
Abstract
PNU-101017 is a novel, imidazoquinoline amide and benzodiazepine receptor partial agonist that has high affinity for the GABAA receptor subtypes containing the α1 and α3 or α5 subunits. At each of these receptors, the compound is a partial agonist with approximately 50% of the intrinsic activity of the full agonist diazepam. In view of the previously demonstrated anti-ischemic effects of some GABA agonists, the purpose of this study was to determine the ability of PNU-101017 to salvage selectively vulnerable neuronal populations in the gerbil forebrain ischemia model. In an initial set of experiments, male gerbils were pretreated 30 minutes before ischemia induction (5 minutes) with PNU-101017 (3, 10, or 30 mg/kg intraperitoneally) and again 2 hours after reperfusion. In vehicle (0.05 N HCl)-treated gerbils, the loss of hippocampal CA1 neurons at 5 days was 80%. PNU-101017 was shown to produce a dose-related increase in CA1 neuronal survival; at either 10 or 30 mg/kg, the loss of CA1 neurons was only 21% (P < 0.005 versus vehicle). A second experiment, examined the therapeutic window for PNU-101017 using the dose level of 30 mg/kg intraperitoneally. Administration of the first of two doses (2 hours apart) at the time of reperfusion resulted in an identical decrease in CA1 damage at 5 days to that seen with preischemic treatment (P < 0.003 versus vehicle). Even with a delay of the initial dosing until 4 hours after reperfusion, PNU-101017 reduced CA1 neuronal loss to only 32% (P < 0.01 versus vehicle). In a third experiment in which the duration of the ischemic insult was increased to 10 minutes and the brains were not analyzed until 28 days after ischemia, daily PNU-101017 dosing for the full 28 days still significantly preserved CA1 neurons, although less effectively than in the milder 5 minute-ischemia model. The loss of dopaminergic nigrostriatal neurons was also reduced. The neuroprotective effect of PNU-101017 was not associated with any overt CNS depression and it did not correlate with hypothermia. This benzodiazepine-receptor partial agonist may have potential for the treatment of global cerebral ischemia.
Selective vulnerability of certain neuronal populations to brief episodes of cerebral ischemia seems to involve a complex cascade of biochemical events including, predominantly, the massive release of the excitatory amino acid neurotransmitters glutamate and aspartate (Benveniste et al., 1984; Hagberg et al., 1985; Meldrum et al., 1985), intracellular calcium overload (Meldrum et al., 1985; Siesjo and Bengtsson, 1989), and oxygen radical mechanisms (Siesjo et al., 1989; Hall and Braughler, 1989). However, whereas the cascade is multifactorial, there is overwhelming evidence that the fundamental insult is excitotoxic in nature. The initial trigger for the degeneration process is the massive liberation of excitatory neurotransmitters which can be exacerbated by persistent intracellular calcium overload and free radical injury to glutamatergic nerve terminals (Pelligrini-Giampietro et al., 1990).
In view of this excitotoxic hypothesis, one possible neuroprotective approach would be the application of compounds that counteract the excessive postischemic excitatory neurotransmission by enhancing inhibitory GABAergic transmitter function. To that end, several GABA-mimetic or GABA-potentiating compounds have been tried in models of global cerebral ischemia. These include the barbiturates thiopental (Bleyaert et al., 1978), methohexital (Kuroiwa et al., 1990), and pentobarbital, (Yoshida et al., 1983; Sternau et al., 1989) which potentiate Cl- channel opening and GABA binding, the benzodiazepine receptor agonist diazepam (Sternau et al., 1989; Huff and Schwartz, 1991; Voll and Auer, 1991; Schwartz et al., 1994, 1995), the GABAA receptor agonists muscimol (Sternau et al., 1989) and chlormethiazole (Cross et al., 1991; Shuaib et al., 1995), the GABAb receptor agonist baclofen (Sternau et al., 1989), and the GABA transaminase inhibitor valproic acid (Sternau et al., 1989). Each of these has been shown to exert some neuroprotective effect, generally assessed in regards to hippocampal CA1 neurons, when used as a preischemic treatment or within the first hour of reperfusion. In the gerbil forebrain ischemia model, only chlormethiazole has been shown to retain efficacy when treatment is delayed until 4 hours after reperfusion (Cross et al., 1991; Shuaib et al., 1995).
PNU-101017 (Fig. 1) is a novel imidazoquinoline benzodiazepine receptor agonist (VonVoigtlander et al., 1995; Im et al., 1996). This compound is unique compared with GABA mimetics studied thus far in models of cerebral ischemia in that it is a partial agonist at the benzodiazepine site on the GABAA receptor. In vitro studies using rat α1, β2, and γ2 receptors expressed in human embryonic kidney cells have shown that PNU-101017 potentiates GABA-stimulated Cl− currents at low concentrations, whereas at higher concentrations (>1 µmol/L), the compound acts as an inverse agonist at a second site on the GABAA receptor. This latter action attenuates GABA-stimulated Cl− currents with a total suppression of agonistic activity occurring at 40 µmol/L. The agonistic property is antagonized by the benzodiazepine-neutral antagonist flumazenil indicating that the facilitation of GABA-stimulated Cl− conductance is mediated by the benzodiazepine receptor (Im et al., 1996). In rat cortical membranes, the Ki for inhibition of flunitrazepam binding is 3.4 nmol/L (Sethy and Wu, 1996). The compound has good brain penetration, as studied in rats, with a 15 mg/kg oral dose level producing greater than 50% occupancy of cortical benzodiazepine receptors (Sethy and Wu, 1996). In rodent in vivo experiments, PNU-101017 doses ranging from 0.3 to 30 mg/kg have been shown to possess anxiolytic and anticonvulsant effects that are blocked by flumazenil (Sethy and Wu, 1996; Tang et al., 1996). In contrast, PNU-101017 is devoid of sedative, physical dependence-producing, and ethanol-potentiating properties (Tang et al., 1997). In the present investigation, the potential neuroprotective efficacy of the compound has been examined in the context of the gerbil forebrain ischemia model. As described herein, PNU-101017 effectively protects the highly vulnerable hippocampal CA1 neurons in gerbils subjected to 5 minutes of forebrain ischemia even when initial dosing is delayed until 4 hours after reperfusion in the absence of overt CNS-depressant properties. The compound is still effective in salvaging a portion of the CA1 population in a more severe 10- minute version of the gerbil model and partially protects nigrostriatal dopamine neurons.
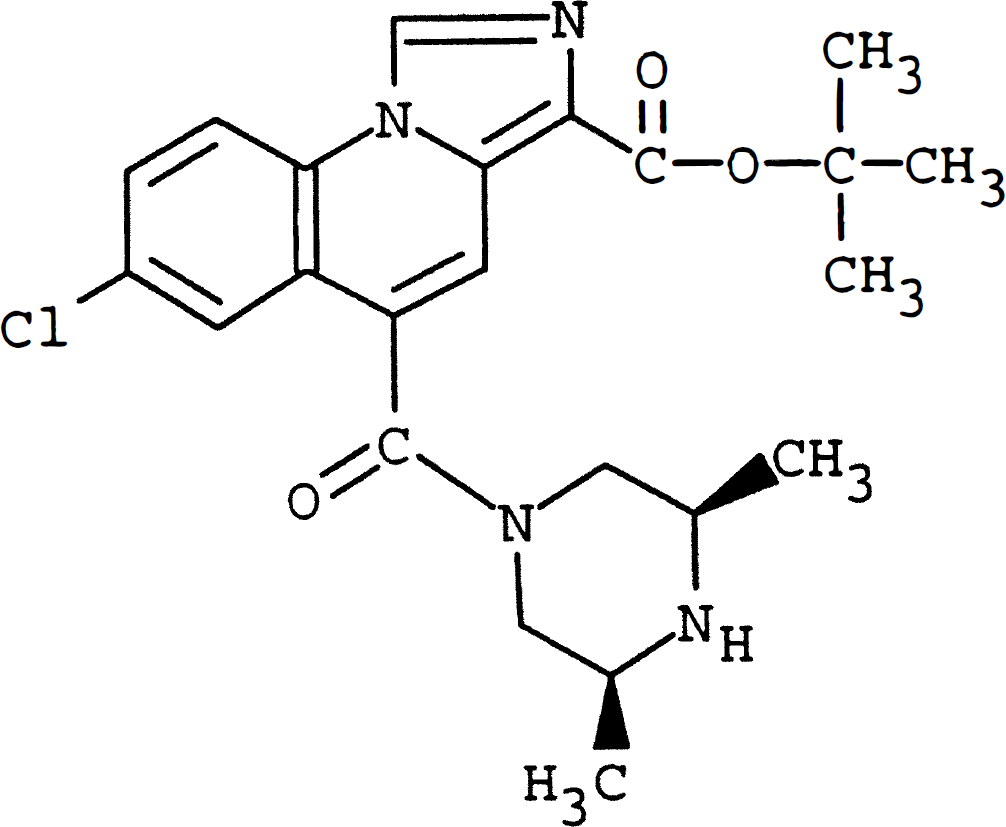
Chemical structure of the benzodiazepine receptor, partial agonist imidazoquinoline PNU-101017.
METHODS
All experiments were performed in strict compliance with the National Institutes of Health Guide for the Care and Use of laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Pharmacia & Upjohn Laboratories.
Ischemia procedure
Male Mongolian gerbils, weighing 50 to 60 g, were obtained from Tumblebrook Farms (West Brookfield, MA, U.S.A.) and anesthetized with methoxyflurane. A 1- to 2-cm midline throat incision provided access to both carotid arteries, which were clamped with microaneurysm clamps. The clamps were removed after 5 (or 10 minutes) of near-complete ischemia. The animals were placed in a warming box with an ambient temperature maintained at 37°C during the ischemic insult and until the animals regained their righting reflex after reperfusion. Previous studies from this laboratory showed that this warming procedure successfully maintains both rectal and brain temperatures of untreated animals at a minimum of 35.5°C while the animals are kept in the chamber (Hall et al., 1993).
PNU-101017 dosing
In an initial dose-response experiment, gerbils were treated with vehicle (0.05 N HCl) or PNU-101017 (3, 10 or 30 mg/kg) intraperitoneally 30 minutes before ischemia and again 2 hours after reperfusion. The tested dose range was selected based on previous rodent studies that documented the anxiolytic actions of the compound and its high level of cortical benzodiazepine receptor occupancy (Sethy and Wu, 1996; Tang et al., 1996). The administration of the second dose at 2 hours after reperfusion was consistent with previous dosing with other effective compounds we have tested in the gerbil (Hall and Pazara, 1988; Hall et al., 1993). The 0.05 N HCl vehicle has been used previously for intraperitoneal dosing with other test compounds and found to be devoid of toxicity or acute distress production (Hall et al., 1988). Killing and histopathology were performed on the fifth day after ischemia.
In a second therapeutic window experiment, gerbils were administered an initial 30 mg/kg intraperitoneal dose of PNU-101017 immediately after, or at 4 or 24 hours after reperfusion. In each case, a second dose was administered 2 hours after the first. Killing and histopathology were again performed on day 5.
In the third experiment, the duration of ischemia was prolonged to 10 minutes and the histopathology was not assessed until 28 days after the ischemic insult. In this instance, the 30 mg/kg intraperitoneal doses on the day of ischemia (30 minutes before plus 2 hours after) were followed by a 30 mg/kg intraperitoneal dose once a day for 28 days.
Hippocampal CA1 histology
Five-minute forebrain ischemic model.
After 5 days, the animals were deeply reanesthetized with methoxyflurane and perfused intracardially with phosphate-buffered saline (pH 7.2) until the effluent was cleared of blood (2 minutes), followed by perfusion with 10% formaldehyde, 10% acetic acid, and 80% methanol. The brains were removed and stored overnight in 10% formaldehyde, 10% acetic acid, and 80% methanol. They were then blocked and embedded in paraffin. Five-micron thick cross-sections were taken through the dorsal hippocampus (2 to 4 mm posterior to Bregma; Paxinos and Watson, 1986) and mounted on gelatin-coated slides and stained with cresyl violet (Hall and Pazara, 1988). Under light microscopy (320x magnification), all normal-appearing CA1 pyramidal neurons in a 315 µ length of the medical CA1 region were counted bilaterally and averaged. Two adjacent sections were examined per animal and the counts averaged. In animals that showed significant asymmetry in regards to the CA1 neuronal counts (>50% difference between hemispheres; about 10% of the animals), only the hemisphere with the lower count (i.e., more severely affected) was included in the analysis.
Ten-minute forebrain ischemic model.
In the 10 minute-ischemic experiments, the animals were deeply reanesthetized with methoxyflurane at 28 days after ischemia and their brains perfusion-fixed as described above. Fifty-micron thick serial sections through the substantia nigra were prepared using a sliding microtome and then processed as free-floating sections for tyrosine hydroxylase immunocytochemistry (anti-tyrosine hydroxylase antibodies obtained from Eugene Tech International, Inc., Ridgefield Park, NJ, U.S.A.) (Hall et al., 1996). The number of tyrosine hydroxylase-positive neurons was manually counted in a rectangular area 0.6 × 1.8 mm at 20x encompassing the pars reticulata and pars compacta of the substantia nigra in a single cross section 5.2 mm posterior to Bregma (Paxinos and Watson, 1986). The raw numbers were converted to the number of tyrosine hydroxylase-positive neurons per square millimeter. The two sides were averaged for each animal. Fifty-micron thick sections through the dorsal hippocampus were also prepared and stained with cresyl violet for an assessment of CA1 neuronal preservation as described above.
Statistical analysis.
The neuronal counts were consistently performed in a blind manner as to the identification of vehicle- or PNU-101017-treated gerbils. All data are expressed as mean ± SD. Statistical evaluation was performed using an analysis of variance followed by a Student's t-test with Bonferroni correction for multiple comparisons where there were either multiple dose groups or treatment initiation times.
RESULTS
Dose-response assessment of the effects of PNU-101017 pretreatment on 5 day postischemic CA1 neuronal survival after 5 minutes of ischemia
It should be noted that PNU-101017 did not potentiate the duration of postanesthetic/postischemic CNS depression typically observed in the gerbils. In vehicle-treated animals (N = 6), the mean time to regaining the righting reflex after methoxyflurane anesthesia plus a 5-minute ischemic episode was 17.2 ± 7.7 minutes while in a PNU-101017-treated group, the recovery time was 19.6 ± 9.5 minutes (not significant versus vehicle group).
Fig. 2 shows the dose-related effect of PNU-101017 (30 minute pretreatment plus 2 hours posttreatment) enhancing the 5-day postischemic survival of hippocampal CA1 neurons. In vehicle (0.05 N HCl)-treated gerbils, the loss of CA1 neurons at 5 days was 80%. PNU-101017 was shown to produce a dose-related increase in CA1 neuronal survival. The 3 mg/kg intraperitoneal dose (×2) reduced CA1 loss to 41% (P < 0.05 versus vehicle). At either 10 or 30 mg/kg, the loss of CA1 neurons was decreased to 21% (P < 0.005 versus vehicle).
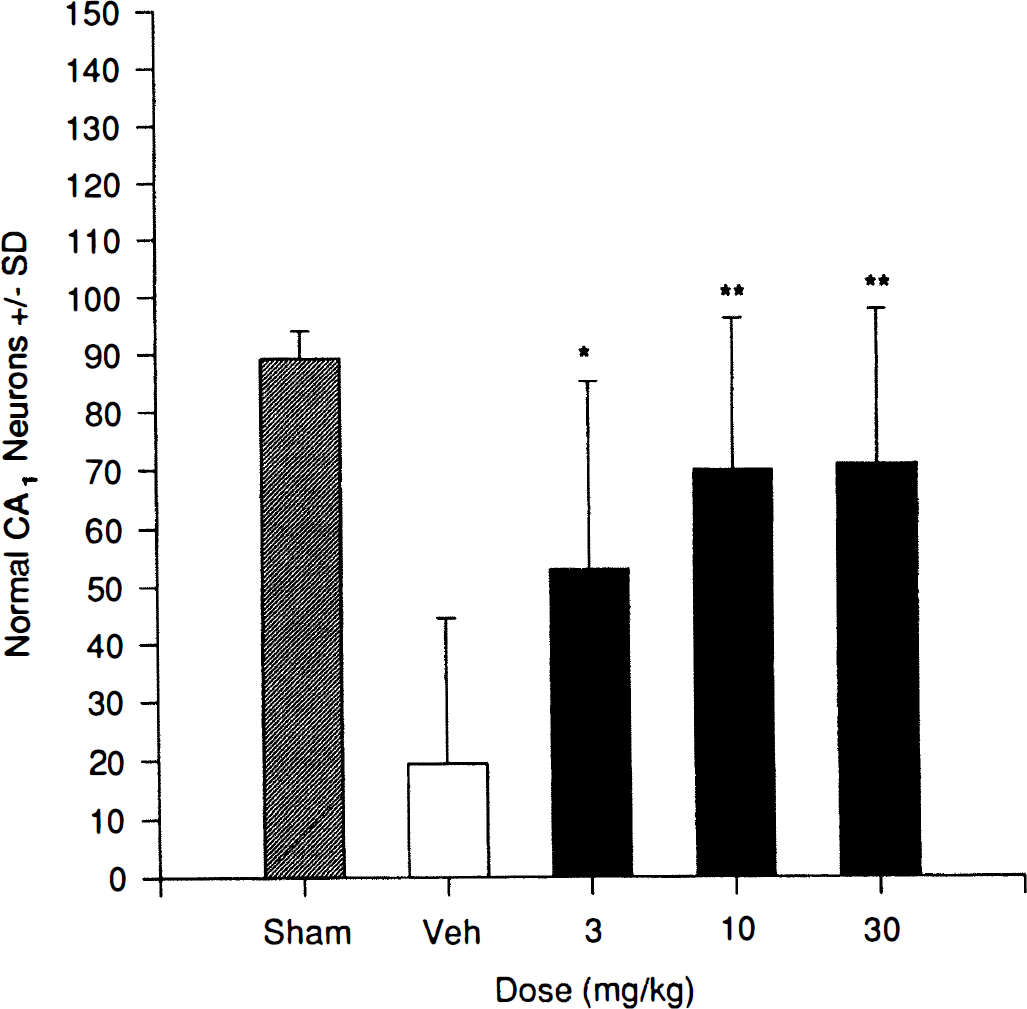
Dose-response relationship for PNU-101017 preservation of hippocampal CA1 neurons assessed at 5 days after a brief episode (5 minutes) of near-complete forebrain ischemia in gerbils. Indicated intraperitoneal doses were administered at 30 minutes before ischemia and repeated at 2 hours after reperfusion. Sham, group not subjected to ischemia; Veh, vehicle-treated group. Values are mean ± SD for 8 to 10 animals per group. *P < 0.05 versus vehicle-treated group. **P < 0.005 versus vehicle.
Dose-response assessment of the effects of PNU-101017 pretreatment on intraischemic and early postreperfusion body temperature
In view of the fact that benzodiazepines such as diazepam can produce profound hypothermia which probably contributes to the neuroprotective action of the compound (Schwartz et al., 1994), the possible hypothermic property of the benzodiazepine receptor partial agonist PNU-101017 was examined in regards to changes in rectal temperatures in ischemic gerbils under conditions identical to our experiments discussed above. Table 1 provides the results of these experiments. The baseline (i.e., postanesthesia) rectal temperatures typically range from 35 to 37°C. Pretreatment with PNU-101017 showed a dose-related trend toward lower temperatures, but this was not close to statistical significance. During ischemia and the initial minute of reperfusion, rectal temperature in the the high dose (30 mg/kg intraperitoneally) group was significantly lower than in vehicle-treated animals despite the fact that the animals were maintained in a warming box. This difference lost significance by 5 minutes after reperfusion. In contrast, the mean temperatures in the 10 mg/kg dose, which produced neuroprotection equivalent to that of the higher dose (Fig. 2), was statistically indistinguishable from that of the vehicle-treated group over the experimental time course.
Effects of PNU-101017 pretreatment on rectal temperature in gerbils

Effects of PNU-101017 pretreatment (10 or 30 mg/kg intraperitoneally at 30 minutes before ischemia) on intraischemic and early postreperfusion rectal temperature in gerbils subjected to a 5 minute episode of near-complete forebrain ischemia. Values are mean ± SD for 5 animals per group.
P < 0.05 versus vehicle-treated group at same time point.
Therapeutic window for PNU-101017 preservation of hippocampal CA1 neurons after 5 minutes of ischemia
Fig. 3 shows the therapeutic window of PNU-101017 using the 30 mg/kg intraperitoneal dose level. Administration of the first of two doses (2 hours apart) at the time of reperfusion decreased CA1 neuronal loss at 5 days to only 20% (P < 0.003 versus vehicle). This was essentially equivalent to the effects of pretreatment at this dose level (Fig. 2). Even delaying the initial dosing until 4 hours after reperfusion still significantly reduced CA1 loss to 32% (P < 0.01 versus vehicle). However, witholding treatment until 24 hours after reperfusion did not result in significant preservation of CA1 neurons.
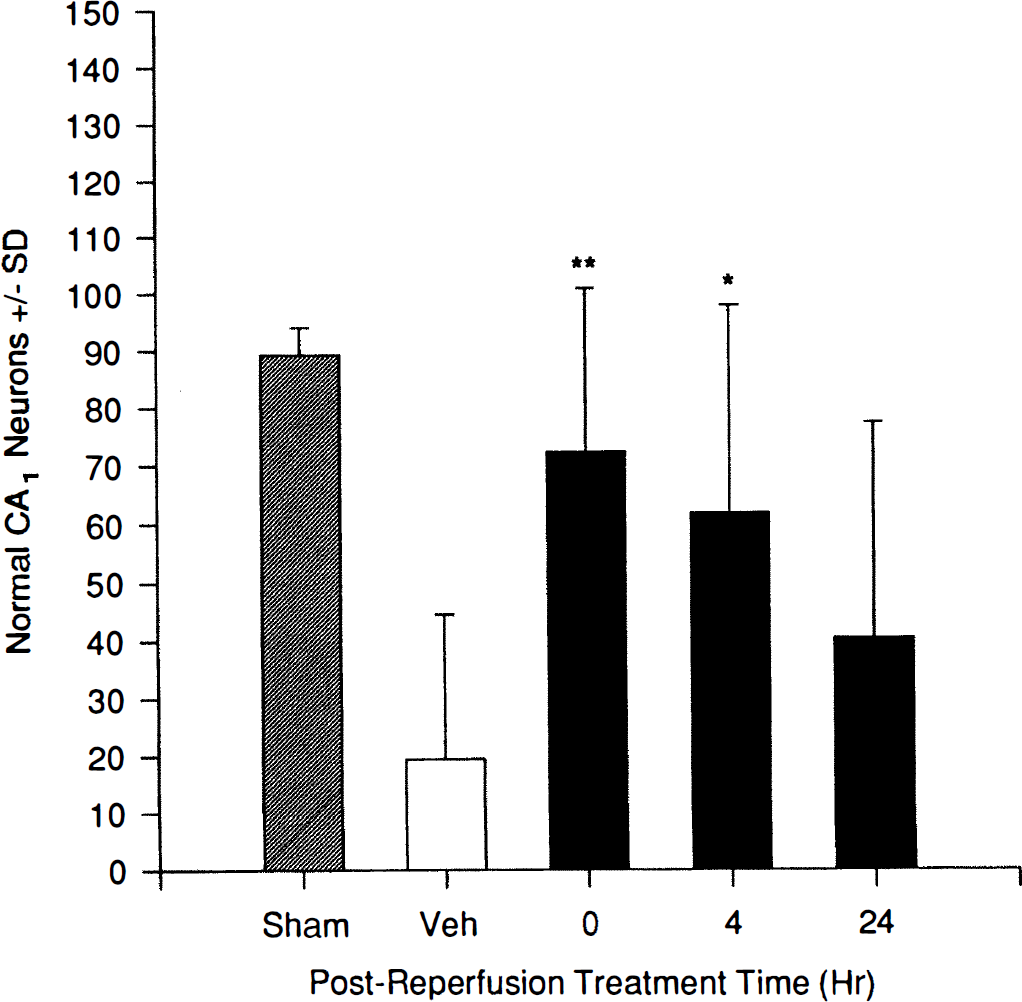
The therapeutic window for PNU-101017 preservation of hippocampal CA1 neurons assessed at 5 days after a brief (5 minutes) episode of near-complete forebrain ischemia in gerbils. A 30 mg/kg intraperitoneal dose was administered immediately or at 4 or 24 hours after reperfusion and repeated in each case 2 hours later. Sham, group not subjected to ischemia; Veh, vehicle-treated group. Values are mean ± SD for 8 to 10 animals per group. **P < 0.005 versus vehicle-treated group; *P < 0.01.
Fig. 4 shows examples of the CA1 histological damage observed 5 days after ischemic insult and the protective effects of a 30 mg/kg dose level of PNU-101017 when administered either immediately after reperfusion or 4 hours later. Fig. 5 shows the medial CA1 region from the same four examples at higher power.
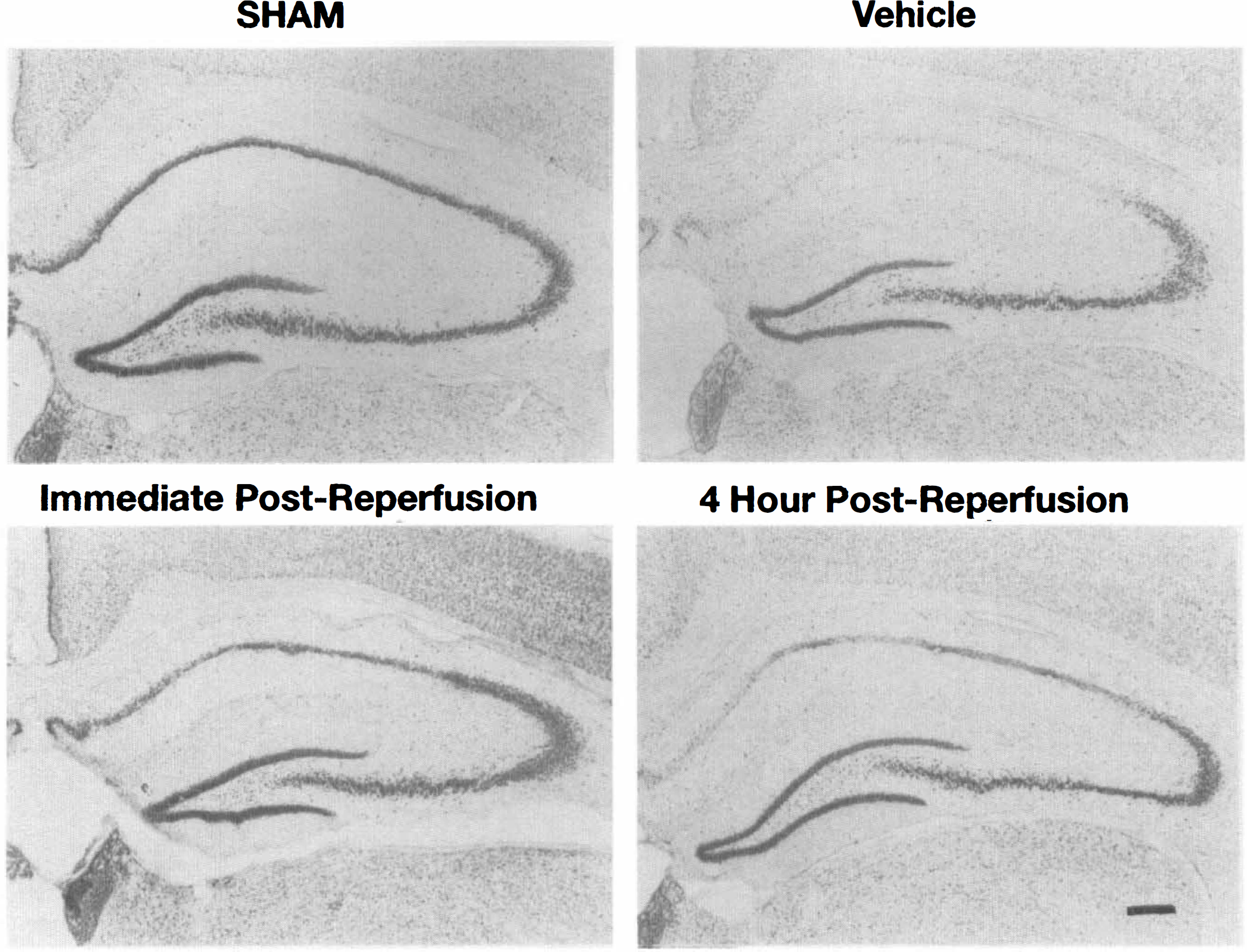
Low-power photomicrographic examples (cresyl violet stained) of the hippocampal CA1 region in gerbils not subjected to ischemia (Sham control), subjected to ischemia 5 days earlier and treated with only vehicle, and subjected to ischemia 5 days earlier and treated with PNU-101017 (30 mg/kg intraperitoneally at the time of reperfusion (plus 2 hours later) or at 4 hours after reperfusion (plus 2 hours later). The latter example is actually somewhat conservative compared with the greater mean CA1 preservation reflected in Fig. 3.
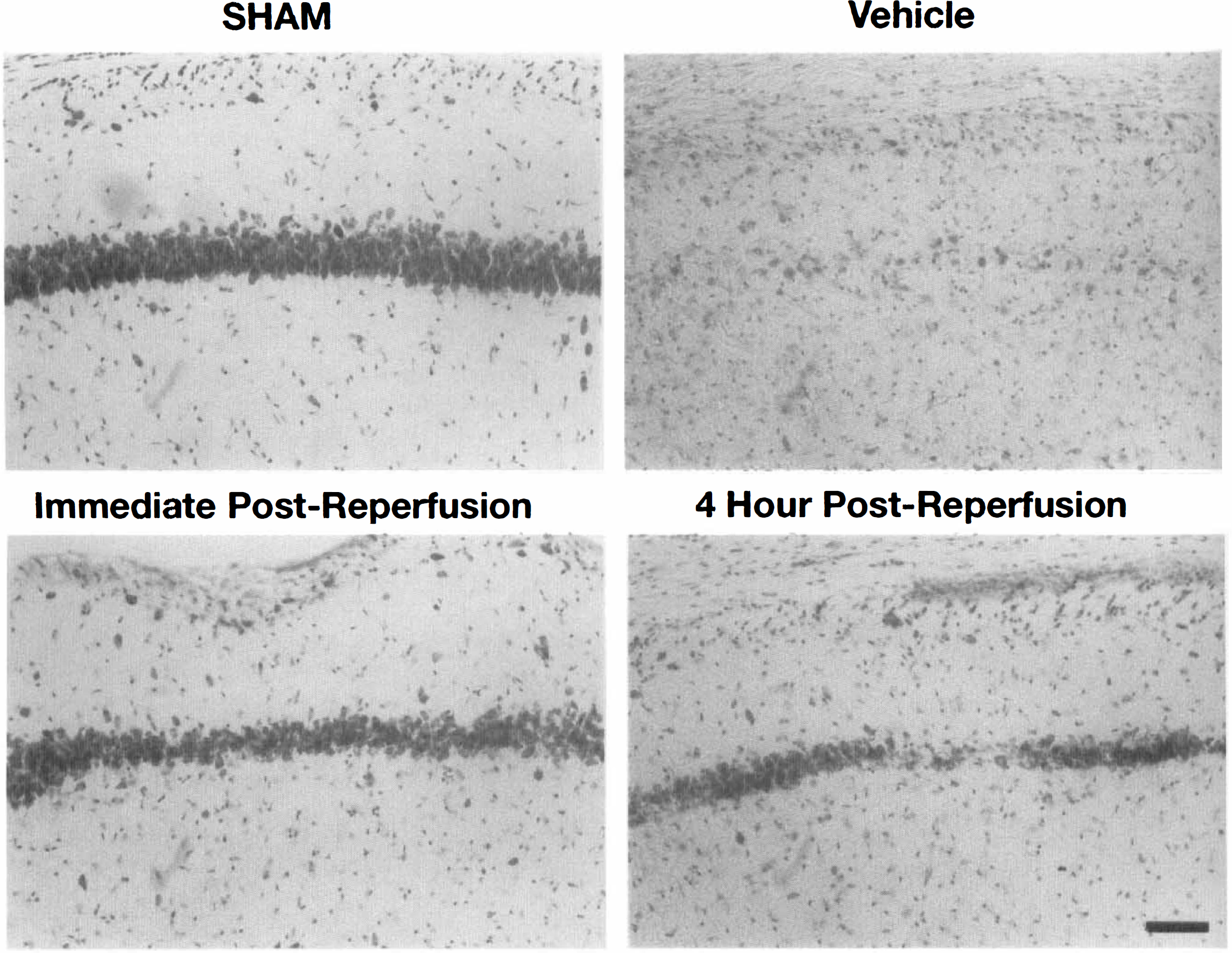
High-power photomicrographic examples (cresyl violet stained) of the hippocampal CA1 region in gerbils not subjected to ischemia (Sham control), subjected to ischemia 5 days earlier and treated with only vehicle, and subjected to ischemia 5 days earlier and treated with PNU-101017 (30 mg/kg intraperitoneally at the time of reperfusion (plus 2 hours later) or at 4 hours after reperfusion (plus 2 hours later). The latter example is actually somewhat conservative compared with the greater mean CA1 preservation reflected in Fig. 3.
Effect of PNU-101017 on 28-day postischemic CA1 and nigrostriatal neuronal survival after 10 minutes of ischemia
Additional experiments examined the efficacy of PNU-101017 in gerbils subjected to a 10-minute episode of near-complete forebrain ischemia with histopathology being assessed at 28 days after ischemia. Fig. 6 shows that the neuroprotective effect is much more modest than that seen with only 5 minutes of forebrain ischemia (compare with Figs. 2 and 3). Nevertheless, a significantly greater number of CA1 neurons was observed in the PNU-101017-treated animals compared with the vehicle-treated group. In vehicle-treated animals, the loss of CA1 neurons was 85% whereas in the PNU-101017-treated group the loss was 68%. This shows that the compound is capable of neuroprotection in the face of a more prolonged ischemia and that the CA1 protection is still manifested at 28 days after the insult.
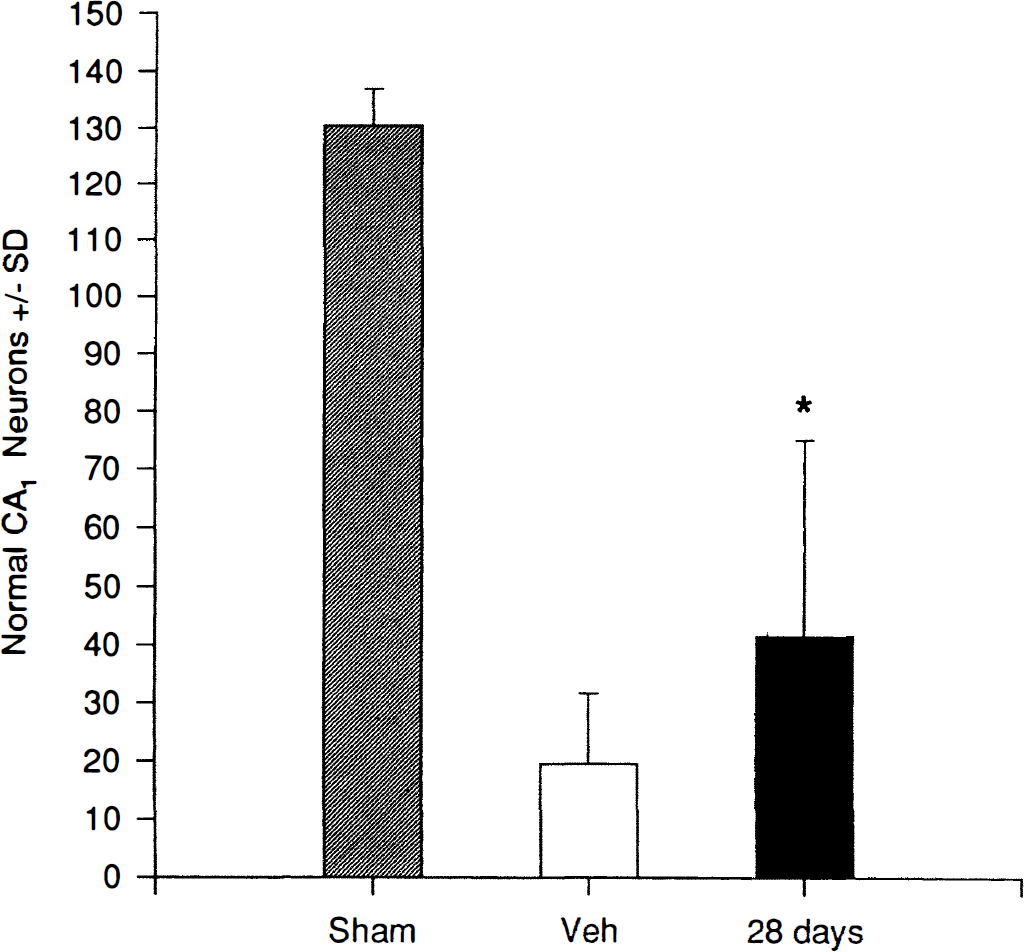
PNU-101017 preservation of hippocampal CA1 neurons assessed at 28 days after a more prolonged episode (10 minutes) of near-complete forebrain ischemia in gerbils. Indicated intraperitoneal doses were administered at 30 minutes before ischemia and repeated at 2 hours after reperfusion and then daily for a total of 28 days. Sham, group not subjected to ischemia; Veh, vehicle-treated group. Values are mean ± SD for 8 to 10 animals per group. * P < 0.01 versus vehicle-treated group. Note: The cell counts in the sham group are higher in these experiments in comparison to those in Figs. 2 and 3 because of the use of thicker (50 µm) sections which, on up and down focusing, yield higher counts.
In addition to CA1 degeneration, the 10-minute version of the ischemic model includes a progressive loss of approximately 40 to 50% of the nigrostriatal dopaminergic neuronal population (Hall et al., 1996). Similar nigrostriatal degeneration has been observed with shorter, but repetitive, episodes of forebrain ischemia in the gerbil (Schuaib et al., 1992; Saji and Volpe, 1993; Saji et al., 1994). PNU-101017 treatment significantly reduced, although not completely, the degeneration of nigral dopamine neurons measured at 28 days after ischemia (Fig. 7). Histological examples of nigral cell loss in vehicle- and PNU-101017-treated gerbils undergoing 10 minutes of ischemia are shown in Fig. 8.
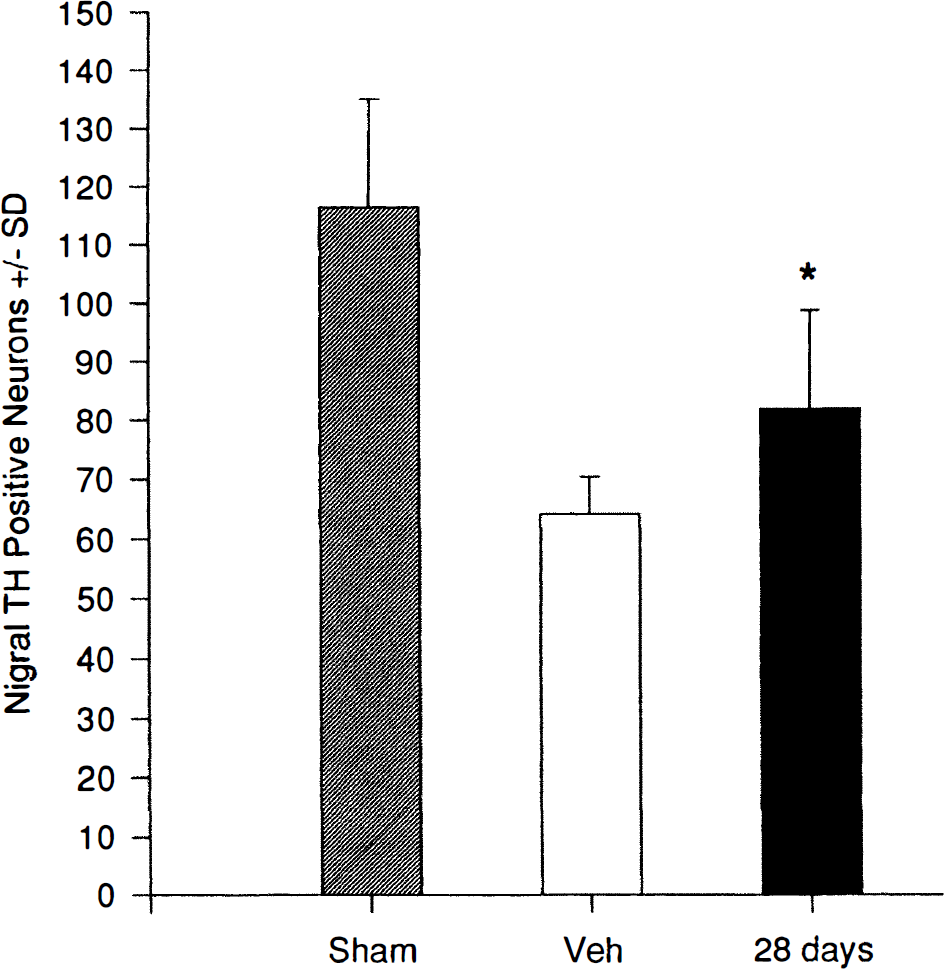
PNU-101017 preservation of nigrostriatal dopamine neurons assessed at 28 days after a more prolonged episode (10 minutes) of near-complete forebrain ischemia in gerbils. Indicated intraperitoneal doses were administered at 30 minutes before ischemia and repeated at 2 hours after reperfusion and then daily for a total of 28 days. Sham, group not subjected to ischemia; Veh, vehicle-treated group. Values are mean ± SD for 8 to 10 animals per group. * P < 0.01 versus vehicle-treated group.
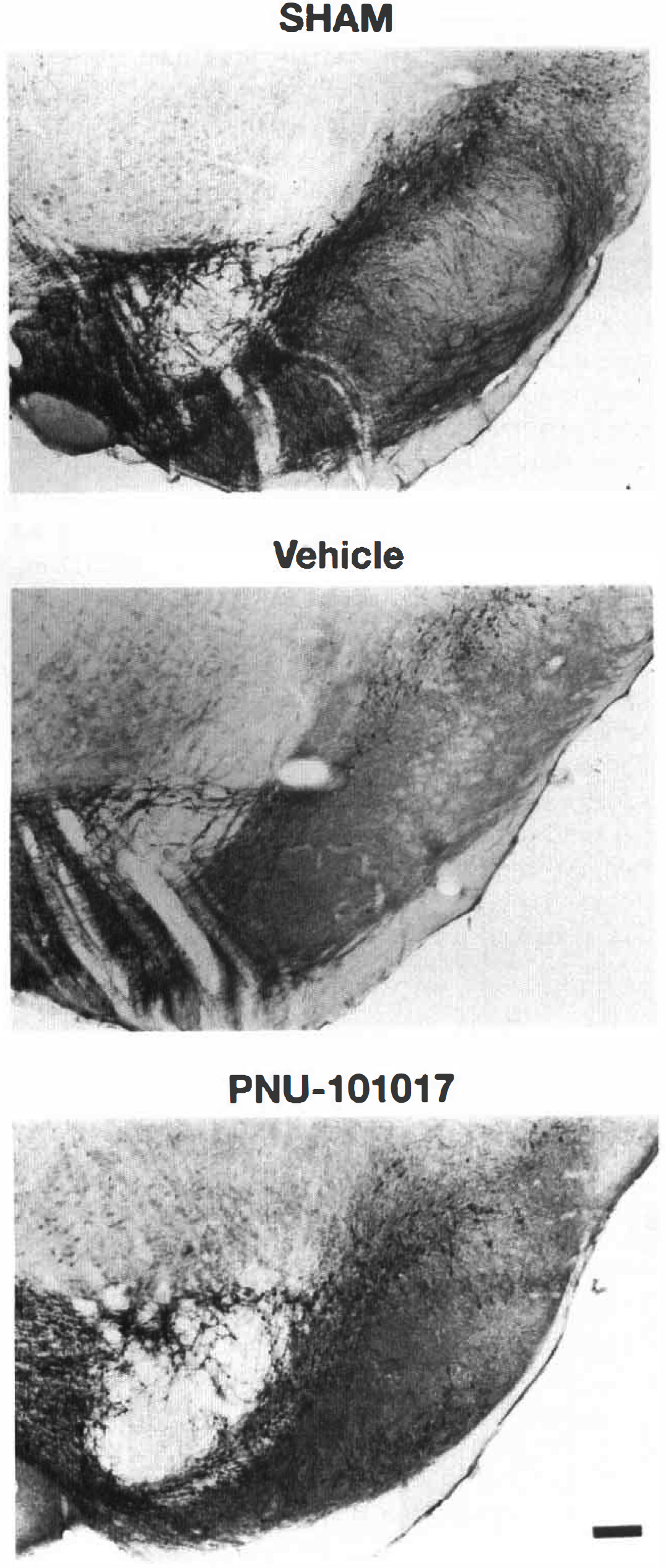
Typical photomicrographic examples (320x; tyrosine hydroxylase-immunostained) of nigral dopamine neurons in a sham, nonischemic animal. The loss of nigral neurons in an animal that received vehicle treatment for 28 days after a 15-minute episode of forebrain ischemia and the partial preservation of nigral neurons in an ischemic animal treated with PNU-101017 (30 mg/kg intraperitoneally at the time of reperfusion plus 2 hours later and then daily for 28 days.)
DISCUSSION
The results show that the novel, benzodiazepine receptor partial agonist PNU-101017 can effectively decrease injury to the highly vulnerable hippocampal CA1 neurons after a 5-minute episode of near-complete forebrain ischemia in gerbils. Whereas the extent of CA1 loss in this model is typically 80% by 5 days, PNU-101017 pretreatment (10 or 30 mg/kg intraperitoneally ×2) resulted in a reduction of the neuronal loss to only 20%. The efficacy of the benzodiazepine receptor partial agonist is still evident after a 10-minute ischemic duration, albeit less than that observed after 5 minutes of ischemia. Furthermore, in the experiments using the more severe, prolonged model, the partial CA1 preservation was still apparent and significant at 28 days after ischemia. This indicates that the preservation of neurons at the assessment on the fifth day after ischemia is predictive of a lasting neuroprotective effect.
PNU-101017 was also significantly effective in attenuating degeneration of the nigrostriatal dopamine neurons. The mechanism of degeneration of this neuronal population is believed to involve initial ischemic injury to the nigrostriatal dopamine nerve terminals in the striatum. The terminal damage seems to be caused by increased dopamine release, metabolism by monoamine oxidase B, and the consequent production of oxygen radicals. If this oxygen radical-mediated injury to the striatal terminals is sufficiently intense, it can trigger a retrograde degeneration of the nigral dopaminergic cell bodies (Hall et al., 1996). Consistent with this scenario, antioxidant agents such as the pyrrolopyrimidine lipid peroxidation inhibitor PNU-101033E (Andrus et al., 1997) or dopaminergic agonists, which can decrease dopamine turnover such as pramipexole (Hall et al., 1996), also protect the nigrostriatal neurons in the present model. However, another mechanistic factor in the nigrostriatal degeneration concerns the postischemic loss of nigral GABAergic neurons (Saji et al., 1994) that may normally inhibit (i.e., tone down) the firing rate of the nigrostriatal neurons. The GABA agonist PNU-101017 may exert its neuroprotective effect by replacing this lost inhibitory function.
The degree of CA1 protection for PNU-101017 compares quite favorably with other GABA-mimetics that have been shown to be effective in the gerbil forebrain ischemia paradigm used in this study. These include the ultrashort-acting barbiturate methohexital (Kuroiwa et al., 1990), the intermediate-acting barbiturate pentobarbital (Sternau et al., 1989), the benzodiazepine-receptor full agonist diazepam (Sternau et al., 1989; Huff and Schwartz, 1991) and the GABA agonists baclofen (Sternau et al., 1989), valproic acid (Sternau et al., 1989), muscimol (Sternau et al., 1989), and most recently, chlormethiazole (Cross et al., 1991; Shuaib et al., 1995). The protective effects of PNU-101017 also equal those reported for the glutamate AMPA receptor antagonist NBQX (Sheardown et al., 1990; Judge et al., 1991).
Perhaps most interesting is the finding that PNU-101017 retains protective efficacy equal to that seen with pre- or immediate posttreatment even when treatment is delayed until 4 hours after reperfusion. With the exception of chlormethiazole (Shuaib et al., 1995), none of the GABA agonists thus far tested in the gerbil forebrain ischemic model have a therapeutic window beyond the first 2 hours after reperfusion. For example, methohexital infusion loses efficacy if delayed beyond 40 minutes after reperfusion (Kuroiwa et al., 1990). In another study, CA1 protective efficacy with pentobarbital, baclofen, or diazepam was only seen when these agents were administered before ischemia induction (Sternau et al., 1989). Seemingly at odds with these results is the report that thiopental was still protective in a model using a monkey undergoing 16 minutes of forebrain ischemia that was delayed until 60 minutes after reperfusion (Bleyaert et al., 1978). However, profound hypothermia was probably a complicating factor in that study because no attempt was made to regulate body or brain temperature. Similarly, postreperfusion treatment with diazepam has been reported by another group to still be effective in the gerbil (Schwartz et al., 1994), but the postreperfusion delay used was only 30 minutes and substantial hypothermia (4 to 6°C) was allowed to take place. Thus, the postischemic actions of diazepam may have been largely caused by hypothermia induction within a time frame in which hypothermia is still able to be protective. The same group has also published data in the rat forebrain ischemic model purporting CA1 protection with diazepam even when delayed until 2 hours after reperfusion which they admit may be partly caused by hypothermia (Schwartz et al., 1995). However, hypothermia cannot provide the entire explanation for the neuroprotective action of diazepam because, in the same study, intrahippocampal injection of the drug significantly protected the CA1 neurons without producing hypothermia.
Whereas PNU-101017 is also capable of producing mild hypothermia, this does not logically explain the observed neuroprotection. First of all, the neuroprotective efficacy of the 10- and 30-mg/kg dose levels was equivalent, whereas only the higher dose produced a significant decrease in body temperature. Secondly, PNU-101017 was statistically just as neuroprotective when its administration was delayed until 4 hours after reperfusion as when it was first administered before ischemia. As already noted, the CA1 protective influence of hypothermia in rodent forebrain ischemia models has only been shown when cooling is instituted within the first 30 minutes after reperfusion (Busto et al., 1989). Therefore, it is implausible that a hypothermic mechanism is operative when treatment is delayed until 4 hours after ischemia. Thirdly, Dietrich et al. (1993) have shown that intraischemic but not postischemic hypothermia (30°C) produces chronic CA1 protection. Thus, the CA1 and nigrostriatal protection observed with PNU-101017 extended to 28 days is not a consequence of the very modest hypothermic effects the compound.
Also, the neuroprotective action of PNU-101017 is not secondary to any overt depression of CNS excitability. Although the compound is an effective benzodiazepine receptor partial agonist, it has been previously shown to be devoid of sedative properties (Tang et al., 1997). This was confirmed in the present results that show that neuroprotective doses of PNU-101017 did not significantly prolong recovery from the rather profound CNS depression associated with methoxyflurane anesthesia plus a 5-minute episode of near-complete forebrain ischemia. Therefore, whereas the neuroprotective activity of the compound is no doubt caused by its ability to enhance GABA transmission and thus counteract excitotoxic degenerative mechanisms, the degree of GABAergic potentiation required is apparently separable from the high degree of CNS depression associated with neuroprotective doses of most full benzodiazepine agonists or other GABA potentiating agents.