
Abstract
Delayed hippocampal neurodegeneration after transient global ischemia is mediated, at least in part, through the activation of terminal caspases, particularly caspase-3, and the subsequent proteolytic degradation of critical cellular proteins. Caspase-3 may be activated by the membrane receptor-initiated caspase-8–dependent extrinsic pathway and the mitochondria-initiated caspase-9–dependent intrinsic pathway; however, the precise role of these deduced apoptosis-signaling pathways in activating caspase-3 in ischemic neurons remains elusive. The authors cloned the caspase-9 gene from the rat brain and investigated its potential role in mediating ischemic neuronal death in a rat model of transient global ischemia. Caspase-9 gene expression and protease activity were extremely low in the adult brain, whereas they were developmentally upregulated in newborn rats, especially at postnatal 12 weeks, a finding consistent with the theory of an essential role for caspase-9 in neuronal apoptosis during brain development. After 15-minute transient global ischemia, caspase-9 was overexpressed and proteolytically activated in the hippocampal CA1 neurons at 8 to 72 hours of reperfusion. The temporal profile of caspase-9 activation coincided with that of cytochrome c release and caspase-3 activation, but preceded CA1 neuronal death. Immunoprecipitation experiments revealed that there was enhanced formation of Apaf-1/caspase-9 complex in the hippocampus 8 and 24 hours after ischemia. Furthermore, intracerebral ventricular infusion of the relatively specific caspase-9 inhibitor N-benzyloxycarbonyl-Leu-Glu-His-Asp-fluoro-methylketone before ischemia attenuated caspase-3–like activity and significantly enhanced neuronal survival in the CA1 sector. In contrast, inhibition of caspase-8 activity had no significant effect on caspase-3 activation or neuronal survival. These results suggest that the caspase-9–dependent intrinsic pathway may be the primary mechanism responsible for the activation of caspase-3 in ischemic hippocampal neurons.
Activation of terminal caspases, particularly caspase-3, may play an important role in mediating neuronal cell death after transient focal or global ischemia (Graham and Chen, 2001). Caspase-3 gene expression and activity are increased selectively in injured neurons that are destined to die after ischemia (Gillardon et al., 1997; Chen et al., 1998; Himi et al., 1998; Namura et al., 1998; Ni et al., 1998; Ouyang et al., 1999). Furthermore, pharmacologic inhibition or molecular blockage of caspase-3 and caspase-7 activity significantly decreases infarct size or attenuates the loss of hippocampal CA1 neurons after transient cerebral ischemia (Hara et al., 1997; Chen et al., 1998; Himi et al., 1998; Fink et al., 1998; Gillardon et al., 1999; Xu et al., 1999). Caspase-3 is thought to mediate ischemic neuronal death through proteolytic degradation of critical intracellular functional proteins such as poly(ADP-ribose)polymerase, DNA protein kinase, and an inhibitor of caspase-activated deoxyribonuclease (Chen et al., 1998; Shackelford et al., 1999; Cao et al., 2001b). Despite emerging evidence suggesting a pivotal role for caspase-3 in the final execution of ischemic neuronal death, the precise mechanism by which caspase-3 is activated in ischemic neurons is not fully understood.
Among the prime molecular pathways leading to caspase-3 activation in mammalian cell apoptosis are the so-called intrinsic (caspase-9–dependent) and extrinsic (caspase-8–dependent) pathways. The intrinsic pathway is initiated when cytochrome c is released from the injured mitochondria and subsequently complexes with Apaf-1 in the presence of dATP or its analogues, forming the large apoptosome complex that recruits and activates caspase-9, and then activates caspase-3 (Li et al., 1997). In the extrinsic pathway, caspase-8 is activated through the Fas or tumor necrosis factor-α membrane-receptor systems (Scaffidi et al., 1998). Activated caspase-8 then activates caspase-3 through direct proteolytic action or through enhancing the intrinsic pathway by activating the mitochondria-damaging proapoptotic Bcl2 family member Bid (Wang et al., 1996; Gross et al., 1999). Although the relative contributions of these pathways in ischemic neuronal death, especially in hippocampal CA1 neurodegeneration, are currently unclear, there are some indications that both pathways may be involved. Several recent studies suggest that Fas-associated apoptosis may occur in brain and spinal cord as a result of ischemia (Martin-Villalba et al., 1999; Felderhoff-Mueser et al., 2000; Matsushita et al., 2000). However, cytochrome c and caspase-9 are released from the mitochondria in selectively vulnerable neurons after transient global or focal ischemia (Fujimura et al., 1998, 1999; Krajewski et al., 1999; Sugawara et al., 1999; Cao et al., 2001a), leading to the speculation that the intrinsic pathway may contribute to ischemic neuronal death. Furthermore, the demonstrable effect of manganese superoxide dismutase in preventing cytochrome c release after ischemia indicates a strong causal link between mitochondrial dysfunction and caspase activation (Fujimura et al., 1999; Noshita et al., 2001).
Given that mitochondrial dysfunction plays a critical role in sensing and transmitting cell death signals for the activation of terminal caspases in both intrinsic and extrinsic pathways (Kroemer and Reed, 2000; Loeffler and Kroemer, 2000), and that mitochondrial release of apoptogenic factors is prominent in ischemic neuronal injury, we hypothesized that the caspase-9–dependent pathway may mediate caspase-3 activation and neuronal cell death after transient cerebral ischemia. Therefore, in this study, we cloned the rat caspase-9 complimentary DNA (cDNA) from a brain cDNA library and investigated its role in hippocampal CA1 neurodegeneration in the rat model of transient global ischemia.
MATERIALS AND METHODS
The rat model of global ischemia and peptide infusion
Transient global ischemia (15 minutes) was induced in male Sprague-Dawley rats (300 to 325 g) using a previously described four-vessel occlusion method (Chen et al., 1998). Blood pressure, blood gases, and blood glucose concentration were monitored and maintained in the normal range throughout the experiments. Rectal temperature was continually monitored and kept at 37°C to 37.5°C using a heating pad and a temperature-regulated heating lamp. Brain temperature was monitored using a 29-Ga thermocouple implanted in the left caudate-putamen and kept at 35.8°C ± 0.2°C during ischemia and at 37°C to 37.5°C thereafter. Electroencephalograms were monitored to ensure isoelectricity within 10 seconds after the induction of ischemia. Sham operations were performed in additional animals using the same anesthesia and surgical exposure procedures, except that the arteries were not occluded.
In selective experiments, rats were subjected to intracerebral ventricular infusion of the caspase-3/7 inhibitor N-benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoro-methylketone (z-DEVD.fmk), caspase-9 inhibitor N-benzyloxycarbonyl-Leu-Glu-His-Asp-fluoro-methylketone (z-LEHD.fmk), or caspase-8 inhibitor N-benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoro-methylketone (z-IETD.fmk) at the doses indicated in each study (see Results) using procedures described previously (Cao et al., 2001b). For the measurement of caspase-3–like or caspase-9–like protease activity in peptide-infused or vehicle-infused brains, the brains were quickly removed 8 or 24 hours after ischemia and processed for the enzyme assay. For histologic outcome experiments, rats were killed 3 or 7 days after ischemia and brains were perfused and processed for paraffin imbedding and sectioning described elsewhere (Chen et al., 1998). Coronal sections at the level of dorsal hippocampus were selected and stained with cresyl violet. Surviving hippocampal CA1 neurons were quantified by two investigators blinded to the experimental conditions, with the assistance of a computerized scanning program (MCID; Imaging Research Inc., St. Catharine's, Ontario, Canada).
Complimentary DNA cloning of rat brain caspase-9
Rat brain caspase-9 cDNA was cloned from a rat brain cDNA library using the rapid amplification of cDNA elongation (RACE) strategy described elsewhere (Chen et al., 1998, 2000; Cao et al., 2001b). Briefly, cerebella were dissected from four 1-week-old male Sprague Dawley rats. Polyadenylated RNA was isolated using the polyA-tract-1,000 messenger RNA (mRNA) isolation system (Promega, Madison, WI, U.S.A.) and used as a template for cDNA synthesis. The cDNA library was constructed using a Marathon cDNA amplification kit (Clontech, Palo Alto, CA, U.S.A.) according to the manufacturer's instructions and subsequently used as a template for RACE. A cDNA fragment encoding the rat brain caspase-9 was first generated by reverse transcription polymerase chain reaction according to the conserved sequences in human and mouse caspase-9: 5′-TCTCGGCGGGATCAGGCCAGGCAGC-3′ (sense) and 5′-AGGAGGGACTGCAGGTCTTCAG-3′ (antisense). Based on the sequence of this cDNA fragment, 5′- and 3′-RACE primers were designed as 5′-CTGGTCATAGATCT-TGAGACTCGAG-3′ and 5′-AGAACCTCTGGCCTGAGCC-GAGTAG-3′. The 5′-RACE–amplified and 3′-RACE–amplified fragments were subcloned into pGEM-T easy vector (Promega) and then sequenced on both strands (University of Pittsburgh Sequencing Facility). The full-length cDNA of caspase-9 was further obtained using polymerase chain reaction based on the obtained 5′-end and 3′-end sequences.
To confirm that the cDNA contains the full open-reading frame, we performed in vitro transcription and translation to detect its protein product. The Kozak sequence was added to the full-length caspase-9 cDNA before the start code using polymerase chain reaction, and then the polymerase chain reaction product was subcloned into the pcDNA3.1 plasmid in forward orientation. Coupled transcription and translation was carried out using the triamcinolone and nystatin quick-coupled transcription/translation systems (Promega) according to the manufacturer's instructions. In brief, 1 μg NotI linearized pcDNA-caspase-9 was incubated at 30°C for 2 hours in 40 μL triamcinolone and nystatin quick master mix and 20 μCi S-methionine (ICN, Costa Mesa, CA, U.S.A.). Two microliters of the reaction mixture was electrophoresed on 12% polyacrylamide gel, transferred to a polyvinyl difluoride membrane, and exposed to x-ray film (Kodak, Rochester, NY, U.S.A.) with intensifying screens. In a separate reaction, S-methionine was replaced by cold methionine, and the reaction product was electrophoresed and subjected to Western blot analysis using an anticaspase-9 antibody (Chemicon, Temecula, CA, U.S.A.).
To determine whether the cloned cDNA encodes a functional caspase, caspase-9 cDNA was transfected into human 293 cells using procedures described previously (Cao et al., 2001a), and cell viability was evaluated after 24 hours. In brief, the caspase-9 cDNA was inserted into the downstream cloning site of the expression vector pcDNA-GFP (green fluorescent protein), designated as pcDNA-GFP-caspase-9. Four micrograms pcDNA-GFP-caspase-9 or the empty vector was transfected into 293 cells using lipofectamine (Invitrogen, Carlsbad, CA, U.S.A.) according to the manufacturer's protocol. The transfected cells were counterstained with DNA dye (Hoechst 33258), and the viability was evaluated using inverted fluorescence microscopy.
Northern blot analysis
Total RNA was prepared from rat tissues using the RNAgent total RNA isolation system (Promega) according to the manufacturer's instructions. The RNA from the following three sets of tissues was analyzed: (1) various organ tissues from adult male rats, including the heart, liver, spleen, lung, kidney, whole brain, intestine, stomach, testis, and skeletal muscle; (2) cerebella of rat brains of different ages, including embryonic day 17 and postnatal weeks 1, 2, 4, and 12 (adult); and (3) hippocampi of adult rat brains subjected to 15-minute global ischemia followed by 8, 24, or 72 hours of reperfusion.
Total RNA (30 μg) was electrophoresed on a 1% agarose-formaldehyde gel and blotted onto a ζ-probe GT nylon membrane (Bio-Rad, Hercules, CA, U.S.A.). The membrane was hybridized overnight at 42°C with the caspase-9 cDNA probe (4 × 106 cpm/mL) labeled with 32P using a random primer labeling kit (Boehringer Mannheim, Indianapolis, IN, U.S.A.). To control for sample loading, the membranes were stripped and rehybridized with a 32P-labeled 28S ribosomal RNA or β-actin probe. Data were analyzed in a manner described previously (Chen et al., 1998).
In situ hybridization
Rats were anesthetized and decapitated 4, 8, 24, and 72 hours after 15-minute global ischemia or 24 hours after sham operation (n = 4 per time point). Frozen coronal sections (15-μm thick) were cut on a cryostat at 20°C. The S-labeled caspase-9 complimentary RNA probe (300 bp) was prepared in both the sense and antisense orientation using the procedures described by Chen et al. (1998). The sections were hybridized with the labeled probe (1×10 cpm/mL) in hybridization cocktail for 18 hours at 55°C. The slides were washed, dehydrated, air dried, and exposed to Kodak film for 3 weeks. The relative changes in mRNA expression were quantified by determining the ratio of the optical density of the specified regions in ischemic brains to controls using the MCID system (Chen et al., 1998).
Western blot analysis and immunoprecipitation
Western blotting was performed using the standard method described by Chen et al. (2000). For the detection of cytochrome c release, cytosolic and mitochondrial protein extracts were prepared from the hippocampi at 4, 8, 24, and 72 hours after ischemia or 24 hours after sham operation (n = 3 per group), and immunoreacted with the monoclonal antibody against cytochrome c at a dilution of 1:1000 (Pharmingen, San Diego, CA, U.S.A.). For the detection of caspase-9, total protein extracts from the hippocampi were immunoreacted with the polyclonal antibodies recognizing the full-length caspase-9 (Chemicon) or the mature active form of caspase-9 (Cell Signaling Technology Inc., Beverly, MA, U.S.A.) according to the manufacturers' instructions.
Immunoprecipitation was performed to examine protein–protein interactions between Apaf-1 and caspase-9 after ischemia using a procedure described previously (Cao et al., 2001a). Briefly, cytosolic protein was isolated from the hippocampi at 8 or 24 hours after ischemia or 24 hours after sham operation (n = 3 per time point, two brains per sample) using RIPA A (150 mmol/L NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, and 50 mmol/L Tri-HCl pH 8.0) buffer. Equal amounts of protein from each experimental condition (300 μg per sample) were subjected to immunoprecipitation using the rabbit anti–Apaf-1 antibody (Chemicon). The resulting immunoprecipitates were then analyzed by immunoblotting with anticaspase-9 (Chemicon) and anti–Apaf-1 antibodies, respectively.
Immunohistochemistry
Animals were anesthetized with 8% chloral hydrate 24 or 72 hours after 15-minute ischemia or 24 hours after sham operation (n = 4 per time point). Rats were perfused with 200 mL heparinized 0.9% saline followed by 500 mL 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (pH 7.4). The brains were removed and processed for paraffin embedding and cutting, and coronal sections at the levels of dorsal hippocampus were selected for immunohistochemical staining. Immunohistochemical staining for caspase-9 was performed using an affinity-purified rabbit polyclonal antibody (dilution, 1:50; Cell Signaling) that recognizes the active forms of rat caspase-9. The procedures for immunohistochemistry were the same as described elsewhere (Chen et al., 1998; Cao et al., 2001b). For the assessment of nonspecific staining, alternating sections from each experimental condition were incubated without the primary antibody.
Measurement of caspaselike activity
Measurement of caspase-3–like, caspase-9–like, and caspase-8–like protease activity in cell extracts was performed as described previously (Cao et al., 2001a). The hippocampus was dissected 2, 4, 8, 24, and 72 hours after ischemia or 24 hours after sham operation (n = 4 per group), and protein extracts were prepared in a manner described elsewhere (Cao et al., 2001a). One hundred micrograms extracted protein was incubated for 1 hour at 37°C with reaction buffer containing 25-mmol/L HEPES (pH 7.5), 10% sucrose, 0.1% 3-[(3-cholamidopropyl) dimethylammonio]-1-propane sulfonate, 5-mmol/L chlorophenothane, and 5-mmol/L edetic acid in a total volume of 150 μL. The reaction mixture also contained 25-μmol/L fluorogenic peptide cleavage substrate (Medical & Biological Laboratories, Watertown, MA, U.S.A.). Three different substrates were used for the assays: Ac-DEVD-AFC for caspase-3–like activity, Ac-LEHD-AFC for caspase-9–like activity, and Ac-IETD-AFC for caspase-8–like activity. Data were expressed as fold change over sham controls or as absolute units (one unit corresponds to the caspaselike activity that cleaves 1 pmol AFC at 37°C at saturating substrate concentrations). To detect nonspecific protease activity, in parallel experiments the protein extracts were incubated in the reaction buffer with inhibitors (5 μmol/L each) for caspase-3 (DEVD-CHO), caspase-9 (LEHD-CHO), or caspase-8 (IETD-CHO) at room temperature for 30 minutes before the addition of assay substrates, and the values were subtracted from those obtained without the inhibitors (Cao et al. (2001a)
Data analysis
All data are presented as mean ± SD. Comparisons of caspase activities, caspase-9 protein expression, cytochrome c release, or CA1 cell survival were made between experimental groups using analysis of variance and post hoc Scheffé tests. A level of P < 0.05 was considered statistically significant.
RESULTS
Molecular cloning of rat caspase-9
Using a rat brain cDNA library as the template for RACE, we cloned two cDNA species that encode full-length caspase-9 (GenBank accession numbers AF262319 and AF286006). The two species shared the identical open-reading frame of 454 amino acids (Fig. 1a). In addition, AF286006 showed an extended 325-bp untranslation sequence at the 3′ end (data not shown). Sequence analysis revealed that the deduced amino acid sequence of rat caspase-9 was 71.4% and 90.1% identical to the published sequences of human (Fujita et al., 1999) and mouse caspase-9 (Duan et al., 1996), respectively.
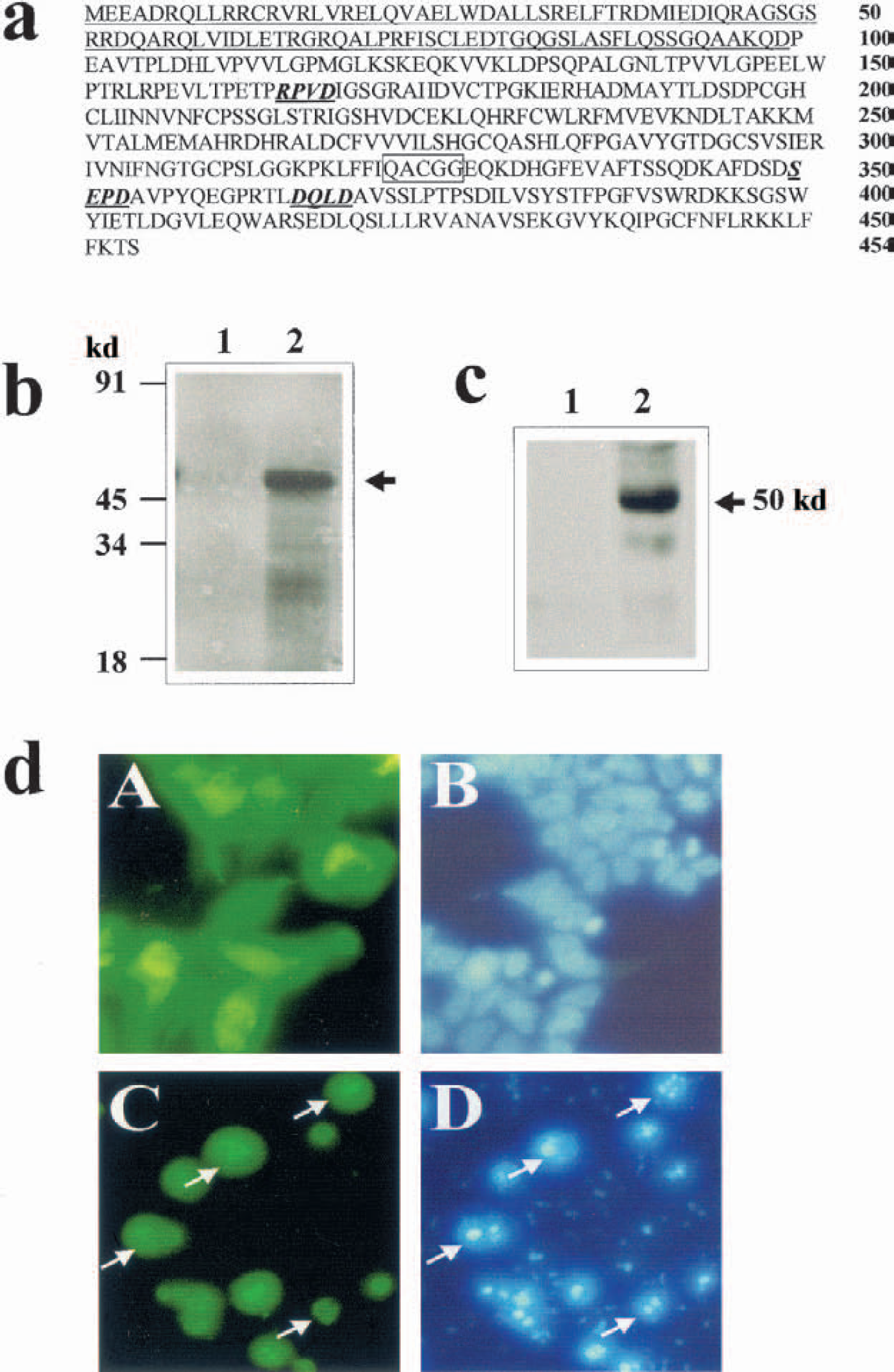
Cloning of rat caspase-9.
The in vitro transcription/translation assay generated a radiolabeled protein product at approximately 51 kd, which is consistent with the predicted size of rat caspase-9 (Fig. 1b). Furthermore, the translated protein was recognized by the anticaspase-9 immunoblotting, thus confirming the validity of the deduced amino acid sequence for rat caspase-9 (Fig. 1c).
To characterize the functional properties of rat caspase-9, we constructed an expression vector (pcDNA-GFP-caspase-9) expressing the GFP-caspase-9 fusion protein. Transfection of pcDNA-GFP-caspase-9 in the human 293 cells resulted in marked induction of apoptosis within 24 hours, characterized by chromatin condensation and advanced nuclear fragmentation (Fig. 1d). Notably, the GFP-caspase-9 fusion protein migrated from the cytosol into the nucleus in the majority of apoptotic cells. This phenomenon was strikingly similar to that which was seen after GFP-caspase-3 transfection (data not shown).
Tissue expression of caspase-9 during rat brain development
Northern blot analysis was performed using the random primer-labeled complimentary RNA probe to examine the distribution of caspase-9 mRNA expression in rat tissues. The Northern blots detected two major mRNA transcripts of approximately 2.5 and 1.7 kb (Fig. 2A). Further prolonged electrophoresis also detected two minor bands of each size below the 2.5-kb and 1.7-kb markers (data not shown). These observations are consistent with the notion that multiple forms of caspase-9 mRNA species may be expressed in mammalian cells due to the differential splicing of the gene (Fujita et al., 1999; Seol and Billiar, 1999).
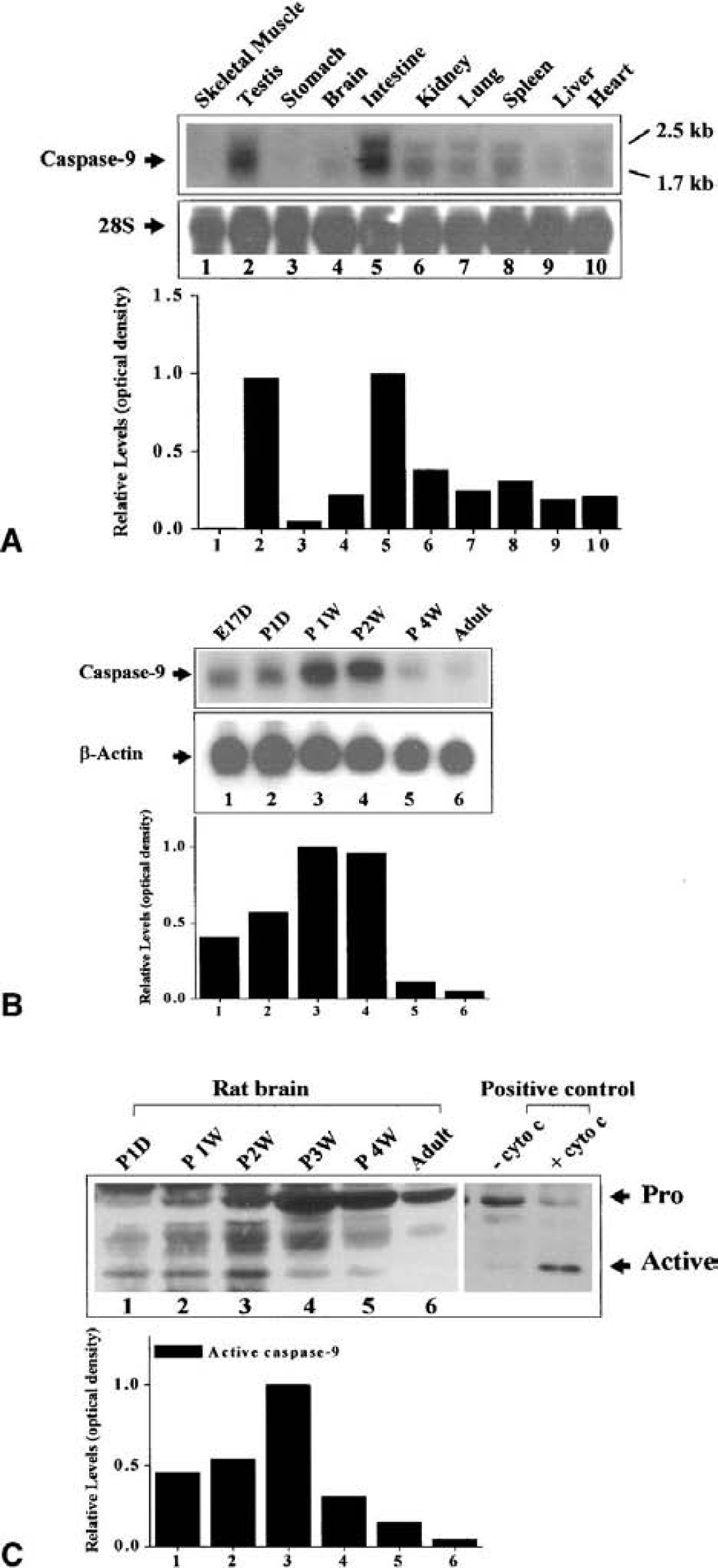
Characterization of caspase-9 expression in the rat.
Caspase-9 was widely distributed in adult rat tissues, with the highest expression in testis and intestine, followed by the kidney, spleen, and lung, and with substantially lower levels in skeletal muscle, stomach, brain, and liver (Fig. 2B).
Because caspase-9 is required for the induction of apoptosis in neuronal progenitor cells during brain development (Kuida et al., 1998), we characterized the temporal profiles of caspase-9 expression at both mRNA and protein levels in the rat cerebellum of various ages. Caspase-9 mRNA was expressed at moderate levels in embryonic cerebellum, but the levels were increased at postnatal day 1 and peaked at postnatal 12 weeks (Fig. 2B). The expression of caspase-9 mRNA subsided to moderate or low levels at postnatal 4 weeks and in adult rats. Furthermore, using Western blot analysis, we detected endogenously activated caspase-9 in the newborn rat cerebellum (Fig. 2C). The temporal profile of endogenous activation of caspase-9 was generally consistent with the known time course of developmental neuronal apoptosis in the brain. In day-1, day-7, and day-14 postnatal brains, the majority of caspase-9 molecules were in their active forms (approximately 20 kd). A very low level or no active caspase-9 was detected at postnatal 4 weeks or in adult brain.
Caspase-9 activation in hippocampal neurons after ischemia
To investigate the potential role of caspase-9 in ischemic neuronal cell death, we examined the expression of caspase-9 at both mRNA levels in normal and ischemic brains, focusing on the hippocampus, where neurons are particularly vulnerable to transient global ischemia. Using Northern blot analysis and in situ hybridization, we detected increased caspase-9 mRNA expression (in both 2.5-kb and 1.7-kb species) in ischemic brains (Fig. 3a). The levels were increased 2.3-fold at 8 hours, 2.2-fold at 24 hours, and 2.5-fold at 72 hours after ischemia. As determined in the in situ autoradiographs, induced caspase-9 mRNA expression was localized in the CA1 sector (Fig. 3b), which was strikingly similar to induced caspase-3 mRNA expression (Chen et al., 1998).
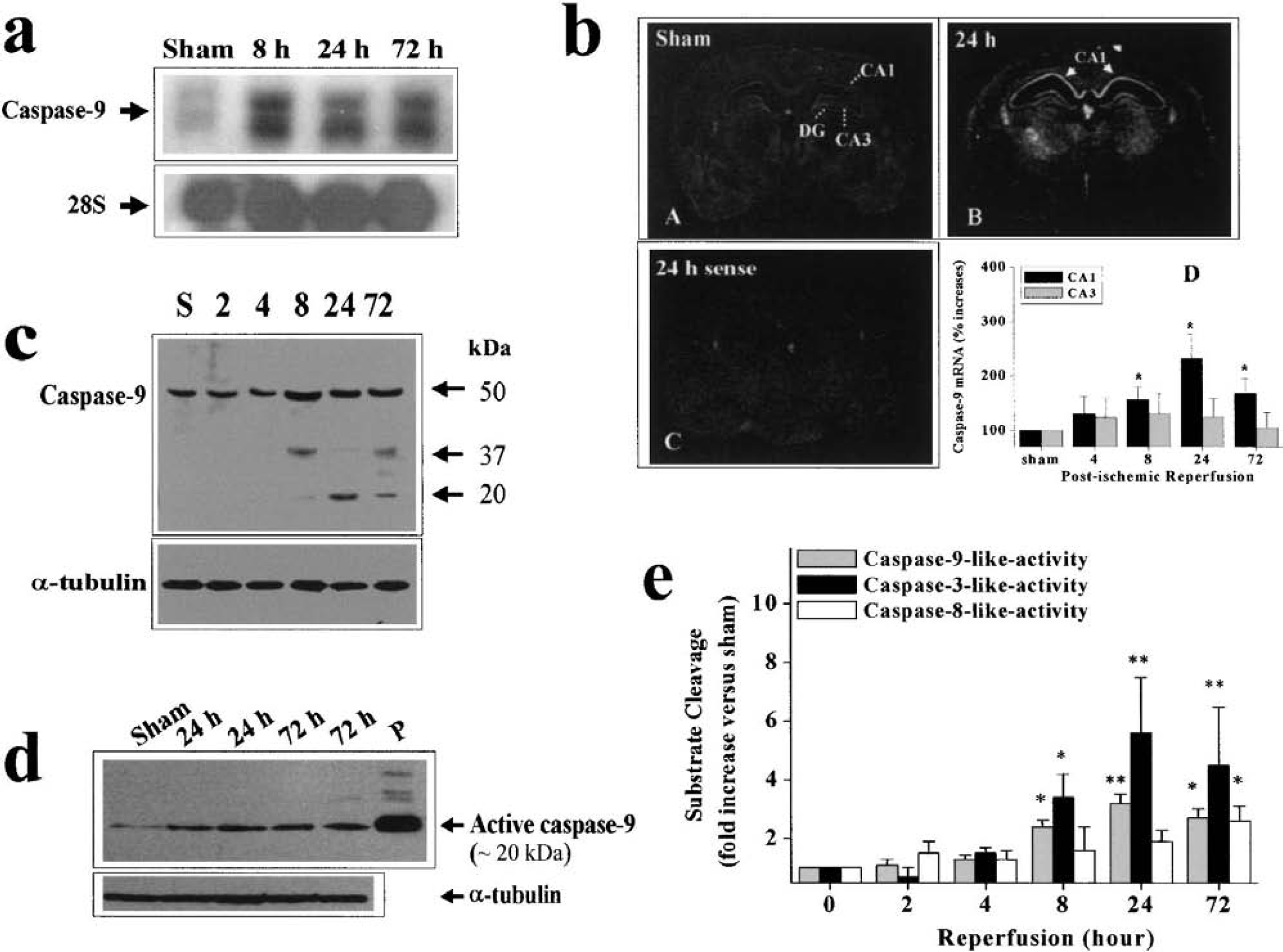
Activation of caspase-9 after cerebral ischemia.
Using Western blot analysis, we detected significantly increased levels of active caspase-9 in hippocampal cell extracts from ischemic brains (Fig. 3c). The time course for caspase-9 activation preceded that of CA1 neuronal degeneration in this model, and was similar to that of caspase-3 activation under similar ischemic conditions (Chen et al., 1998). The activation of caspase-9 in the hippocampus was also confirmed using an antibody that specifically recognizes the mature active form of caspase-9 (Fig. 3d). To further verify the findings by Western blots, we performed caspase activity assays based on the cleavages of the fluorogenic substrates preferred by caspase-9–like, caspase-3–like, or caspase-8–like activity in the hippocampal cell extracts. As shown in Fig. 3e, significantly increased caspase-9–like and caspase-3–like activity was detected as early as 8 hours after ischemia and peaked 24 to 72 hours after ischemia. In contrast, a moderate but significant increase in caspase-8–like activity was detected only at the 72-hour time point, which is consistent with our recent observation that Bid (a specific caspase-8 substrate) cleavage was a late event after global ischemia (Cao and Chen, unpublished data, 2001).
To further characterize the cellular distribution of caspase-9 activation after ischemia, we performed immunofluorescent staining in brain sections obtained 24 and 72 hours after ischemia. As determined using an antibody against the active form of caspase-9, caspase-9 activation was localized predominantly in the CA1 pyramidal neurons after ischemia (Fig. 4). Further characterization of the subcellular localization of active caspase-9 showed a reperfusion-duration–dependent alteration. Although caspase-9 immunofluorescence exhibited a predominant cytosolic localization at the 24 -hour time point, it was localized mainly in the cell nucleus and colocalized with terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling staining at the 72-hour time point.
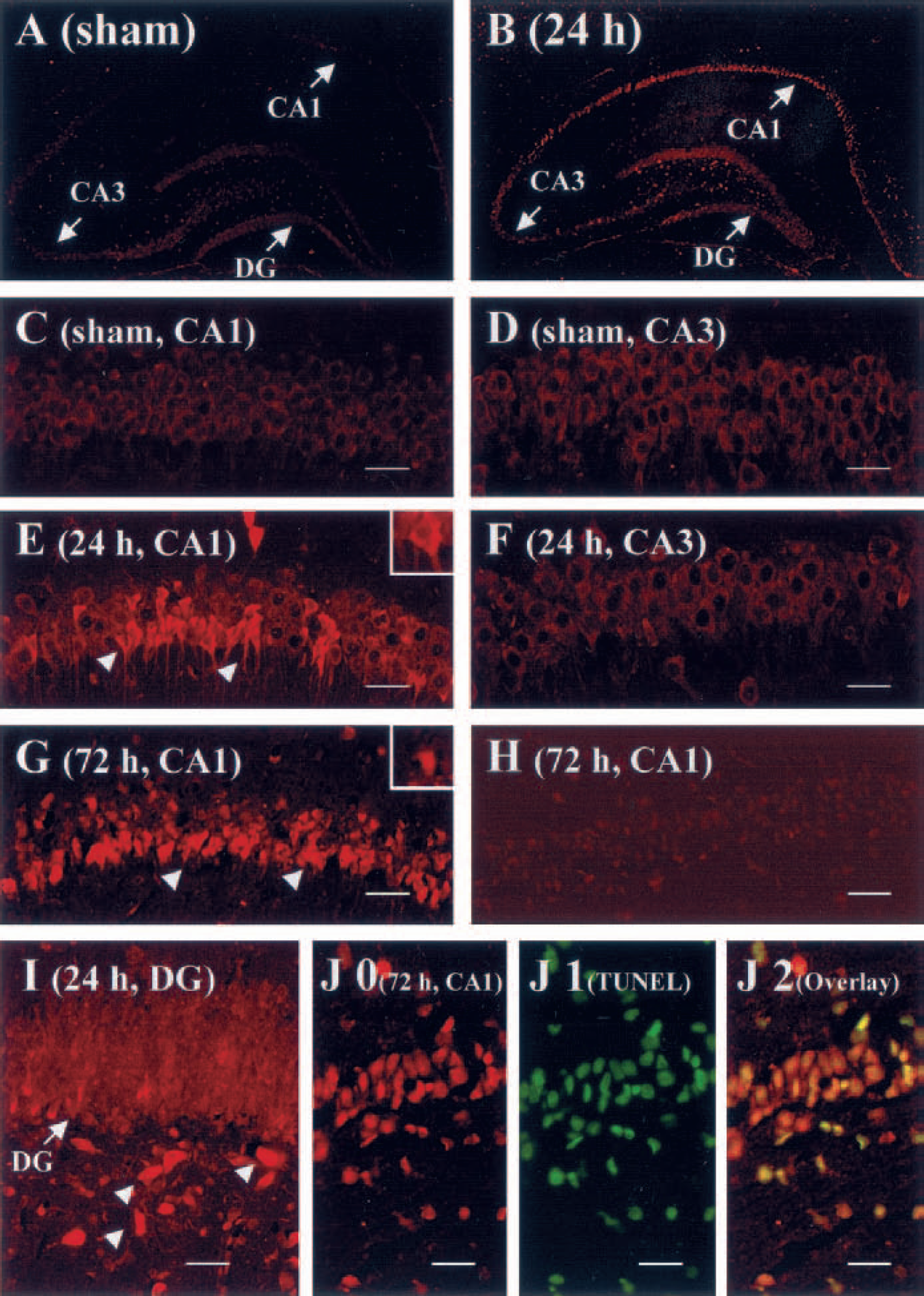
Activation of caspase-9 in CA1 neurons after ischemia and immunofluorescent images of caspase-9 in the hippocampus after ischemia. Immunostaining was performed using the antibody specifically recognizing the active form of caspase-9 (Fig. 3D) on paraffin-embedded coronal sections obtained 24 or 72 hours after ischemia or after sham operation.
Evidence supporting an enhanced caspase-9/Apaf-1 interaction after ischemia
In the intrinsic pathway for caspase-3 activation, caspase-9 and Apaf-1 form heterodimers through the N-terminal caspase recruitment domain present in both molecules (Li et al., 1997). This reaction appears to take place in the cytosol and requires the addition of cytochrome c and dATP (or ATP). Thus, the detection of caspase-9/Apaf-1 protein dimerization in the cytosolic extracts represents a valid indicator of the formation of apoptosome, which is essential for the activation of caspase-3 via the intrinsic pathway (Li et al., 1997). To detect caspase-9/Apaf-1 dimerization in the ischemic hippocampus, cytosolic protein was extracted from the hippocampus 8 and 24 hours after ischemia or sham operation and subjected to immunoprecipitation using a monoclonal antibody against Apaf-1, followed by Western blot analysis of caspase-9 and APAF-1 (Fig. 5A). Although Apaf-1 immunoreactivity was present at similar levels across the samples studied, increased caspase-9 immunoreactivity was detected in the immunoprecipitates 8 and 24 hours after ischemia in experimental samples, but not in sham controls. These results suggest that more Apaf-1/caspase-9 complexes were formed in the postischemic hippocampal cells.
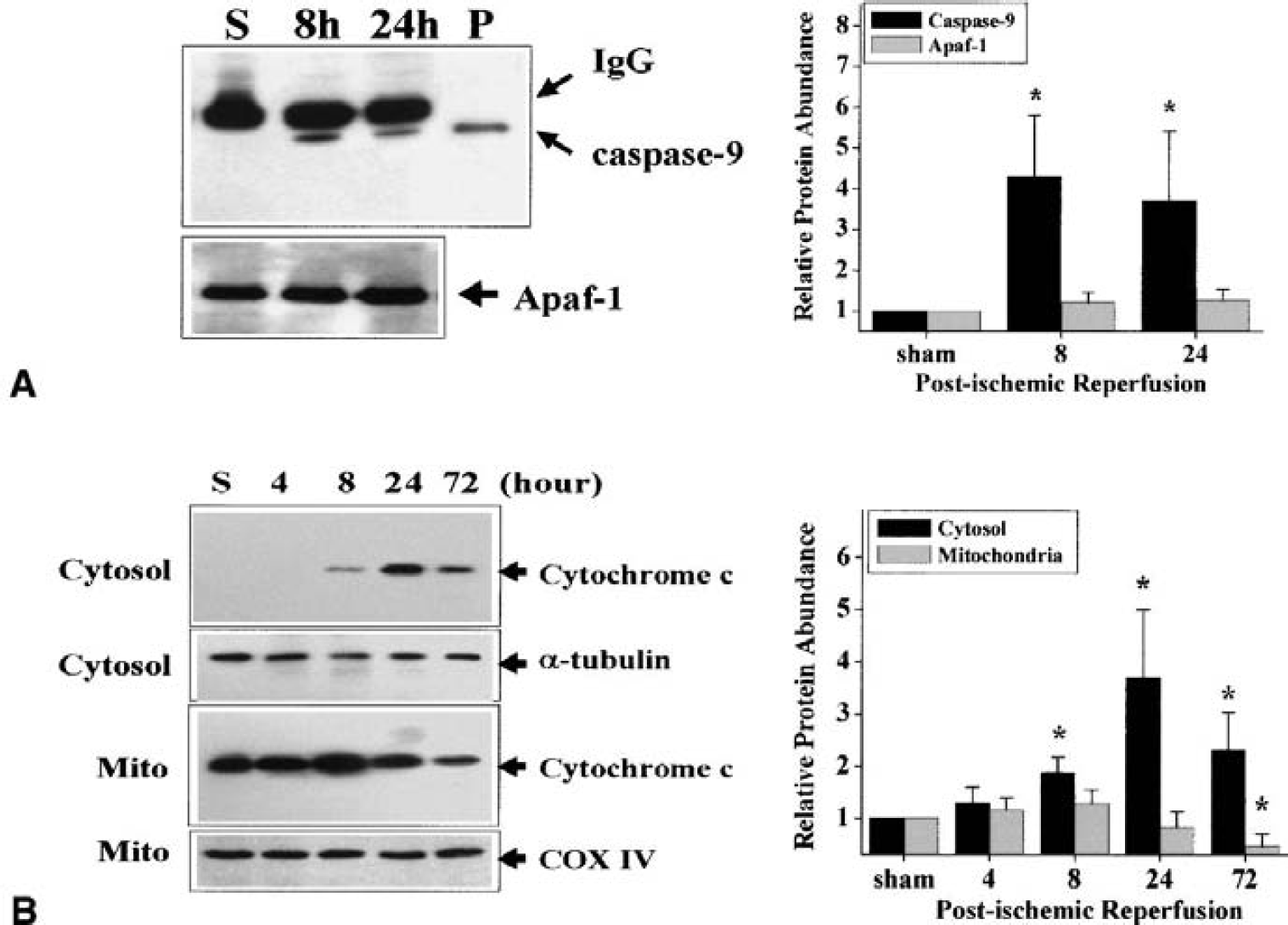
Increased caspase-9 and Apaf-1 interaction after ischemia.
Western blots were performed using fractionated proteins to delineate the subcellular translocation of cytochrome c in the postischemic hippocampus. Consistent with the observations by Sugawara et al. (1999) in a similar rat model of transient global ischemia, increased cytochrome c immunoreactivity was detected in the cytosolic fraction 8 to 72 hours after ischemia (Fig. 5B). In contrast, the mitochondrial cytochrome c was decreased 72 hours after ischemia, suggesting that a partial loss of cytochrome c from this organelle occurred in postischemic cells.
Caspase-9 inhibition enhances CA1 neuronal survival after ischemia
To investigate the role of caspase-9 (representing the intrinsic pathway) or caspase-8 (representing the extrinsic pathway) in mediating caspase-3 activation after ischemia, in vivo caspase inhibition studies were performed using the relatively selective inhibitors z-LEHD-fmk (for caspase-9) and z-IETD-fmk (for caspase-8). The peptides were infused twice (1.5 μg each infusion) into the ventricles through an implanted cannula 30 minutes before and 2 hours after ischemia. In preliminary studies, this dose of inhibitors completely suppressed caspase-9–like and caspase-8–like activity, respectively, in the hippocampus 24 and 72 hours after ischemia (data not shown), respectively. Using this dosage regimen, the effect of the peptide inhibitors on hippocampal caspase-3 activity was studied at 8 and 24 hours after ischemia. As shown in Fig. 6A, z-LEHD-fmk infusion almost completely attenuated caspase-3 activation at both time points after ischemia, whereas z-IETD-fmk infusion had no significant effect on caspase-3–like activity.
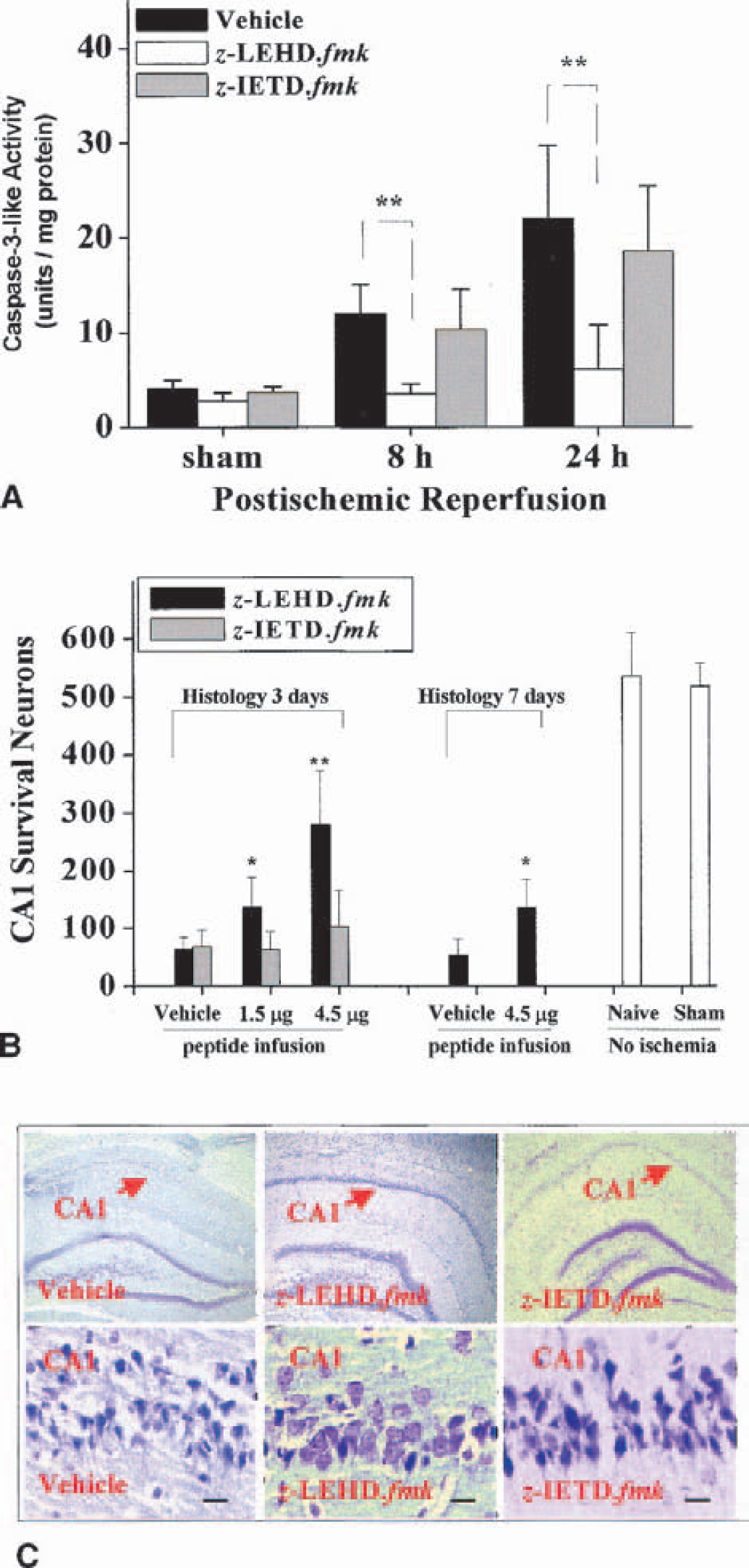
Protective effect of in vivo inhibition of caspase-9–like protease activity.
The effect of caspase inhibitors on histologic outcomes in the CA1 was studied using the triple-infusion regimen (Chen et al., 1998), in which peptides were infused in three equal dosages 30 minutes before and 2 and 24 hours after ischemia, resulting the total amounts of peptide of 1.5 or 4.5 μg in each animal. As determined 3 and 7 days after ischemia, z-LEHD.fmk significantly promoted neuronal survival (as determined by cresyl-violet staining) in CA1 (Figs. 6B and 6C). In contrast, z-IETD-fmk offered marginal protection in CA1 3 days after ischemia, but the difference in cell survival between peptide-treated and vehicle-treated animals did not reach statistical significance (P > 0.05).
DISCUSSION
Caspase-9 is an important initiation caspase and an essential component of the mitochondria-dependent intrinsic apoptosis-signaling pathway for the activation of terminal execution caspases, particularly caspase-3 (Li et al., 1997). This signaling pathway has been elucidated in neuronal apoptosis induced by trophic factor, potassium withdrawals, or DNA damage (Deshmukh et al., 2000; Kermer et al., 2000; D'Sa-Eipper et al., 2001; Gerhardt et al., 2001), but its role in ischemic neuronal death had not been established. The results described in this study strongly suggest a role for caspase-9 in mediating the caspase-3–dependent pathway of neuronal death in the hippocampal CA1 after transient global ischemia. This conclusion is based on the following experimental evidence: (1) rat caspase-9 is a potent cell death inducer, and it is overexpressed and proteolytically processed (activated) in CA1 neurons after ischemia; (2) increased formation of the Apaf-1/caspase-9 heterodimers is evident in the postischemic hippocampus; (3) the temporal profiles of caspase-9 activation appear to coincide with those of cytochrome c release and caspase-3 activation, but precede CA1 cell death; and (4) inhibition of caspase-9–like protease activity attenuates caspase-3 activation and promotes CA1 cell survival after global ischemia.
The role of caspase-9 as a potent apoptosis-inducing protease is well established in various types of cells and tissues (Hu et al., 1998, 1999; Benedict et al., 2000). Caspase-9 activity appears to be essential for the induction of neuronal apoptosis during brain development. The caspase-9 gene knockout is lethal during the embryonic stage, and clearly shows a neuronal phenotype similar to Apaf-1 or caspase-3 knockouts due to the defection in brain cell apoptosis (Cecconi et al., 1998; Kuida et al., 1998; Yoshida et al., 1998). In the present study, we cloned the rat caspase-9 gene from a rat brain cDNA library and subsequently characterized its gene expression in developing brain under physiologic conditions and in the adult brain after transient global ischemia. The expression of caspase-9 is developmentally regulated in the brain, with higher levels expressed during postnatal 12 weeks, and markedly downregulated in adulthood. Furthermore, based on the immunoblotting detection of active caspase-9 peptides in brain cell extracts, we found that caspase-9 is markedly activated during brain development, but remains at extremely low levels in the adult brain. These patterns of caspase-9 regulation in the brain are similar to that of Apaf-1, an essential factor for the activation of caspase-9 (Yakovlev et al., 2001). These observations are consistent with the notion that caspase-9 and Apaf-1 may be the essential components of the endogenous machinery for the execution of neuronal apoptosis in the brain.
These data show that the expression and protease activity of caspase-9 were induced in hippocampal neurons after ischemia. Caspase-9 mRNA levels were increased in the hippocampus 8 to 72 hours after ischemia, and the increased mRNA appeared to localize predominantly in the CA1 pyramidal neuronal layer. Furthermore, in support of its speculative role in mediating ischemic cell death, caspase-9 was proteolytically activated in the postischemic hippocampus, as determined by Western blot analysis, protease activity assays, and immunofluorescent detection of the active caspase-9 peptides. The time course of caspase-9 activation was consistent with that of cytochrome c release (8 to 72 hours) but preceded CA1 cell death (72 hours). These results support the recent speculation that mitochondrial dysfunction and cytochrome c release may constitute a critical step in the molecular cascade leading to apoptosis of vulnerable neurons after transient cerebral ischemia (Fujimura et al., 1999; Sugawara et al., 1999). At least three mechanisms have been proposed concerning caspase-9 activation in mammalian cell apoptosis, including the cytochrome c/Apaf-1–dependent pathway, the Akt phosphorylation-dependent pathway, and direct proteolytic activation by caspase-3. Although the Akt phosphorylation site has been identified in caspase-9 in certain species, this function-regulating site does not appear to exist in the mouse caspase-9 (Fujita et al., 1999) or rat caspase-9 as predicted based on their deduced amino acid sequences, and thus is unlikely the target for direct regulation by Akt in relevance to this study. The present study does not rule out a role for the possible caspase-3/caspase-9 feedback amplification loop in ischemic neurons, because active caspase-3 is a potent activator of caspase-9 in vitro and in vivo (Fujita et al., 2001). However, results from the in vivo caspase inhibition studies did not support such a speculation because the caspase-3/7 inhibitor z-DEVD-fmk failed to prevent caspase-9 activation in the ischemic hippocampus (data not shown). Because cytochrome c release and enhanced formation of the Apaf-1/caspase-9 heterodimeric complex were reproducibly detected in the brain after ischemia, the cytochrome c/Apaf-1 pathway may be a significant and possibly the primary mechanism responsible for the activation of caspase-9 in the postischemic hippocampus.
A major objective of the present study was to determine whether the Apaf-1/caspase-9 intrinsic pathway mediates the activation of the terminal caspases (caspase-3) and subsequent CA1 cell death after global ischemia. Several recent studies have shown that both caspase-9 (representing the intrinsic pathway) and caspase-8 (representing the extrinsic pathway) are activated in the brain after transient focal ischemia (Velier et al., 1999; Benchoua et al., 2001; Culmsee et al., 2001; Dubois-Dauphin et al., 2001; Morita-Fujimura et al., 2001; Noshita et al., 2001). However, a direct functional linkage between these deduced upstream caspases and terminal caspases in ischemic neurons has not been fully established. Based on the temporal correlation analysis, it was suggested that in focal ischemia the activation of caspase-3 during the early phase of injury might be related to the caspase-8–dependent extrinsic pathway, whereas caspase-9 activation might contribute to caspase-3 activation associated with the secondary expansion of infarction (Benchoua et al., 2001). In another study, however, caspase-8 and caspase-3 were expressed in different populations of cortical neurons after ischemia (Velier et al., 1999), opposing a direct role of the extrinsic pathway. These somewhat discrepant observations raised the possibility that the mechanism underlying terminal caspase activation after ischemia may vary depending on the neuronal phenotype and the stage of ischemic injury, and that the differences between ischemia models and between different ischemic regimens (permanent vs. transient) may also contribute to the variables. In the present study, we measured caspase-8–like and caspase-9–like protease activities in the ischemic hippocampus as functions of reperfusion duration. The temporal profile of caspase-9 activation was consistent with its deduced role in mediating caspase-3 activation, at least during the 8-to-24-hour reperfusion period. The relatively late induction of caspase-8 activity in the hippocampus may reflect the changes in the levels of the Fas receptor in this region after ischemia. The Fas receptor is expressed at extremely low levels in the normal nonischemic hippocampus, but levels are increased 24 to 72 hours after ischemia (Cao and Chen, unpublished data, 2001). Furthermore, in vivo caspase inhibition studies showed that intracerebroventricular infusion of z-LEHD.fmk but not z-IETD.fmk abolished the increased caspase-3–like activity and promoted CA1 cell survival in the hippocampus after ischemia (Fig. 6). These results suggest that caspase-9, rather than caspase-8, may play an important role in initiating caspase-3 activation and caspase-3–dependent CA1 neuronal death after transient global ischemia.
It is notable that none of the peptide caspase inhibitors used in this study or in similar studies (Chen et al., 1998; Himi et al., 1998; Gillardon et al., 1999) completely prevented CA1 neuronal loss in the global ischemia model. At the optimal doses (which almost completely inhibited endogenous caspaselike protease activity), the caspase-9 inhibitor rescued up to approximately half of CA1 neurons 3 days after ischemia (Fig. 6). Furthermore, when the observation window for neuroprotection was extended to 7 days of reperfusion, the protective effect offered by caspase inhibition was partially lost. This pattern of protection by the caspase-9 inhibitor was similar to that of the caspase-3/7 inhibitor z-DEVD.fmk (Chen et al., 1998). These results suggest that although caspase-9–like and caspase-3–like protease activities probably play a role in mediating ischemic CA1 neuronal death, other types of caspases could also be involved in the cell death process via caspase-3–dependent and/or caspase-3–independent pathways. A recent study by Kang et al. (2000) suggests that caspase-11 may contribute to focal ischemic brain injury by activating caspase-1 and caspase-3. Alternatively, it is also possible that mechanisms independent of caspase activities could be partially responsible for CA1 neuronal loss after ischemia (Graham and Chen, 2001). Several caspase-independent pro-apoptotic molecules, such as apoptosis-inducing factor and endonuclease G, have recently been identified and characterized (Susin et al., 1999; Widlak et al., 2001). As determined in nonneuronal cells, apoptosis-inducing factor and endonuclease G are released from mitochondria during apoptosis and cause apoptotic nuclear degradation through caspase-independent actions. In a recent study, mitochondrial release and nuclear translocation of apoptosis-inducing factor were detected in CA1 neurons after transient global ischemia (Cao and Chen, unpublished observations, 2001), suggesting that apoptosis-inducing factor could be an additional caspase-independent executioner of cell death in ischemic neurons.
In summary, this study provides new evidence that the caspase-9 gene is induced and its protein product is activated in selectively vulnerable CA1 neurons after transient global ischemia. Furthermore, the results suggest that caspase-9 is the upstream caspase that activates the terminal caspases, especially caspase-3, and is responsible, at least in part, for ischemic CA1 neuronal loss. Future studies using highly selective inhibitors of the Apaf-1/caspase-9–intrinsic apoptosis-signaling pathway, such as the dominant negative mutants of Apaf-1 or caspase-9, are warranted to further confirm this hypothesis and to explore novel strategies for preventing irreversible neuronal injury after cerebral ischemia.
Footnotes
Acknowledgments:
The authors thank Carol Culver for editorial assistance and Pat Strickler for secretarial support.