrity, were markedly decreased in both regions (Figures 1I, 1K, and 1L). The MCAO-induced MMP-9 upregulation and tight junction protein downregulation were robustly prevented by VPA treatment. However, no significant degradation of another tight junction protein, occludin, was observed after ischemia (data not shown). MMP-9, claudin-5, and ZO-1 protein levels were essentially unaffected by MCAO in the contralateral hemisphere.
Valproic Acid-Induced Blood–Brain Barrier Protection Was Associated with Inhibition of Histone Deacetylases and Nuclear Factor-κB
Protein levels of acetylated histone-H3, an index of HDAC inhibition, were decreased in ipsilateral cortex and striatum, and this reduction was blocked by VPA treatment (Figures 2A and 2B). VPA also increased acetylated histone-H3 levels in contralateral cortex and striatum, compared with sham-operated or MCAO group. The MCAO caused nuclear factor-κB (NF-κB) p65 translocation from the cytoplasm to nucleus in ipsilateral cortex and striatum, and VPA markedly inhibited nuclear translocation of p65 in both regions (Figures 2C and 2D). Additionally, another HDAC inhibitor, SB (300 mg/kg), injected at the onset of MCAO, also attenuated Evans blue and IgG extravasation, reduced the increase in MMP-9 activities, and inhibited p65 nuclear translocation 24 hours after reperfusion (Figures 2E–2I).
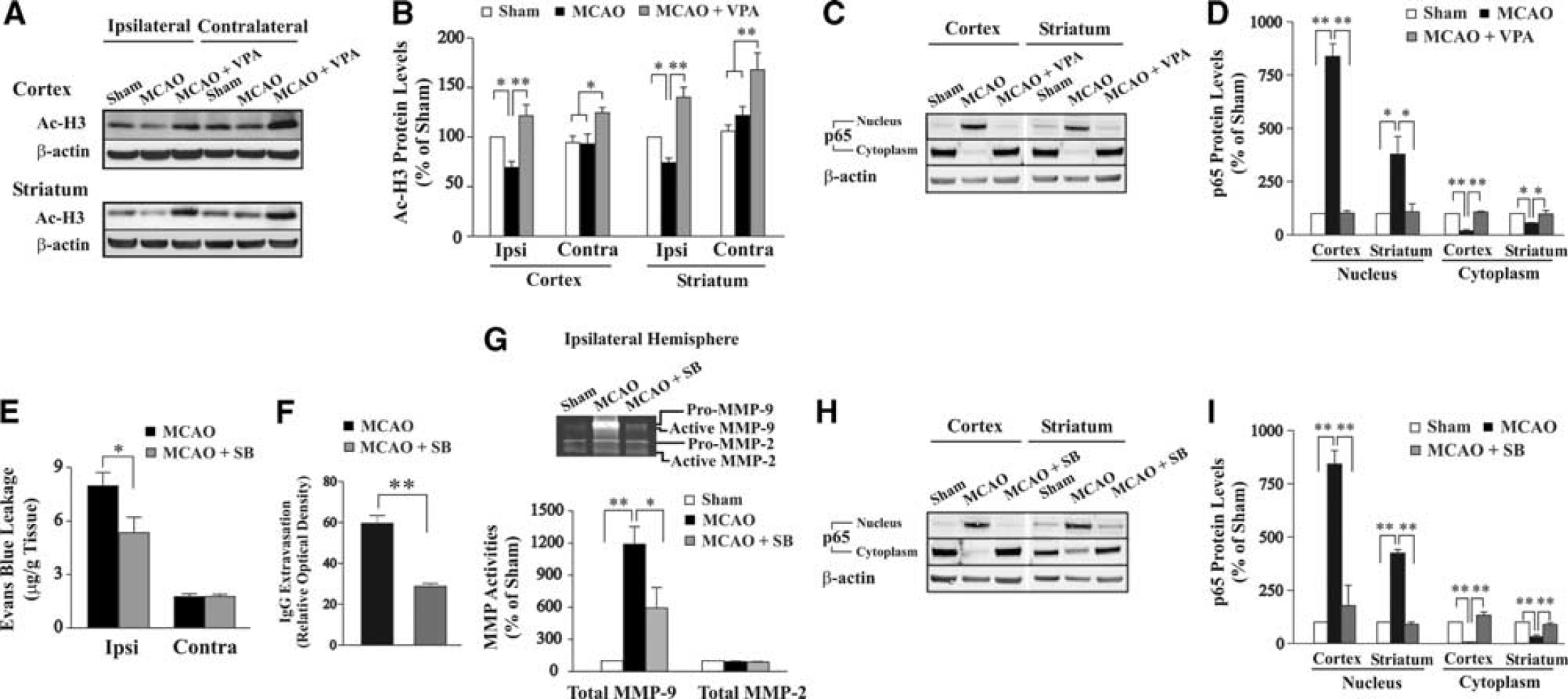
Valproic acid (VPA)-induced blood–brain barrier (BBB) protection was associated with inhibition of histone deacetylases (HDACs) and nuclear factor-κB (NF-κB) activation. (
Discussion
The present study suggests that the protective effects of VPA on brain ischemic injury involve its ability to attenuate BBB disruption and brain edema. This BBB protection may involve suppression of MCAO-induced overexpression of MMP-9 and downregulation of claudin-5 and ZO-1.
Accumulating evidence supports the notion that histone hypoacetylation and transcriptional dysfunction are involved in a large number of neurodegenerative conditions (Chuang et al, 2009). The HDAC inhibition has been shown to be neuroprotective against cerebral ischemia (Kim et al, 2007). In this study, postischemic treatment with clinically relevant doses of VPA substantially attenuated transient MCAO-induced BBB disruption, brain edema, and loss of histone acetylation. VPA-induced BBB protection was dose dependent and persisted at least 72 hours after reperfusion. Moreover, SB, another HDAC inhibitor, mimicked the effects of VPA. Therefore, our results suggest that VPA-induced protection against BBB disruption after transient MCAO is triggered by HDAC inhibition to induce chromatin remodeling.
Abnormal upregulation of both MMP-2 and MMP-9 has been linked to ischemia-induced brain injury and BBB disruption by degrading the basal components of BBB and facilitating immune cell infiltration (Yang et al, 2007). In the acute phase (within hours), both MMP-2 and MMP-9 are responsible for the BBB opening (Fujimura et al, 1999; Heo et al, 1999), whereas at 24 to 48 hours after ischemia, severe BBB disruption is associated with marked elevation of MMP-9 (Cunningham et al, 2005; Park et al, 2009). A very recent report shows that BBB disruption ∼24 hours after stroke onset in humans is associated with increased MMP-9 (Barr et al, 2010). Accordingly, we observed robust upregulation of MMP-9 in both cortex and striatum 24 hours after reperfusion, whereas MMP-2 remained at its basal level. Tight junction proteins are essential components of BBB and substrates of MMPs. It has been reported that MMP-9 knockout mice are resistant to BBB disruption induced by transient focal cerebral ischemia, and this protection is mediated by reduced degradation of ZO-1 (Asahi et al, 2001). Similar findings were also reported by using MMP pharmacological inhibitors (Yang et al, 2007). A recent study using human stroke brain tissue shows that elevated leukocytic MMP-9 is correlated with basal lamina degradation and BBB disruption (Rosell et al, 2008). In our paradigm, VPA strongly inhibited MMP-9 activity as well as protein elevation, and concomitantly restored protein levels of claudin-5 and ZO-1. Interestingly, we were unable to detect significant degradation of occludin after MCAO, indicating differential sensitivity of various tight junction proteins to ischemic insult, as reported previously (Asahi et al, 2001). Taken together, our results suggest that MMP-9 is a key protease interfering with BBB integrity, and that the ability of VPA to block MMP-9 elevation, and claudin-5 and ZO-1 downregulation is involved in BBB protection by this drug. Emerging evidence supports that MMP-9 may participate in neurovascular remodeling after stroke (Rosell and Lo, 2008). The role of MMP-9 in the long-term beneficial effects of VPA in MCAO rats remains to be investigated.
Mechanisms underlying VPA-induced suppression of MCAO-triggered MMP-9 upregulation remain unclear. The HDAC inhibition by VPA is expected to result in histone hyperacetylation and a more relaxed conformation in the promoter regions of a given gene (Chuang et al, 2009). This could cause transcriptional activation or inhibition, depending on whether transcription factor(s) or repressor(s) is recruited to the gene promoter. The NF-κB is a substrate of HDACs, and its hyperacetylation by HDAC inhibition causes a loss of NF-κB transactivation (Faraco et al, 2009). Consistent with these reports, we found that both VPA and SB significantly inhibited MCAO-induced NF-κB activation. Alternatively, MMP-9 gene promoter contains a highly conserved motif that matches the NF-κB p65 binding element and inhibition of NF-κB blocks MMP-9 upregulation in ischemic brain endothelium in vivo and in vitro (Cheng et al, 2006; Van den Steen et al, 2002). It has also been reported that other BBB-stabilizing molecules, such as activated protein C, reduce BBB disruption through inhibiting the NF-κB-dependent MMP-9 pathway (Cheng et al, 2006). Therefore, VPA-induced blockade of NF-κB activation could suppress MMP-9 transcription. To our knowledge, our study is the first to show that postinsult VPA treatment robustly reduces MCAO-induced BBB disruption. This BBB protection likely involves inhibition of HDACs, NF-κB activation, and MMP-9 expression, and may contribute to the overall beneficial effects of VPA in ischemic rodents.
Footnotes
Acknowledgements
The editorial assistance of Ms Ioline Henter is greatly appreciated.
The authors declare no conflict of interest.