
Abstract
Nicotinamide, a ß-nicotinamide adenine dinucleotide (NAD+) precursor and an essential nutrient for cell growth and function, may offer critical insights into the specific cellular mechanisms that determine neuronal survival, since this agent significantly impacts upon both neuronal and vascular integrity in the central nervous system. The authors show that nicotinamide provides broad, but concentration-specific, protection against apoptotic genomic DNA fragmentation and membrane phosphatidylserine exposure during oxidative stress to secure cellular integrity and prevent phagocytic cellular demise. Activation of the protein kinase B (Akt1) pathway is a necessary requirement for nicotinamide protection, because transfection of primary hippocampal neurons with a plasmid encoding a kinase-deficient dominant-negative Akt1 as well as pharmacologic inhibition of phosphatidylinositol-3-kinase phosphorylation of Akt1 eliminates cytoprotection by nicotinamide. Nicotinamide fosters neuronal survival through a series of intimately associated pathways. At one level, nicotinamide directly modulates mitochondrial membrane potential and pore formation to prevent cytochrome c release and caspase-3–and 9–like activities through mechanisms that are independent of the apoptotic protease activating factor-1. At a second level, nicotinamide maintains an inhibitory phosphorylation of the forkhead transcription factor FOXO3a at the regulatory sites of Thr32 and Ser253 and governs a unique regulatory loop that prevents the degradation of phosphorylated FOXO3a by caspase-3. Their work elucidates some of the unique neuroprotective pathways used by the essential cellular nutrient nicotinamide that may direct future therapeutic approaches for neurodegenerative disorders.
Nicotinamide, the amide form of niacin (vitamin B3), is the precursor for the coenzyme ß-nicotinamide adenine dinucleotide (NAD+) and is considered to be necessary for cellular function and metabolism. Yet current interest has focused on the development of nicotinamide as an agent that is critical for preserving cell survival in the brain (Maiese and Chong, 2003). In both neuronal and vascular cell populations, nicotinamide enhances survival during a variety of insults such as chemical retinal degeneration (Kiuchi et al., 2002), free radical exposure (Chong et al., 2002b; Lin et al., 2000), and oxidative stress (Crowley et al., 2000).
The ability of nicotinamide to modulate neuronal survival is intriguing, but further knowledge of the specific cellular mechanisms that determine protection by this agent is required. In this regard, oxidative stress via ischemic free radical exposure or oxygen—glucose deprivation has been established as a significant precipitant of neurodegenerative disorders, such as Alzheimer and Parkinson disease (Metodiewa and Koska, 2000; Simpkins and Jankovic, 2003). Oxidative stress precipitates apoptotic cellular injury that consists of both nuclear DNA degradation and membrane phosphatidylserine (PS) exposure (Chong et al., 2003b; Vincent and Maiese, 1999; Witting et al., 2000). DNA fragmentation, which is considered to be a late component of apoptosis, can be a committed event that permanently destroys cellular integrity (Jessel et al., 2002). Yet membrane PS exposure may play a more critical role in the early evolution of apoptotic injury that involves acute cellular inflammation (Dombroski et al., 2000) and microglial phagocytosis of viable neurons (Chong et al., 2003b; Witting et al., 2000).
One potential pathway that may be central to the neuroprotective capacity of nicotinamide is protein kinase B, also referred to as PKBα or Akt1 after the oncogene ν-Akt. Maximal activity of Akt1 is achieved through phosphoinositide 3 kinase (PI 3-K) and subsequent phosphorylation by phosphoinositide-dependent kinase 1 at Ser473 to confer protection against genomic DNA degradation (Chong et al., 2002a; Wick et al., 2002; Yamaguchi et al., 2001) and membrane PS exposure (Chong et al., 2002a, 2003b; Kang et al., 2003b). This protection against apoptotic injury may be dependent upon the forkhead transcription factor (FOXO3a, FHKRL1), a substrate of Akt1 that can modulate survival in a variety of cell systems. Inhibition of FOXO3a activity may prevent apoptosis through several mechanisms, such as blocking FOXO3a transcription during its association with 14-3-3 protein (Brunet et al., 1999), receptor-dependent repression of FOXO3a function (Li et al., 2003), or through the possible modulation of mitochondrial membrane depolarization and cytochrome c release (Yu et al., 2003).
Central to maintaining neuronal survival is the regulation of mitochondrial membrane potential (Δ Ψm) and the intrinsic cysteine protease pathway. During oxidative stress, mitochondrial permeability increases resulting in cytochrome c release and the activation of executioner caspases (Chong et al., 2003a; Lin et al., 2000; Ueda et al., 2002). Cytochrome c results in the oligomerization of apoptotic protease activating factor-1 (Apaf-1) and leads to the allosteric activation of caspase-9 forming the Apaf-1 apoptosome (Li et al., 1997). Subsequently, caspase-9 proteolytically activates caspase-3 to result in the cleavage of DNA protein kinase (DNA-PK), actin, and fodrin that results in both DNA fragmentation and membrane PS exposure (Chong et al., 2003b; Li et al., 1997; Maiese and Vincent, 2000). Here, we show that nicotinamide functions through two parallel pathways to provide cellular protection after the required activation of Akt1. In one pathway, nicotinamide directly modulates mitochondrial membrane potential to prevent cytochrome c release and caspase-3– and caspase-9–like activities that remain independent of Apaf-1 expression. Alternatively, nicotinamide provides another approach for cellular survival by maintaining an inhibitory phosphorylation of FOXO3a and preventing the degradation of phosphorylated FOXO3a by caspase-3.
MATERIALS AND METHODS
Primary hippocampal neuronal cultures
The hippocampi were obtained from E-19 Sprague-Dawley rat pups and incubated in dissociation medium (90 mmol/L Na2SO4, 30 mmol/L K2SO4, 5.8 mmol/L MgCl2, 0.25 mmol/L CaCl2, 10 mmol/L kynurenic acid, and 1 mmol/L HEPES with the pH adjusted to 7.4) containing papain (10 U/mL) and cysteine (3 mmol/L) for two 20-minute periods. The hippocampi were then rinsed in dissociation medium and incubated in dissociation medium containing trypsin inhibitor (10 to 20 U/mL) for three 5-minute periods. The cells were washed in growth medium (Leibovitz's L-15 medium; Invitrogen, Carlsbad, CA, U.S.A.) containing 6% sterile rat serum (ICN, Aurora, OH, U.S.A.), 150 mmol/L NaHCO3, 2.25 mg/mL of transferrin, 2.5 μg/mL of insulin, 10 nmol/L progesterone, 90 μmol/L putrescine, 15 nmol/L selenium, 35 mmol/L glucose, 1 mmol/L L-glutamine, penicillin and streptomycin (50 μg/mL), and vitamins. The dissociated cells were plated at a density of approximately 1.5 × 103 cells/mm2 in 35 mm polylysine/laminin-coated plates (Falcon Labware, Lincoln Park, NJ, U.S.A.). Neurons were maintained in growth medium at 37°C in a humidified atmosphere of 5% CO2 and 95% room air for 2 weeks. Nonneuronal cells were negligible.
Transfection of dominant-negative Akt1 (dn-Akt1) cDNA construct into neurons
For neuronal clones overexpressing a dominant-negative Akt1 mutant that lacked kinase activity (Kang et al., 2003b), cells were generated by transfecting the neurons with a cDNA construct under the control of a cytomegalovirus promoter (CMV) promoter with cDNA that contains a substitution of methionine (ATG) for lysine (AAG) at residue 179 in pUSEamp and a Myc-His tag at the 3′ end of the mouse Akt1 open reading frame (K179M mutant, Upstate Biotechnology, Lake Placid, NY, U.S.A.) by lipofection with Lipofectamine Plus reagent (Invitrogen, Gaithersburg, MD, U.S.A.). Clones were selected using Myc Tag (anti-Myc rabbit polyclonal immunoglobulin G [IgG], 1:1000; Upstate Biotechnology) with transfection efficiency equal to approximately 28% (n = 20). Individual clones for dn-Akt1 were characterized by the absence of phosphorylated Akt1 expression on Western analysis. Clones also were subjected to immunocytochemistry detection with Myc Tag (anti-Myc rabbit polyclonal IgG, 1:1000; Upstate Biotechnology).
Experimental treatments
Assessment of cell survival
Neuronal injury was determined by bright field microscopy using a 0.4% trypan blue dye exclusion method 24 hours after OGD exposure per our previous protocols (Chong et al., 2003a; Lin et al., 2000; Maiese and Vincent, 2000). The mean survival was determined by counting eight randomly selected nonoverlapping fields with each containing approximately 10 to 20 cells (viable + nonviable). Each experiment was replicated four to six times independently with different cultures.
Assessment of DNA fragmentation
Genomic DNA fragmentation was determined by the terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) assay (Chong et al., 2003a; Lin et al., 2000; Maiese and Vincent, 2000). Briefly, neuronal cells were fixed in 4% paraformaldehyde/0.2% picric acid/0.05% glutaraldehyde and the 3′-hydroxy ends of cut DNA were labeled with biotinylated dUTP using the enzyme terminal deoxytransferase (Promega, Madison, WI, U.S.A.) followed by streptavidin-peroxidase and visualized with 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA, U.S.A.).
Assessment of membrane phosphatidylserine residue externalization
Phosphatidylserine exposure was assessed through the established use of annexin V. Per our prior protocols (Chong et al., 2003b; Maiese and Vincent, 2000; Vincent and Maiese, 1999), a 30-μg/mL stock solution of annexin V conjugated to phycoerythrin (PE; R&D Systems, Minneapolis, MN, U.S.A.) was diluted to 3 μg/mL in warmed calcium containing binding buffer (10 mmol/L HEPES, pH 7.5; 150 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 1.8 mmol/L CaCl2). Plates were incubated with 500 μL of diluted annexin V for 10 minutes. Images were acquired with “blinded” assessment with a Leitz DMIRB microscope (Leica, McHenry, IL, U.S.A.) and a Fuji/Nikon Super CCD (6.1 megapixels) using transmitted light and fluorescent single excitation light at 490 nm and detected emission at 585 nm.
Modulation and assessment of mitochondrial permeability transition pore membrane potential (ΔΨm)
The fluorescent probe JC-1 (Molecular Probes, Eugene, OR, U.S.A.), a cationic membrane potential indicator, was used to assess ΔΨm. Neuronal cells in 35-mm2 plates were incubated with 2 μg/mL JC-1 in growth medium for 30 minutes. After washing, cells were then analyzed immediately under a Leitz DMIRB microscope (Leica) with a dual-emission fluorescence filter with 515 to 545 nm for green fluorescence and emission at 585 to 615 nm for red fluorescence. For modulation of mitochondrial membrane permeability, the exposure of tert-butylhydroperoxide (tBu-OOH; Sigma), atractyloside (Atr; Sigma), and cyclosporin A (CsA; Sigma, St. Louis, MO) were continuous.
Assessment of cysteine protease activity
The activities of caspase-9 and caspase-3 were assessed by determination of the cleavage of their colorimetric substrates (Ac-LEHD-pNA, caspase-9; Ac-DEVD-pNA, caspase-3; Calbiochem) as previously described (Chong et al., 2002b, 2003a,b).
Modulation of cysteine protease activity
Modulation of cysteine protease activity in neuronal cells was performed by using the irreversible and cell-permeable caspase-inhibitors (50 μmol/L 1 hour before OGD) Z-LEHD-FMK for caspase-9 (LEHD) and Z-DEVD-FMK (DEVD) for caspase-3 (BD Pharmingen, San Diego, CA, U.S.A.).
Western blot analysis for Akt1 phosphorylation, total FOXO3a expression, FOXO3a phosphorylation, cytochrome c release, and Apaf-1 expression
Cells were homogenized and after protein determination, each sample (50 μg/lane) was then subjected to 7.5% (Akt1, FOXO3a, Apaf-1) or 12.5% (cytochrome c) sodium dodecyl sulfate polyacrylamide gel electrophoresis. After transfer, the membranes were incubated with primary mouse monoclonal antibodies against phosphorylated Akt1/PKBα (p-Akt1, Ser473, 1:1000; Upstate Biotechnology), cytochrome c (1:2000) (BD Pharmingen), a goat polyclonal antibody against total FOXO3a (FOXO3a, 1:100; Santa Cruz Biotechnologies, Santa Cruz, CA, U.S.A.), a goat polyclonal antibody against phosphorylated FOXO3a (p-FOXO3a, Thr32, 1:100, or Ser253, 1:100; Santa Cruz Biotechnologies), or a polyclonal rabbit antibody against Apaf-1 (1: 2000; BD Pharmingen). After washing, the membranes were incubated with a horseradish peroxidase conjugated secondary antibody (goat anti—mouse IgG, 1:2000, rabbit anti—goat IgG, 1:2000, or goat anti—rabbit IgG, 1:15000; Pierce, Rockford, IL, U.S.A.). The antibody-reactive bands were revealed by chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.).
Statistical analysis
For each experiment involving assessment of neuronal cell survival, DNA degradation, membrane PS exposure, ΔΨm, and caspase activity, the mean and standard deviation were determined from four to six replicate experiments. Statistical differences between groups were assessed by means of analysis of variance (ANOVA) with the post hoc Student's t-test. Results are expressed as the mean ± the standard deviation. Statistical significance was considered at P < 0.05.
RESULTS
Nicotinamide is toxic to neurons only at elevated concentrations
Increasing concentrations of nicotinamide (1 to 50 mmol/L) were administered directly to cultures and cell survival was evaluated by a trypan blue dye exclusion method 24 hours later (Fig. 1A). As compared with neuronal survival in untreated control cultures (86% ± 9%), no significant toxicity over a 24-hour period was present in the cultures containing nicotinamide in the concentrations of 1 to 25 mmol/L (Fig. 1A). Only nicotinamide at 50 mmol/L was toxic and yielded a significant decrease in neuronal survival (55% ± 11%, P < 0.01).
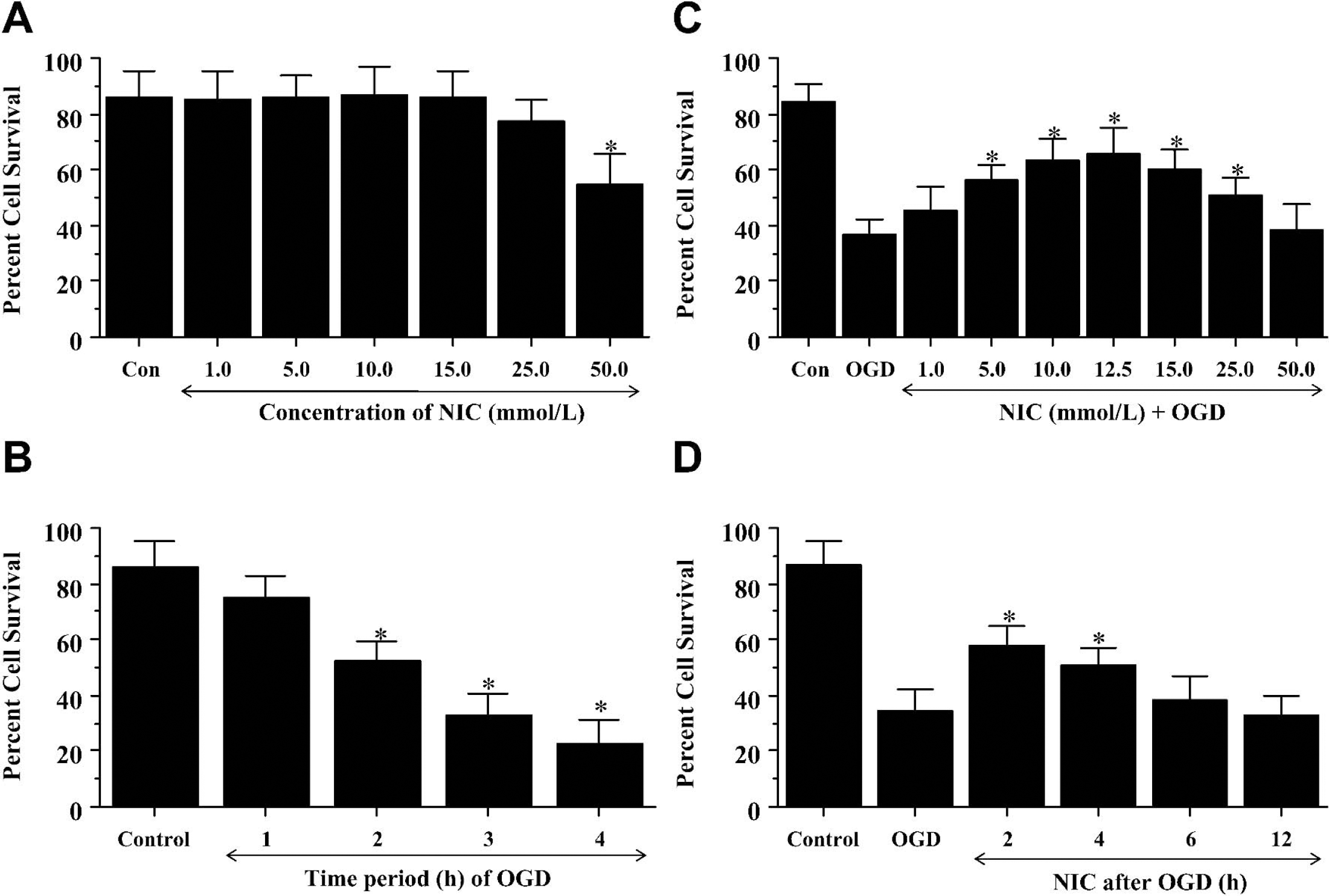
Nicotinamide (NIC) increases neuronal survival after oxidative stress in a concentration specific manner. (
Progressive exposure to oxidative stress yields significant neuronal injury
We examined the toxic effect of progressive time periods of OGD exposure on neuronal cultures. Neuronal survival was determined by trypan blue dye exclusion 24 hours after a 1-hour, 2-hour, 3-hour, and 4-hour period of OGD. As shown in Fig. 1B, a significant decrease in hippocampal neuronal survival was present in cultures exposed to OGD for the duration of 2 hours (52% ± 5%), 3 hours (33% ± 5%), and 4 hours (23% ± 5%), but not for a 1-hour period (75% ± 7%) when compared with untreated control (86% ± 6%). Since an OGD exposure period of 3 hours resulted in survival rate of approximately 35% (65% neuronal cell loss), this duration of OGD toxicity was used for the remainder of the experimental paradigms.
Nicotinamide increases neuronal survival after oxidative stress
Nicotinamide prevents apoptotic genomic DNA fragmentation and membrane phosphatidylserine externalization during oxidative stress
Neurons were exposed to OGD, and either cellular genomic DNA fragmentation was assessed with TUNEL or cellular membrane PS exposure was determined by annexin V 24 hours later. In Fig. 2A, representative pictures showed that OGD resulted in both DNA fragmentation and membrane PS externalization in neurons. In cells exposed to OGD, cell injury is evident by chromatin condensation and significant induction of annexin V label. In contrast, pretreatment with nicotinamide (12.5 mmol/L) 1 hour before OGD significantly reduced nuclear condensation and membrane PS externalization.
To quantitatively determine the ability of nicotinamide to prevent OGD induced DNA fragmentation and membrane PS externalization, nicotinamide (12.5 mmol/L) was administered 1 hour before OGD and assessment was performed 24 hours later. As shown in Fig. 2B, OGD alone resulted in a significant increase in percent DNA fragmentation (56% ± 6%) in neurons when compared with untreated control cultures (8% ± 4%). DNA fragmentation was reduced to 34% ± 5% in cells treated with nicotinamide after OGD exposure. An increase in annexin V label was observed in neurons 24 hours after OGD exposure that reached a level of 63% ± 6% when compared with untreated control cultures of 17% ± 4% (Fig. 2C). Cells treated with nicotinamide displayed a significant reduction in annexin V label to 32% ± 5% 24 hours after OGD.
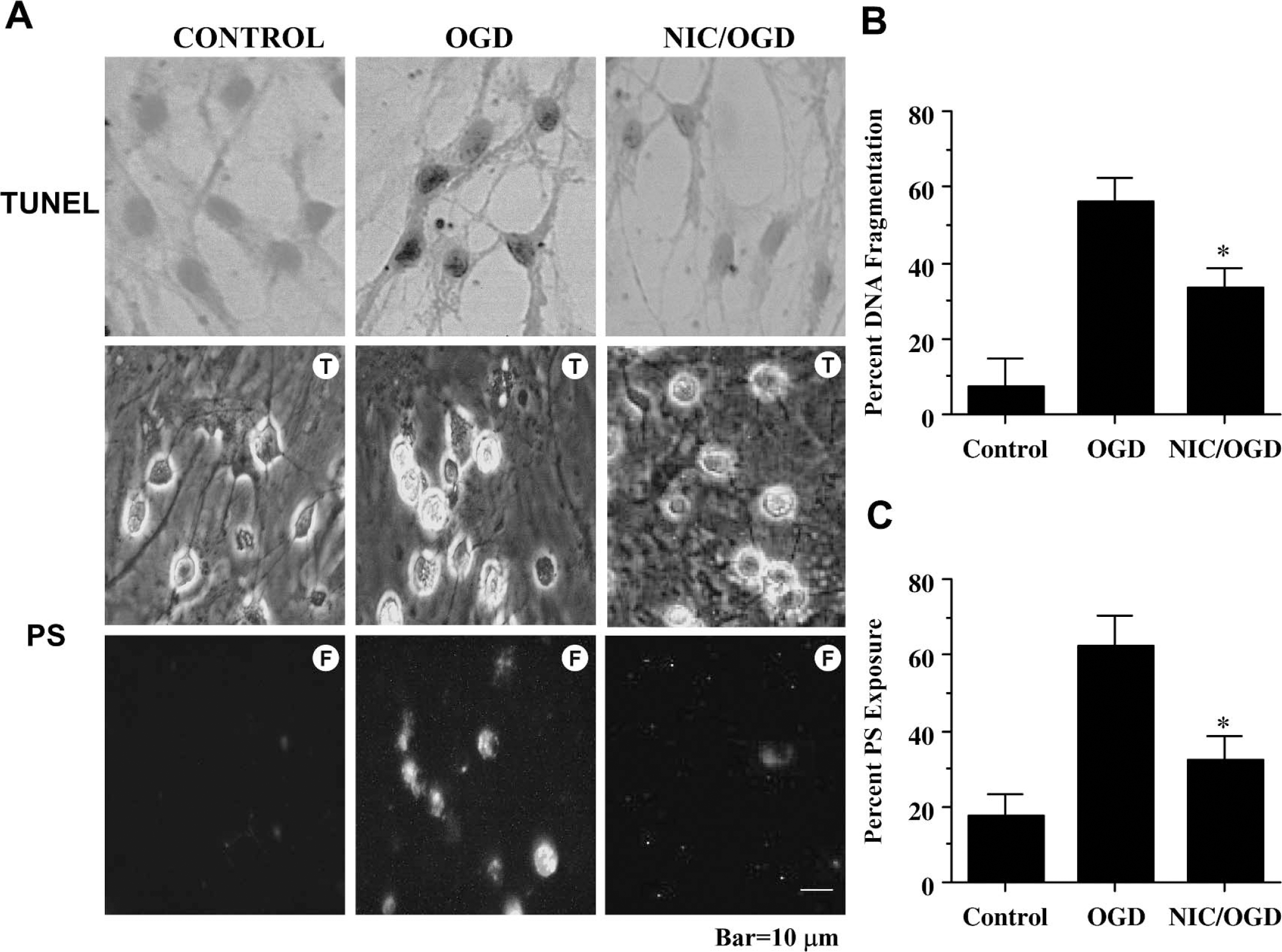
Nicotinamide (NIC) prevents apoptotic genomic DNA fragmentation and membrane phosphatidylserine (PS) exposure during oxidative stress. (
Nicotinamide uses Akt1 to confer neuronal protection
Western blot assay was performed for phosphorylated Akt1 (p-Akt1) (activated form of Akt1) 12 hours after OGD. In Fig. 3A and Fig. 3B, both OGD and nicotinamide independently increased the expression of p-Akt1, but nicotinamide, either alone or in the presence of OGD, increased p-Akt1 expression to a greater degree than application of OGD alone. This increased expression of p-Akt1 was blocked by the agents wortmannin (0.5 μmol/L), which forms a covalent link with the lysine residue of PI 3-K (Wymann et al., 1996), and LY294002 (10 μmol/L), which reversibly competes for ATP binding (Vlahos et al., 1994) (Fig. 3C). In Fig. 3D, application of nicotinamide (12.5 mmol/L) 1 hour before OGD significantly increased neuronal survival to 68% ± 7%. However, coapplication of wortmannin (0.5 μmol/L) or LY294002 (10 μmol/L) at concentrations that block activation of Akt1 during OGD (Fig. 3C) with nicotinamide (12.5 mmol/L) significantly reduced the ability of nicotinamide to protect neurons against OGD, suggesting that nicotinamide required some level of Akt1 activation to offer protection. When administered in the absence of OGD, wortmannin (0.5 μmol/L) and LY294002 (10 μmol/L) were not toxic to neurons (Fig. 3D).
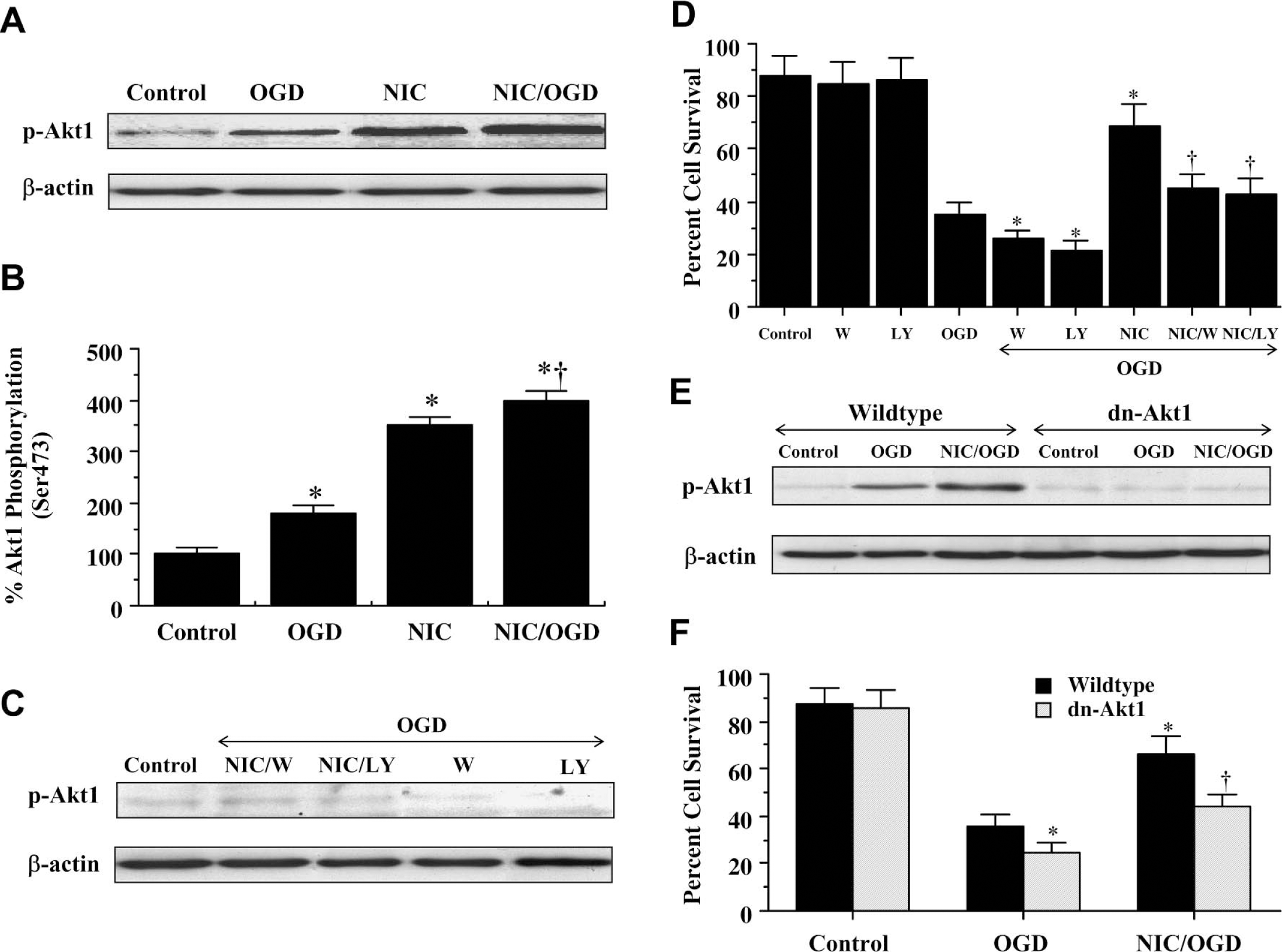
Akt1 is an important component for neuronal protection by nicotinamide (NIC). (
To independently assess whether activation of Akt1 was important for nicotinamide to foster cellular protection, we examined whether the overexpression of a kinase-deficient dominant-negative Akt1 in neuronal cells would alter cellular survival during OGD. Initially, Western blot assay was performed for phosphorylated Akt1 (p-Akt1) 12 hours after OGD exposure in cells that overexpress dn-Akt1 (Fig. 3E). Similar to our previous results, increased expression of p-Akt1 was present during OGD and combined application of nicotinamide and OGD. However, cells with dn-Akt1 overexpression that lacked kinase activity were without expression of p-Akt1 during these treatment protocols. We next assessed whether neuronal protection by nicotinamide was compromised by overexpression of a kinase deficient dn-Akt1 (Fig. 3F). In wild-type cells, cell survival was significantly reduced from 87% ± 7% (untreated control cells) to 36% ± 5% (P < 0.01) after exposure to OGD. This cell injury was significantly enhanced in cells that actively overexpress dn-Akt1, decreasing cell survival to approximately 25% and suggesting that an additional endogenous reserve of Akt1 protein exists to protect against cell injury. In addition, overexpression of dn-Akt1 in neurons significantly impaired nicotinamide to protect against OGD, further supporting the premise that activation of Akt1 is an important component for nicotinamide to offer neuronal protection.
Nicotinamide modulates mitochondrial membrane potential during oxidative stress
Exposure to OGD produced a significant decrease in the red/green fluorescence intensity ratio using a cationic membrane potential indicator JC-1 within 3 hours when compared with untreated control cultures (Fig. 4A,Fig. 4B), suggesting that OGD alters mitochondrial permeability transition pore membrane potential (ΔΨm) and results in mitochondrial membrane potential depolarization. Application of nicotinamide (12.5 mmol/L) 1 hour before OGD exposure significantly increased the red/green fluorescence intensity of the neurons (Fig. 4A, Fig. 4B) and prevented mitochondrial cytochrome c release as shown by Western analysis (Fig. 4C), suggesting that ΔΨm was restored to baseline.
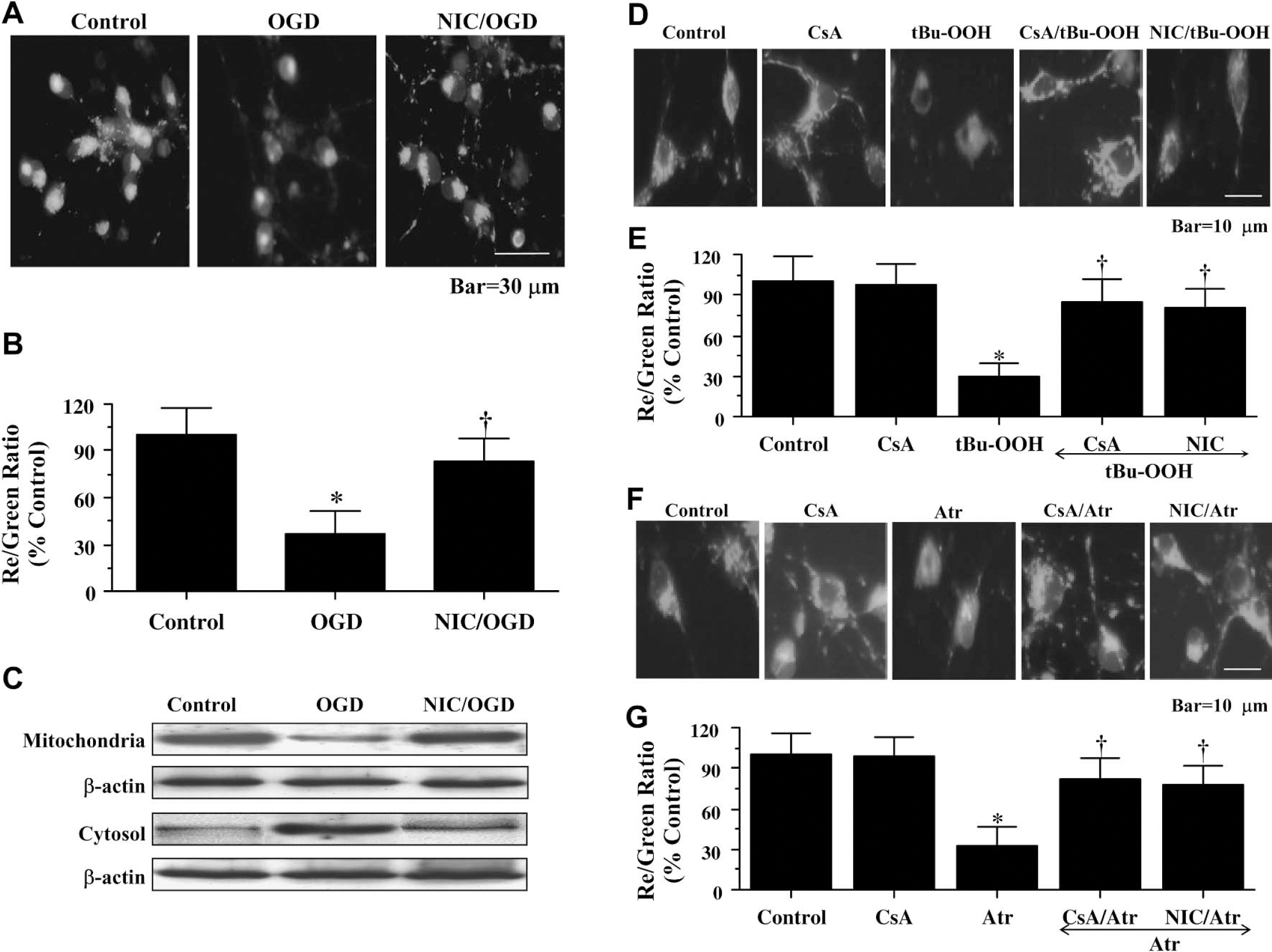
Nicotinamide (NIC) functions at the level of ΔΨm to prevent cytochrome c release. (
Nicotinamide also was able to maintain ΔΨm during the pharmacologic increase of mitochondrial membrane permeability. Cyclosporin A (CsA), an agent that inhibits the assembly of the mitochondrial permeability transition pore complex (PTPC) to prevent mitochondrial membrane potential depolarization (Halestrap et al., 1997), was used as an experimental control to modulate ΔΨm (Figs. 4D–Fig. 4G). CsA (5 μmol/L) prevented mitochondrial membrane potential depolarization during application of tBu-OOH (10 μmol/L) (Fig. 4D and Fig. 4E), an oxidative inducer of mitochondrial membrane permeability that impairs mitochondrial ATP synthesis (Imberti et al., 1993), and during the administration of Atr (5 mmol/L) (Fig. 4G), an agent that binds to the mitochondrial adenosine nucleotide translocator to elicit pore formation (Brown et al., 1997). In a similar manner, nicotinamide (12.5 mmol/L) applied 1 hour before treatment with tBu-OOH (10 μmol/L) (Fig. 4D andFig. 4E) or before treatment with Atr (5 mmol/L) (Fig. 4F andFig. 4G) prevented ΔΨm depolarization, suggesting that nicotinamide maintains ΔΨm directly at the level of mitochondrial membrane pore formation.
Nicotinamide maintains the integrity and phosphorylation of p-FOXO3a during oxidative stress
Western blot assay was performed for phosphorylated FOXO3a (p-FOXO3a) at 3, 6, and 12 hours after OGD exposure. In Fig. 5A, OGD initially significantly increased the expression of p-FOXO3a at the regulatory site of Thr32 (Brunet et al., 1999) over a 3-hour period when compared with control cultures. Beyond 3 hours after OGD exposure, expression of p-FOXO3a was progressively diminished and approached control levels (Fig. 5A). In contrast, nicotinamide alone (Fig. 5B) or in combination with OGD exposure (Fig. 5C) significantly increased the expression of p-FOXO3a and maintained this increase in FOXO3a phosphorylation over a 12-hour course, suggesting that nicotinamide may prevent the degradation of p-FOXO3a (Fig. 5C).
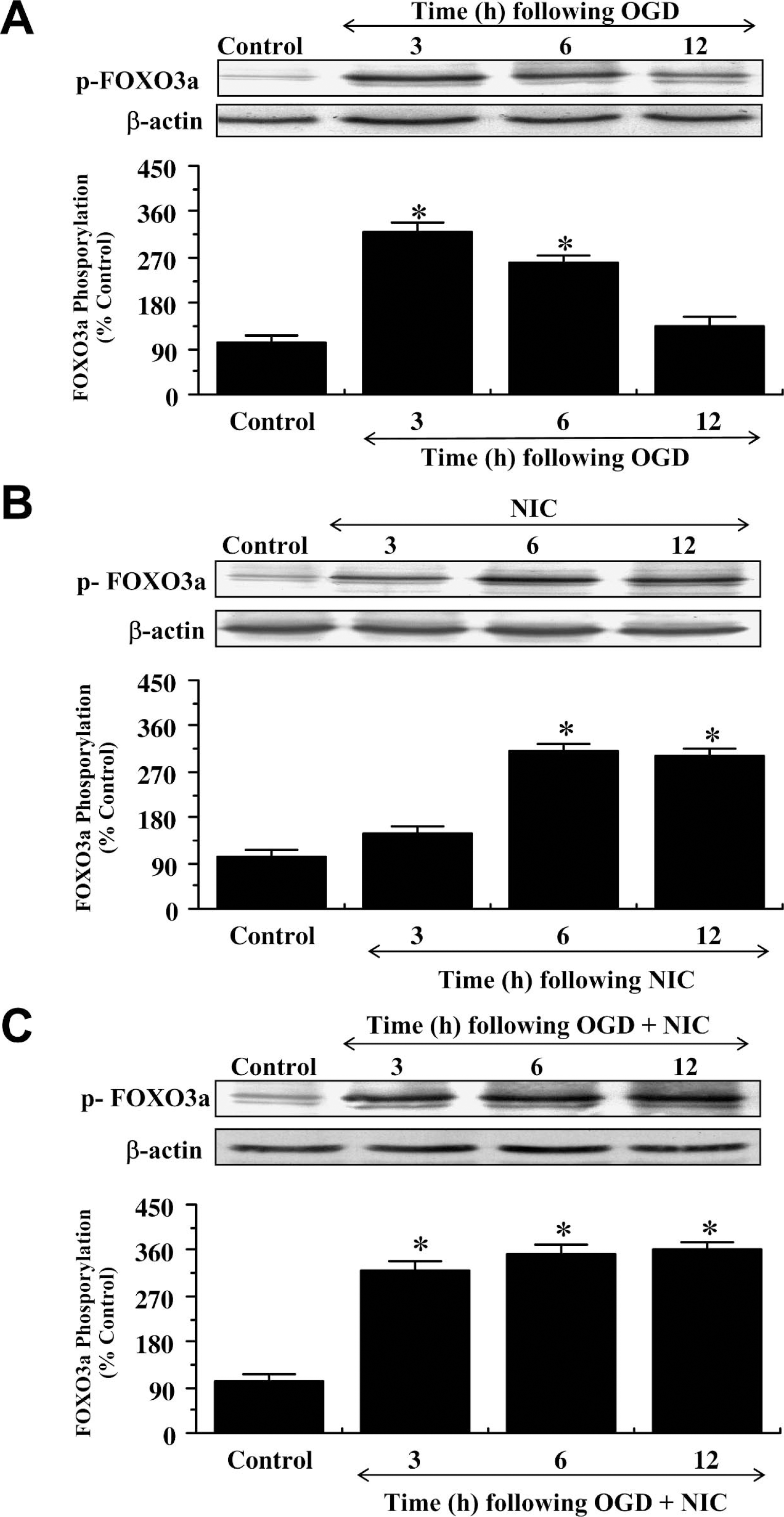
Nicotinamide (NIC) maintains the integrity and phosphorylation of p-FOXO3a (Thr32) during oxidative stress. (
We next examined the expression of p-FOXO3a at the preferential phosphorylation site for Akt of Ser253 (Rena et al., 1999) as well as the expression of total FOXO3a during OGD. Similar to the Thr32 site, OGD significantly increased the expression of p-FOXO3a at the site of Ser253 over a 6-hour period when compared with control cultures, but expression of p-FOXO3a was decreased and reached control levels within 12 hours after OGD (Fig. 6A). However, nicotinamide alone or in combination with OGD exposure (Fig. 6A) significantly increased the expression of p-FOXO3a at Ser253 and maintained this increase over a 12-hour course, further supporting the premise that nicotinamide prevents the degradation of p-FOXO3a. In addition, nicotinamide prevented the loss in the expression of total FOXO3a during OGD exposure over a 12-hour course (Fig. 6B).
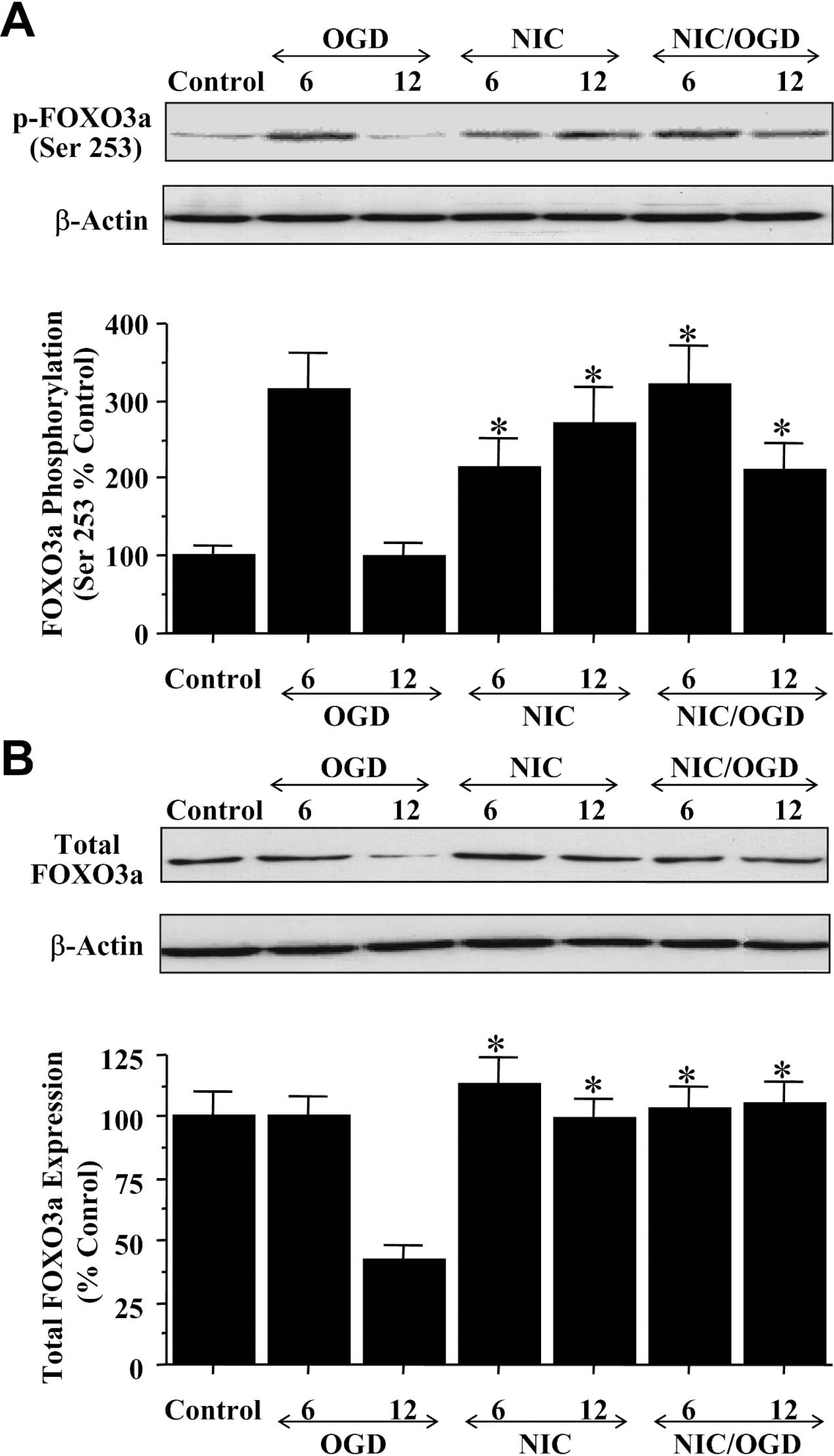
Nicotinamide (NIC) maintains the integrity and phosphorylation of p-FOXO3a at the high-affinity site for Akt of Ser253 during oxidative stress. (
Nicotinamide inhibits caspase-3– and caspase-9–like activities during OGD
Nicotinamide (12.5 mmol/L) was applied to neuronal cultures 1 hour before OGD and data for caspase-9 and caspase-3 activities were obtained at 6 hours and 12 hours after OGD exposure (Fig. 7A and Fig. 7B), because this time period represented the peak activities for these cysteine proteases (Chong et al., 2002b, 2003a,b). Administration of nicotinamide significantly decreased caspase-3–like activity to 0.13 ± 0.04 μmol·min−1·g−1 (P < 0.01, 6 hours) and to 0.15 ± 0.06 μmol·min−1·g−1 (P < 0.01, 12 hours) (Fig. 7A). Nicotinamide treatment also significantly reduced the activity of caspase-9–like activity (0.10 ± 0.04 μmol·min−1·g−1, 6 hours; 0.14 ± 0.06 μmol·min−1·g−1, 12 hours) when compared with cultures treated with OGD alone (0.21 ± 0.07 μmol·min−1·g−1, 6 hours; 0.31 ± 0.09 μmol·min−1·g−1, 12 hours) (Fig. 7B). We next examined whether inhibition of caspase-9– and caspase-3–like activities by nicotinamide were necessary for neuronal survival during OGD. Administration of DEVD (50 μmol/L) or LEHD (50 μmol/L) to inhibit caspase-3– or caspase-9–like activities significantly increased neuronal survival to approximately 65% ± 7% and 64% ± 9%, respectively (Fig. 7C).
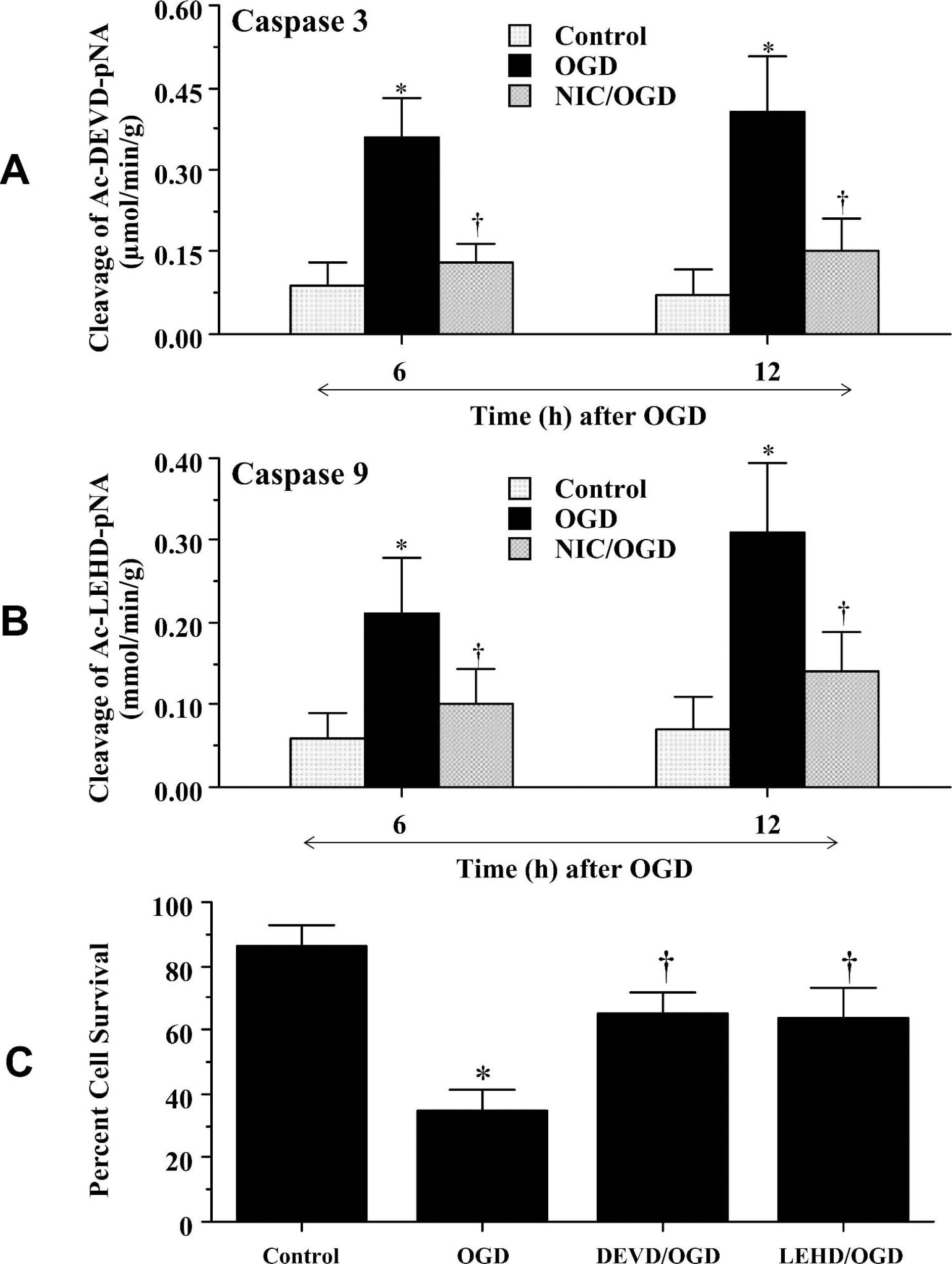
Nicotinamide (NIC) protects neurons from injury through the modulation of caspase-3– and caspase-9—like activities. In panels
Nicotinamide prevents caspase-3–mediated cleavage of p-FOXO3a independent of Apaf-1 expression
Western blot assay was performed for Apaf-1 at 6 and 12 hours after OGD exposure. As shown in Fig. 8A, OGD independently increased the expression of Apaf-1 within 6 hours after the initial insult. Expression of Apaf-1 remained elevated over a 12-hour period. Application of nicotinamide (12.5 mmol/L) 1 hour before OGD did not alter the expression of Apaf-1 over a 12-hour period after OGD exposure (Fig. 8A).
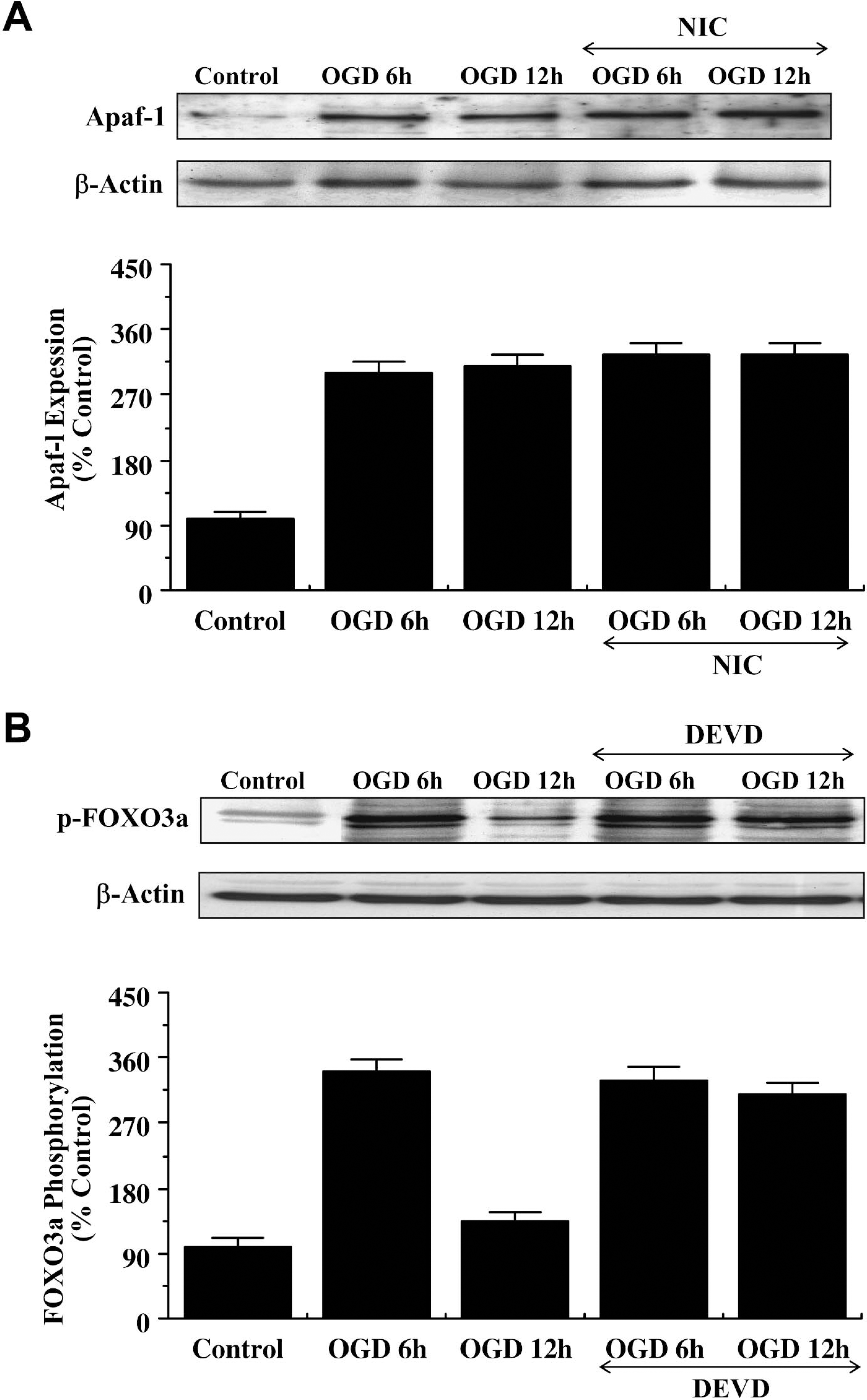
Nicotinamide (NIC) prevents caspase-3 mediated cleavage of p-FOXO3a independent of Apaf-1 expression. (
Given that nicotinamide prevents the loss of p-FOXO3a expression during OGD exposure, we subsequently examined whether potential modulation of caspase-3–like activity by nicotinamide was responsible for the maintenance of p-FOXO3a integrity. Recently, FOXO3a, has been shown to be a substrate for caspase-3–like proteases at the consensus sequence DELD304 A (Charvet et al., 2003). In Fig. 8B, Western blot assay was performed for phosphorylated FOXO3a (p-FOXO3a) at 6 hours and 12 hours after OGD exposure during inhibition of caspase-3–like activity. Consistent with FOXO3a containing a consensus sequence for caspase-3, inhibition of caspase-3–like activity prevented p-FOXO3a degradation in neurons 6 hours and 12 hours after OGD exposure.
DISCUSSION
Interest in novel neuroprotectants for the central nervous system continues to garner significant attention. Yet it is the knowledge of the cellular elements that not only precipitate neuronal injury, but also foster neuronal integrity, that is required for future development of therapeutic strategies against neurodegenerative disorders. We show that nicotinamide prevents OGD-induced apoptotic neuronal injury in a specific concentration range. Administration of nicotinamide in a range of 5.0 to 25.0 mmol/L significantly protected neurons during OGD. This concentration range is similar to other injury paradigms in both animal models (Kiuchi et al., 2002) and in cell culture models (Chong et al., 2002b; Lin et al., 2000; Maiese and Chong, 2003), illustrating loss of protection or toxicity when nicotinamide is administered outside of its optimal concentration range. The mechanisms that determine nicotinamide toxicity remain to be defined, but toxicity of this agent has been described in other cellular systems. Increased levels of nicotinamide can block the function of rat β-cells, decrease DNA content of adult rat islet cells, and induce cell death in fetal rat islet cells (Reddy et al., 1995). Elevated concentrations of nicotinamide can trigger excessive release of choline that may precipitate neuronal injury (Koppen et al., 1993), whereas combination therapy with nicotinamide and methamphetamines can alter cellular thermoregulation related to serotonin release (Hervias et al., 2000). Additional work has identified another toxic aspect of nicotinamide that involves inhibition of the NAD+-dependent histone deacetylase Sir3 to decrease cellular lifespan (Anderson et al., 2003).
Nicotinamide provides protection against two independent components of neuronal apoptosis through the maintenance of intact genomic DNA and the prevention of cellular membrane PS exposure. In several cell systems, exposure of membrane PS residues, even in cells that have undergone repair and are without further injury, can promote cell to cell interactions and lead to the “tagging” of cells for removal by microglia (Chong et al., 2003b; Dombroski et al., 2000; Hoffmann et al., 2001; Maiese and Vincent, 2000). For effective neuroprotective strategies, protection against apoptotic injury should be broad in nature by addressing the separate components of genomic DNA destruction and cellular membrane PS exposure. Metabolites of nicotinamide have been reported to possess antiinflammatory properties (Gebicki et al., 2003). In addition to the preservation of genomic DNA integrity to avert apoptotic cell destruction, prevention of membrane PS exposure by nicotinamide provides an additional level of cellular protection to block inflammatory cell demise.
A series of cellular signal transduction pathways appear to mediate the neuronal protection provided by nicotinamide. In this cellular cascade, the protein Akt1 appears to hold a central role for nicotinamide to preserve neuronal integrity during OGD. In primary neuronal cultures, endogenous activation of Akt1 during OGD provides a minimum level of protection, because inhibition of the PI-3K pathway with wortmannin or LY294002 reduces neuronal survival during OGD. Interestingly, nicotinamide independently increases the phosphorylation and activation of Akt1 to a greater degree than OGD alone, suggesting that nicotinamide may use Akt1 to provide neuroprotection. Subsequent work illustrates that nicotinamide is dependent on the activation of Akt1 to prevent neuronal injury, because prevention of Akt1 phosphorylation with inhibitors of PI-3K activity significantly reduces the ability of nicotinamide to protect neurons. Using the overexpression of a kinase-deficient dominant-negative Akt1, we further support the premise that nicotinamide requires Akt1 to protect neurons from oxidative stress. Neurons with a dominant-negative overexpression that lacked kinase activity suffered a significant loss in cell survival during OGD exposure. During nicotinamide administration in the presence of OGD, absence of Akt1 activity through the overexpression of a kinase-deficient dominant-negative Akt1 significantly reduced the protective capacity of nicotinamide, supporting that activation of Akt1 by nicotinamide during cellular injury is necessary for cellular protection.
Absence of Akt1 activation results in a significant, but not complete reduction in neuronal protection by nicotinamide, suggesting that alternate cellular mechanisms may contribute to the protective capacity of nicotinamide. One pathway that is closely linked to the activation of Akt1 is the maintenance of ΔΨm (Kang et al., 2003a; Kennedy et al., 1999). Loss of ΔΨm represents a significant determinant for cell injury and the subsequent induction of the apoptotic cascade (Chong et al., 2003a; Lin et al., 2000; Ueda et al., 2002). Our studies illustrate that OGD acutely alters ΔΨm and results in mitochondrial membrane depolarization, but that administration of nicotinamide maintains ΔΨm and prevents the release of cytochrome c. Interestingly, nicotinamide appears to act directly at the level of mitochondrial membrane pore formation to prevent cytochrome c release. Nicotinamide blocked the chemical induction of mitochondrial membrane depolarization during application of either tBu-OOH or Atr. Although the precise pathways that are necessary for nicotinamide to modulate mitochondrial membrane pore formation require further analysis, it is possible that nicotinamide may function through the stabilization of cellular energy metabolism because the maintenance of mitochondrial membrane potential is an ATP facilitated process (La Piana et al., 2003). However, alternative mechanisms may play a more significant role for nicotinamide to maintain ΔΨm, such as inhibiting the assembly of the mitochondrial PTPC similar to the action of CsA (Halestrap et al., 1997).
Interestingly, increased activity of Akt1 may constitute only one scenario for nicotinamide to prevent neuronal injury through the maintenance of ΔΨm. Modulation of ΔΨm can occur not only at the level of Akt1, but also through one of its substrates, namely FOXO3a. Activation of the transcription factor FOXO3a has been shown to disrupt ΔΨm and may result in cytochrome c release (Yu et al., 2003). We illustrate that OGD results in an initial inhibitory phosphorylation of FOXO3a at the regulatory phosphorylation sites (Thr32 and Ser253) (Brunet et al., 1999; Rena et al., 1999) within 3 or 6 hours after OGD exposure, but leads to the loss of phosphorylated FOXO3a expression within a subsequent 12-hour period. Nicotinamide may derive its neuroprotective capacity through two separate mechanisms of posttranslational modification of FOXO3a not only by maintaining inhibitory phosphorylation of FOXO3a, but also by preserving the integrity of total FOXO3a and phosphorylated FOXO3a over a 12-hour period. Although we show that brief initial inhibitory phosphorylation of FOXO3a occurs during OGD exposure, the loss of both FOXO3a phosphorylation and the integrity of this transcription factor may function as a significant precipitant of neuronal injury. FOXO3a proteolysis occurs during cell injury yielding an amino-terminal (Nt) fragment that can become biologically active (Charvet et al., 2003). During cell injury and caspase-dependent cleavage of Akt1 (Widmann et al., 1998), it is the activation of FOXO3a Nt fragments that become available and result in apoptotic cellular injury. Nicotinamide, through both the promotion of extended phosphorylation of FOXO3a at regulatory sites that possess high affinity for Akt (Rena et al., 1999) and the inhibition of the proteolytic cleavage of FOXO3a, may maintain mitochondrial integrity and prevent induction of a cysteine protease cascade.
Intimately associated with the disruption in ΔΨm and the release of cytochrome c into the cytosol during neuronal injury is the induction of cysteine protease activity. Oligomerization of Apaf-1 with cytochrome c is critical for the allosteric activation of caspase-9 (Li et al., 1997). Although some “antiapoptotic” proteins, such as erythropoietin (Chong et al., 2003a) and heat-shock protein 70 (Beere et al., 2000), appear to modulate both Apaf-1 expression and cytochrome c release, protection through nicotinamide remains independent from Apaf-1. Expression of Apaf-1 significantly increased within 6 hours after OGD, but application of nicotinamide did not alter this increased expression of Apaf-1 during OGD.
Independent of Apaf-1, downstream cellular pathways that may be responsible for neuroprotection with nicotinamide appear to reside at the level of caspase activity. Both caspase-3 and caspase-9 are associated with the separate apoptotic pathways of genomic DNA cleavage and cellular membrane PS exposure (Chong et al., 2003a; Lin et al., 2000; Takahashi et al., 1999). Initial evidence that caspase-3– and caspase-9–like activities play a significant role in neuronal injury and the protection offered by nicotinamide is present in our posttreatment protocols. Application of nicotinamide shows a progressive loss in neuronal protection during a 6-hour period after OGD. This work correlates well with other posttreatment paradigms involving nicotinamide (Lin et al., 2000; Chong et al., 2002b; Kiuchi et al., 2002), and may be a reflection of the gradual increase in caspase-3–and caspase-9–like activities during this same postinjury period. If administered before the induction of the maximal activities of caspase-3 and caspase-9, nicotinamide can then significantly prevent cell injury by inhibiting caspase-3– and caspase-9–like activity. Modulation of caspase-3 activity by nicotinamide also appears to be closely associated with a unique regulatory mechanism that blocks the proteolytic degradation of phosphorylated FOXO3a by caspase-3. Given that FOXO3a has been shown to be a substrate for caspase-3–like proteases at the consensus sequence DELD304 A (Charvet et al., 2003), we show that blockade of caspase-3–like activity prevents the destruction of phosphorylated FOXO3a in neurons during OGD. In light of the dual capacity of nicotinamide to directly inhibit caspase-3–like activity and maintain inhibitory phosphorylation of FOXO3a during OGD, our work suggests that nicotinamide maintains a regulatory “neuroprotective loop” through the independent modulation of caspase-3 and phosphorylated FOXO3a integrity.
In conclusion, we illustrate that nicotinamide, either directly or through a receptor-mediated mechanism, maintains both genomic DNA integrity and membrane PS asymmetry to provide broad cellular protection. Prevention of neuronal injury requires the activation of Akt1 and the subsequent modulation of mitochondrial membrane polarization, cytochrome c release, and caspase-3–and caspase-9–like activities, but remains independent of Apaf-1 expression. Furthermore, nicotinamide maintains inhibitory phosphorylation of FOXO3a and governs a unique regulatory loop for neuronal survival through the prevention of phosphorylated FOXO3a degradation by caspase-3 (Fig. 9).
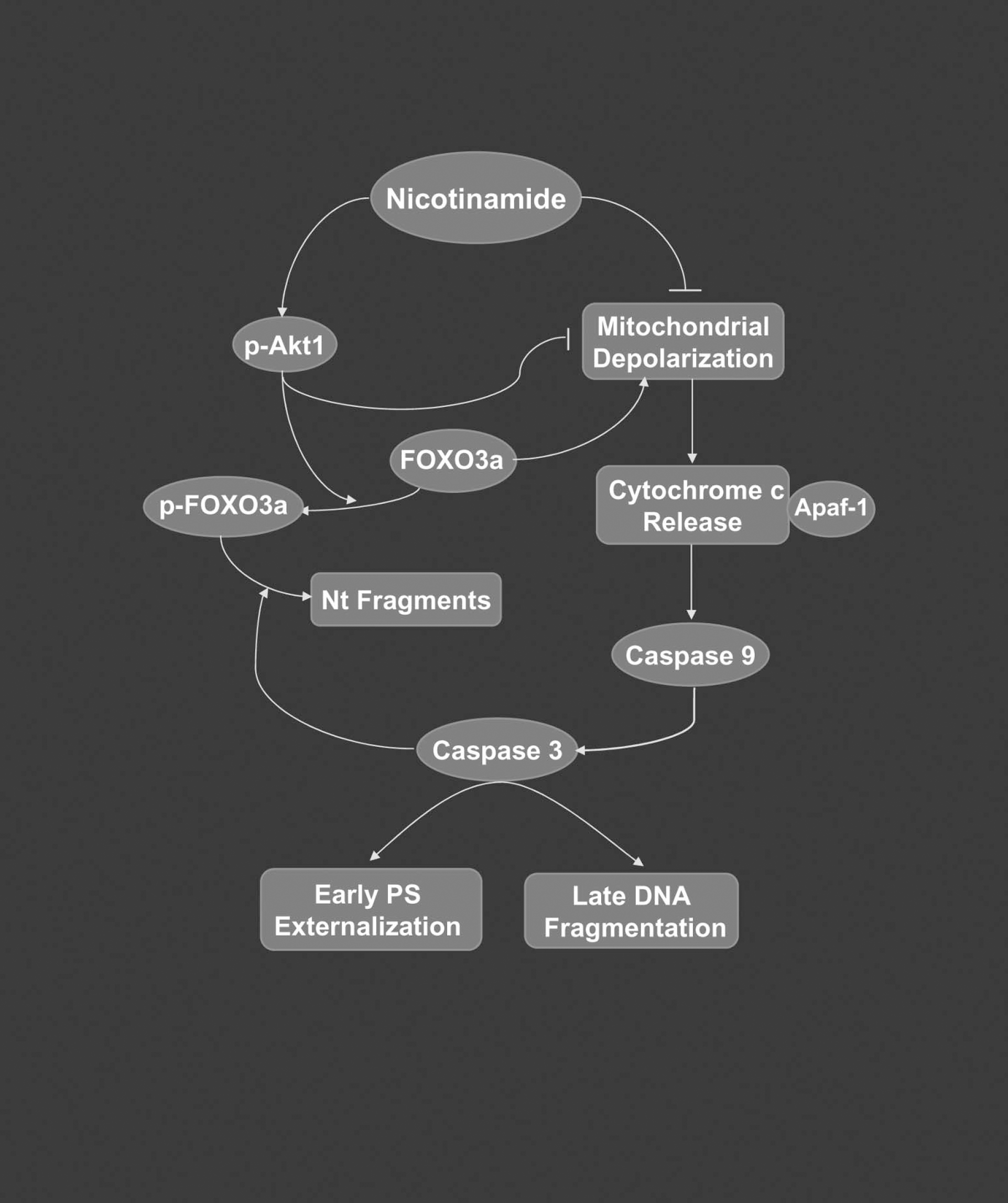
Nicotinamide prevents neuronal injury through a series of pathways that involve Akt1, FOXO3a, ΔΨm, and cysteine protease activity. Nicotinamide maintains cellular integrity and prevents phagocytic cellular removal through the maintenance of genomic DNA integrity and membrane PS asymmetry. Nicotinamide requires the activation of Akt1 and uses at least two parallel pathways to provide cellular protection. At one level, nicotinamide directly modulates ΔΨm and cytochrome c release to inhibit caspase-3– and caspase-9–like activities, but does not appear to involve Apaf-1 expression. Alternatively, nicotinamide maintains inhibitory phosphorylation of FOXO3a at the regulatory sites of Thr32 and Ser253 and oversees a regulatory loop for neuronal survival through the prevention of phosphorylated FOXO3a degradation by caspase-3.