
Abstract
Apoptosis is implicated in neonatal hypoxic/ischemic (H/I) brain injury among various forms of cell death. Here we investigate whether overexpression of heat shock protein (Hsp) 70, an antiapoptotic protein, protects the neonatal brain from H/I injury and the pathways involved in the protection. Postnatal day 7 (P7) transgenic mice overexpressing rat Hsp70 (Tg) and their wild-type littermates (Wt) underwent unilateral common carotid artery ligation followed by 30 mins exposure to 8% O2. Significant neuroprotection was observed in Tg versus Wt mice on both P12 and P21, correlating with a high level of constitutive but not inducible Hsp70 in the Tg. More prominent injury was observed in Wt and Tg mice on P21, suggesting its continuous evolution after P12. Western blot analysis showed that translocation of cytochrome c, but not the second mitochondria-derived activator of caspase (Smac)/DIABLO and apoptosis-inducing factor (AIF), from mitochondria into cytosol was significantly reduced in Tg 24 h after H/I compared with Wt mice. Coimmunoprecipitation detected more Hsp70 bound to AIF in Tg than Wt mice 24 h after H/I, inversely correlating with the amount of nuclear, but not cytosolic, AIF translocation. Our results suggest that interaction between Hsp70 and AIF might have reduced downstream events leading to cell death, including the reduction of nuclear AIF translocation in the neonatal brains of Hsp70 Tg mice after H/I.
Keywords
Introduction
Perinatal complications because of anoxia and reduced cerebral blood flow (CBF) are often associated with adverse neurologic outcomes, including epilepsy, cerebral palsy, and mental retardation (Vannucci, 1990; Vexler and Ferriero, 2001). The mechanisms of brain injury after hypoxia/ischemia (H/I) are thought to include energy depletion, excitotoxicity, oxidative stress, inflammation, and apoptosis (Barks and Silverstein, 1992; Cheng et al, 1998; Hagan et al, 1996; Liu et al, 1996a; Martin et al, 1997; McDonald et al, 1988). The susceptibility of the immature brain to H/I largely depends on the temporal and regional status of critical developmental processes (Vexler and Ferriero, 2001). During normal development, apoptosis, the molecularly regulated cell death event, is prevalent in many brain regions (Kuan et al, 2000a, b; Northington et al, 2001a, b).
Apoptosis can also be induced by ischemia, including neonatal H/I injury, and by many other acute and chronic neurodegenerative processes (Love, 2003; Northington et al, 2001a; Vexler and Ferriero, 2001). In the intrinsic pathway of apoptosis involving mitochondria, which are pivotal regulators of cell death, a universal downstream event is the activation of caspases (Adrain et al, 2001; Du et al, 2000; Hakem et al, 1998; Keane et al, 2001; Li et al, 1997; Liu et al, 1996b; Saito et al, 2003; Verhagen et al, 2000). In response to multiple proapoptotic signals, the outer membrane of mitochondria becomes permeabilized, resulting in the release of molecules normally confined to the mitochondrial intermembrane space (Desagher and Martinou, 2000; Kroemer, 2003; Martinou and Green, 2001; Zamzami and Kroemer, 2003). Those proapoptotic proteins translocate from mitochondria to cytosol in a process that is controlled by the Bcl-2 family proteins (Kroemer and Reed, 2000; Kuwana et al, 2002; Martinou and Green, 2001). One of those proapoptotic proteins is cytochrome c. After being released into the cytosol, cytochrome c binds to apoptotic protease activating factor-1 (Apaf-1) in the presence of dATP, promoting the oligomerization of Apaf-1 itself. Concurrently or subsequently, this complex recruits procaspase-9, forming the complex called the apoptosome (Beere et al, 2000; Li et al, 1997). The assembly of apoptosome permits the autoactivation of procaspase-9, which is followed by the recruitment and activation of procaspase-3. Caspase-3 cleaves the inhibitor of caspase-activated deoxyribonuclease and activates DNase, leading to DNA fragmentation (Li et al, 1997; Slee et al, 1999). Another known mitochondrial proapoptotic protein is Smac/DIABLO, which is translocated from mitochondria into the cytosol concurrently with cytochrome c. Smac/DIABLO counteracts the effects of many inhibitor-of-apoptosis proteins (IAPs) and finally promotes caspase activation (Vaux and Silke, 2003; Verhagen et al, 2000). A caspase-independent apoptotic pathway has also been identified in experimental models of stroke (Cregan et al, 2004; Ferrer and Planas 2003; Plesnila et al, 2004; Postler et al, 2000; Zhao et al, 2004), including the activation of apoptosis-inducing factor (AIF). Like cytochrome c and Smac/DIABLO, AIF is normally confined to the mitochondrial intermembrane and translocates into the nucleus after the induction of apoptosis. Nuclear import of AIF triggers caspase-independent nuclear changes, including large-scale (~50 kDa) DNA fragmentation and peripheral chromatin condensation.
Although the final outcome for neurons expressing heat shock protein (Hsp) 70 after various injuries is controversial (Lee et al, 2001; Plumier et al, 1997; Ran et al, 2004), overexpression of Hsp70 has been shown to provide neuroprotection against cerebral ischemia both in vitro (Kelly et al, 2001; Lee et al, 2001; Lowenstein et al, 1991; Papadopoulos et al, 1996; Rordorf et al, 1991) and in vivo (Hoehn et al, 2001; Rajdev et al, 2000; Tsuchiya et al, 2003a; Yenari et al, 1998). The mechanism of neuroprotection has been attributed to the function of Hsp70 as a molecular chaperone protein that antagonizes apoptosis in both caspase-dependent and - independent pathways. Hsp70 inhibits apoptosis by binding to Apaf-1 and prevents the formation of the apoptosome, blocking the activation of caspase-9 (Beere et al, 2000; Saleh et al, 2000). Previously, we showed that overexpression of Hsp70 was associated with the reduction of cytochrome c release from mitochondria (Tsuchiya et al, 2003b), presumably leading to a reduction in apoptotic events downstream. Recent work has shown that Hsp70 may also prevent AIF-induced chromatin condensation in a caspase-independent apoptotic pathway by binding directly to AIF (Ravagnan et al, 2001).
In order to understand the molecular basis of the neuroprotective and antiapoptotic effect of Hsp70 in the developing brain, we studied the activation of mitochondrial apoptotic pathways in mice over-expressing Hsp70 in a model of neonatal H/I injury. We show that high constitutive expression of Hsp70 protects the brain from H/I in the neonatal period and differentially affects these apoptotic pathways. Our data provide new insights into the molecular mechanism of Hsp70-related neuroprotection in cerebral ischemia.
Materials and methods
Animals and Animal Care
Mice were housed and cared for according to the guidelines issued by the National Institutes of Health and the local institutional Animal Care and Use Committee. Transgenic mice overexpressing rat Hsp70 (Tg) and transgenic negative wild-type (Wt) littermates bred on CB6F1 background were kindly provided by Dr WH Dillman (University of California, San Diego, USA) and genotyped as described previously (Rajdev et al, 2000; Tsuchiya et al, 2003b). Hsp70 Tg mice were produced by using a chimeric transgene that consisted of the rat-inducible Hsp70 gene inserted into a pGAGGS vector under the control of human cytomegalovirus enhancer and chicken-actin promotor. There was no apparent phenotypic difference between Hsp70 Tg and Wt mice.
Neonatal Hypoxia/Ischemia Model
The Rice-Vannucci neonatal adaptation of the Levine procedure was used to induce H/I injury (Rice et al, 1981). Postnatal 7-day-old (P7) Hsp70 Tg (n = 102) and Wt (n = 98) mice weighing 4 to 5 g were anesthetized with 2% to 2.5% isoflurane in 30% O2 balanced with 70% N2O. Pups were subjected to right common carotid artery (CCA) occlusion by electrical coagulation on a warm mattress maintained at 37°C. Pups were returned to their dams immediately after surgery for 90 mins and were then exposed to 8% humidified O2 and balanced N2 for 30 mins in Plexiglas chambers partially submerged in a 38°C water bath. After hypoxia exposure, the pups were returned to their dams until euthanized. Multiple litters of mice were used throughout this study in each experiment to dilute any litter effect.
Cortical Cerebral Blood Flow and Body Temperature
In a separate group of Hsp70 Tg and Wt pups that were not included in the histologic evaluation, bilateral cortical CBF was monitored with laser Doppler flowmetry (Vasamedic, St Paul, MN, USA) during and 10 mins after H/I insult as described previously (Aden et al, 2003; Liu et al, 1999). To obtain reliable measurements, the pups were exposed to 8% O2 through a snug-fitting, nonrebreathable face mask containing continuous 2% isoflurane. After right CCA ligation, a midline scalp incision was made for securing the CBF probe to the skull surface. The probe was positioned 2 mm posterior to bregma and 2 mm lateral to the midline. During 30 mins of hypoxia, pups were kept on a warm mattress maintained at 37°C. After the completion of the hypoxia exposure, monitoring was continued for 10 mins more. The scalp incision was sutured, and pups were returned to their dams. Because anesthesia during the exposure to hypoxia might have influenced the severity of brain damage in these pups, they were not included in the histologic evaluation of brain damage. Rectal temperature was measured in both Hsp70 Tg and Wt mice pups during the 30 mins while mice were in the hypoxic chamber and for 30 mins thereafter (BIOPAC System Inc., Santa Barbara, CA, USA).
Measurement of Brain Injury and Dimensions
At 5 or 14 days after H/I, pups were anesthetized with an intraperitoneal injection of ketamine (80 mg/kg; Parke-Davis, Morris Plains, NJ, USA) and xylazine (15 mg/kg; Butler, Columbus, OH, USA) and transcardially perfused with 4% para-formaldehyde (PFA) in 100 mmol/L phosphate buffer (PB), pH 7.4 (PB). Brains were removed, postfixed for 2 h in 4% PFA-PB, and placed in 20% sucrose for 24 h. Cryostat cut 30-μm coronal sections were obtained.
Sections with 300-μm intervals spanning the entire neuroaxis were stained with hematoxylin and eosin (H&E) and evaluated by two investigators in a masked fashion with a well-developed brain damage scoring system (Sheldon et al, 1998). In brief, eight regions of brains were scored including the anterior, middle, posterior cortex, CA1, CA2, CA3, dentate gyrus of the hippocampus, and caudate putamen, with the contralesional hemisphere serving as a reference for uninjured tissue. Each region was given a rating from 0 to 3, in which 0 = no detectable neuronal cell loss; 1 = small focal areas of neuronal cell loss; 2 = columnar damage in the cortex involving predominantly layers II-IV, moderate cell loss; and 3 = cystic infarction and gliosis. The score from each region was then summed to generate a full range score from 0 to 24 in each animal; scores were averaged between 2 evaluators. We observed no cell injury in the hemisphere that was exposed to hypoxia only without CCA ligation, and thus designated it as the uninjured (UI) hemisphere (as in Figures 4A and 4C). Naïve mice without hypoxia or CCA ligation were used as controls (as in Figures 3B, 4B, 5, 6, and 7).
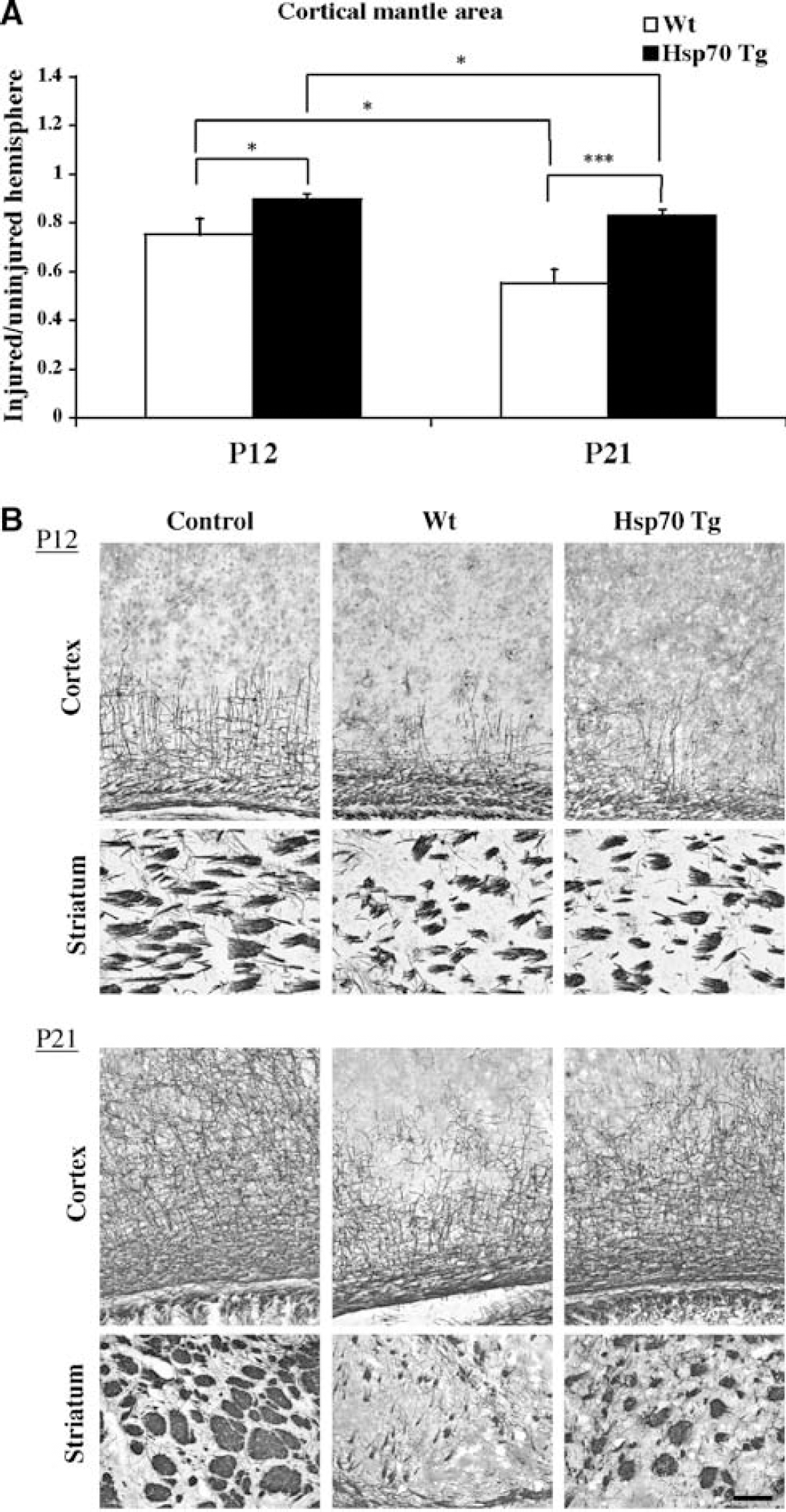
Progression of injury occurs in the gray and white matter between P12 and P21 in both transgenic (Tg) and wild-type (Wt) mice after hypoxic/ischemia (H/I) insult. (
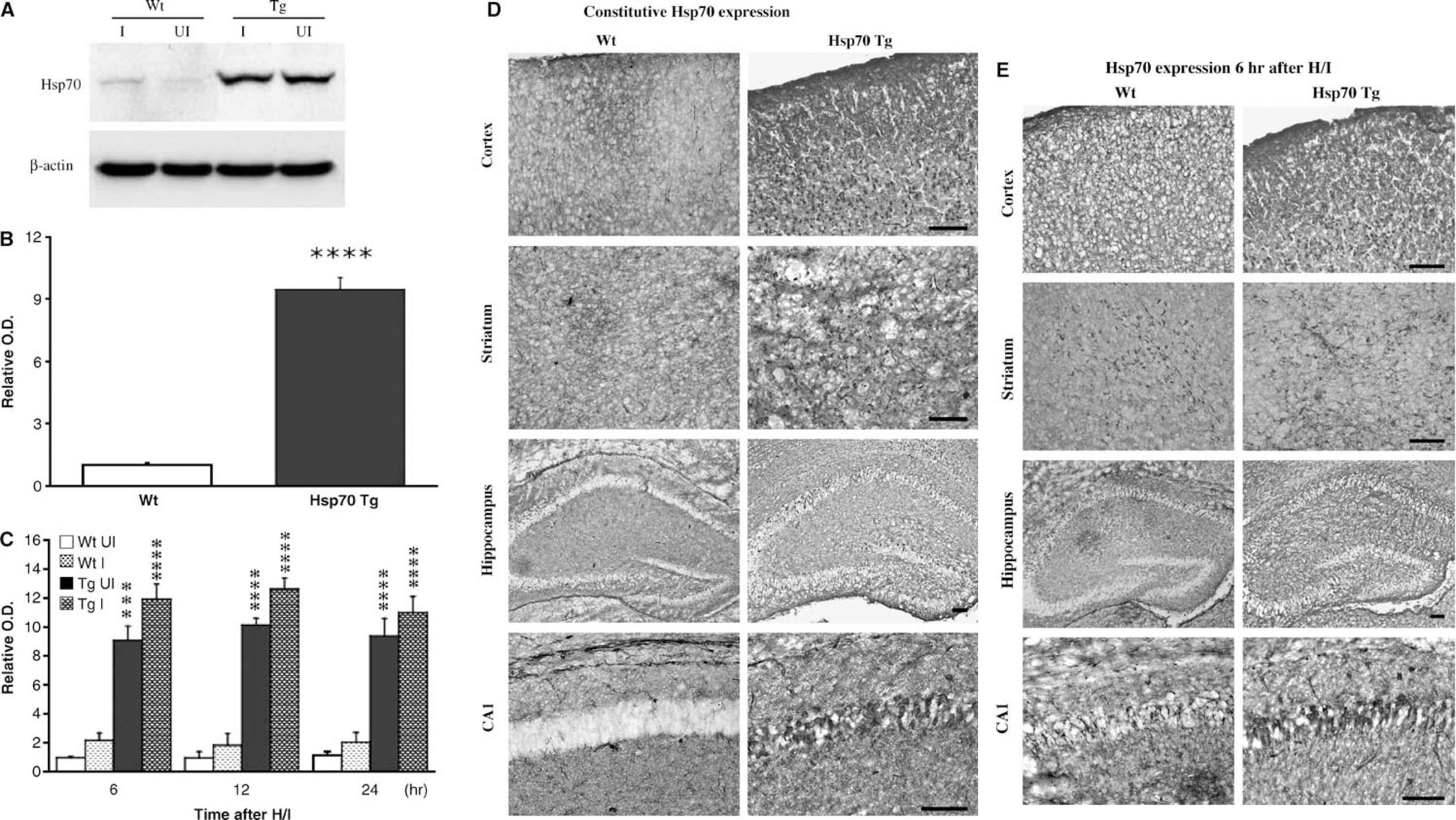
Constitutive and inducible Hsp70 protein expression in neonatal Hsp70 transgenic (Tg) and wild-type (Wt) mice before and after hypoxic/ischemia (H/I) injury. (
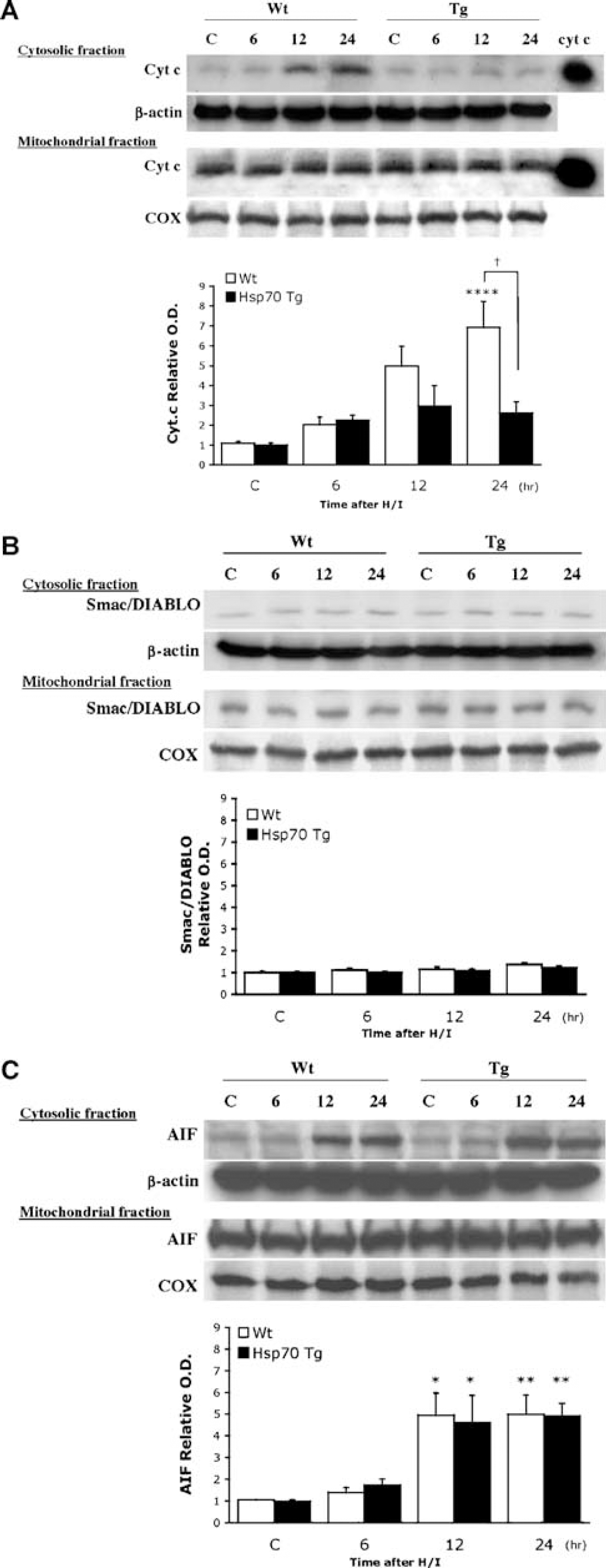
Translocation of proapoptotic proteins from mitochondria to cytosol in Hsp70 transgenic (Tg) and wild-type (Wt) littermates 6 h (n = 8 in Tg; n = 6 in Wt), 12 h (n = 7 in Tg; n = 5 in Wt) and 24 h (n = 7inTg;n = 7 in Wt) after hypoxic/ischemia (H/I) injury. Brains from naive Wt mice served as controls (C) (n = 4 each genotypes). β-Actin and cytochrome oxidase (COX) were used as internal controls for the cytoslic and mitorchondrial fractions, respectively. 0.2 μg of purified cytochrome c protein (cyt c) was used as a positive control. OD, optical density. (
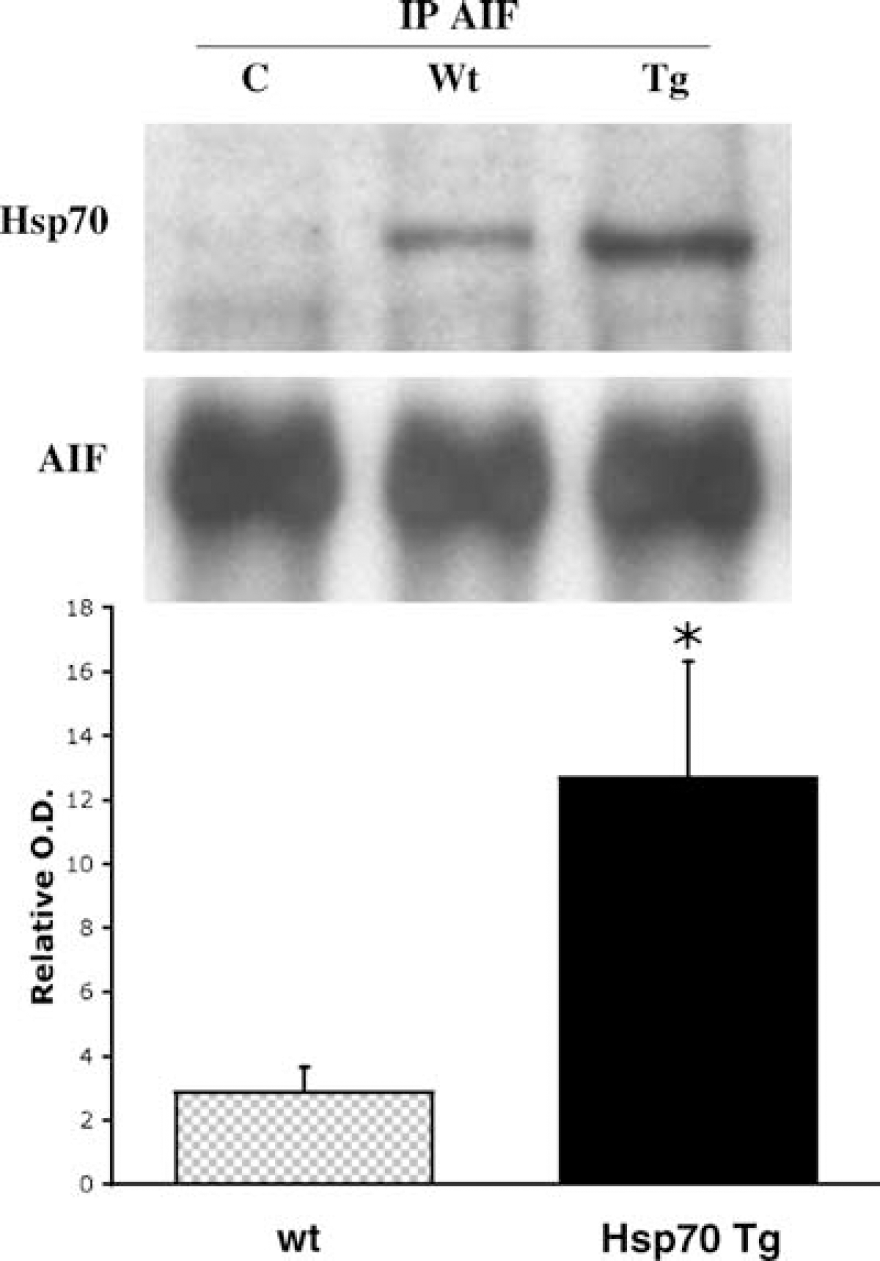
Increased binding of apoptosis-inducing factor (AIF) with Hsp70 in Hsp70 transgenic (Tg) mice at 24 h after hypoxic/ischemia (H/I) injury. Representative Western blots probed with Hsp70 and AIF as indicated from immunoprecipitated lysates with an antibody against AIF in injured brains of Hsp70 Tg and wild-type (Wt) mice at 24 h after H/I. Little or no Hsp70 was detected in the immuno pull-down from brain lysates of naive Wt control mice (C). Significantly more Hsp70/AIF was detected in Hsp70 Tg (n = 5) as compared with Wt mice (n = 4) (*P<0.05). Apoptosis-inducing factor signals did not differ among control, Hsp70 Tg, and Wt mice.
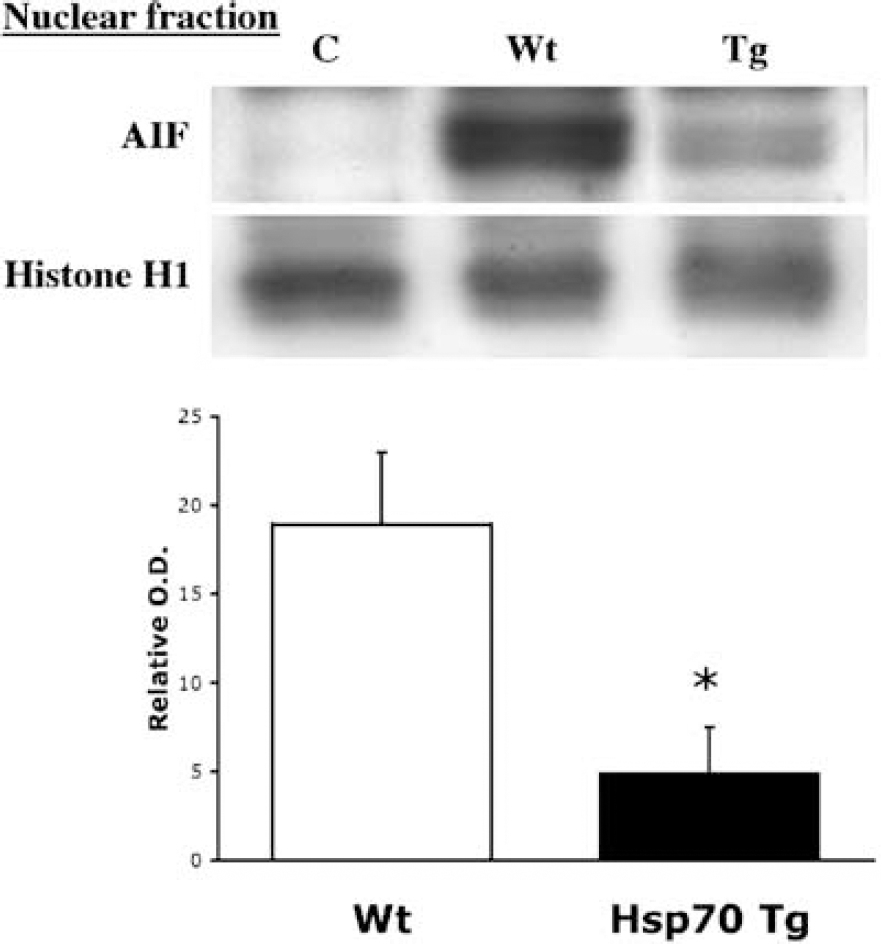
Reduced apoptosis-inducing factor (AIF) nuclear translocation in the Hsp70 Tg mice at 24 h after hypoxic/ischemia (H/I) insult. Western blots detected significantly less AIF translocation into nuclear fraction in Hsp70 transgenic (Tg) (n = 5) as compared with wild-type (Wt) (n = 11) mice (*P<0.05) at 24 h after H/I. Little or no AIF was detected in the nuclear fraction of brain lysates from naive Wt control mice (C). Histone H1 was used as an internal control for nuclear protein concentration.
To evaluate the progression of injury, areas of cortical mantle (Fan et al, 2003) were measured in H&E-stained sections by using Image J program (version 1.31, NIH image) and expressed as a ratio of injured to uninjured hemisphere to rule out variance in brain dimensions among individuals. Only brains that were sectioned in perfect symmetry between two hemispheres were used for the evaluation.
Immunohistochemistry
Immunohistochemistry was performed to evaluate Hsp70 and myelin basic protein (MBP) expression as described (Raber et al, 2004) by using mouse anti-Hsp70 monoclonal (1:1,000; Stressgen, Victoria, BC, Canada, USA) and rabbit anti-MBP (1:400; Chemicon, Temecula, CA, USA). Biotinylated sheep anti-mouse or goat anti-rabbit Ig (1:200; Amersham Life Science, Arlington Heights, IL, USA) were used as secondary antibodies for 2 h, followed by 2 h in avidin-biotin complex solution (ABC) (Vector Laboratories, Youngstown, OH, USA) in PB. Sections were then visualized with 0.05% diaminobenzine-tetrachloride (DAB Fast; Sigma, St Louis, MO, USA) in 0.01% H2O2, and 0.04% NiCl2.
Subfractionation of Cellular Proteins from Brain Samples for Western Blot Analysis
Protein samples for Western blot analysis were prepared as described with some modification (Yin et al, 2002). Injured and uninjured hemispheres were dissected out from the brains at 6, 12, and 24 h after H/I and were processed separately. Whole-cell lysates were obtained by gently homogenizing the brain sample with a tissue pestle grinder (Kontes, Vineland, NJ, USA) for 10 strokes in 5 volumes of buffer A (20 mmol/L HEPES-KOH, 250 mol/L sucrose, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol (DTT), 0.1 mmol/L PMSF and proteinase inhibitor cocktail tablets; pH 7.9). Samples were further centrifuged at 750g at 4°C for 15 mins to separate into supernatant A and pellet A. Pellet A containing the nuclear fraction was resuspended in 90 μL of buffer B (20 mmol/L HEPES-KOH, 1.5 mmol/L MgCl2, 20 mmol/L KCl, 0.2 mmol/L EDTA, 0.5 mmol/L DTT, 0.2 mol/L PMSF and proteinase inhibitor cocktail tablets; pH 7.9), and mixed with 30 μL of buffer C (20 mmol/L HEPES-KOH, 1.2 mol/L KCL, 0.2 mmol/L EDTA, 0.5 mmol/L DTT, 0.2 mmol/L PMSF, and proteinase inhibitor cocktail tablets; pH 7.9). The samples were placed on ice for 30 mins during extraction and centrifuged at 12,000g. Supernatants containing nuclear fraction were transferred and stored at −70°C. Supernatant A containing cytosolic/mitochondrial proteins was further centrifuged at 16,000g for 30 mins at 4°C to separate supernatant B from pellet B. Supernatant B was used as the cytosolic fraction, and pellet B was used as the mitochondrial fraction after resuspension in buffer A. Protein concentrations were determined by the Bradford method (Bio-Rad, Hercules, CA, USA).
Western Blots Analysis
Equal amounts of protein samples (10 μg of each protein) or purified cytochrome c from rat heart at 0.1 μg (Sigma) as a positive control were loaded in each lane and electrophoresed on a 7.5% to 15% sodium dodecyl sulfate polyacrylamide gel, and transferred onto a nitrocellulose membrane. The membrane was incubated overnight at 4°C with primary mouse anti-Hsp70 (1:1,000; Stressgen), rabbit anti-cytochrome c (1:1,000; Santa Cruz Biotechnology, Santa Cruz CA, USA), rabbit anti-Smac/DIABLO (1:2,000; Chemicon), rabbit anti-AIF (1:8,000; Sigma), mouse anti-β-actin (1:8,000; Sigma), mouse anti-cytochrome oxidase (COX) subunit IV (0.2 μg/mL; Molecular Probes, Eugene, OR, USA) or mouse anti-histone H1 (1:1,000; Santa Cruz Biotechnology). Blots were developed with peroxidase-conjugated secondary antibodies against appropriate species, and then signals were visualized with ECL Plus reagents (Amersham) followed by exposure to X-ray film. Films were scanned and optical density (OD) was determined with Image J program. β-Actin, COX IV, and histone H1 were used as an internal control for the cytosolic, mitochondrial, and nuclear fraction, respectively. Blots of the cytosolic fraction were also probed with antibodies against COX IV and histone H1 to cross check and ensure proper fractionation. To compare protein level in the Western blots, OD ratios were obtained by correcting the OD of each sample with the OD of appropriate internal standard.
Coimmunoprecipitation
Whole-cell lysates from brains at 24 h after H/I were obtained as described earlier. Whole cell lysates in buffer A were preincubated with 10 μL of protein A agarose (Oncogene, Boston, MA, USA) for 3 h at 4°C to remove nonspecific binding proteins. After centrifugation at 12,000g for 20 secs, the supernatant was incubated overnight at 4°C with 5 μL of anti-AIF antibody (Sigma) and then for 3 h at 4°C with 10 μL of protein A agarose. Pellets were precipitated by centrifugation at 12,000g for 20 secs and washed 3 times with buffer A. After boiling for 3 mins to dissociate the immune complexes, the samples were again centrifuged at 12,000g for 20 secs and the supernatant was used for Western blot analysis.
Statistical Analysis
The Mann-Whitney test was used for nonparametric comparisons of brain damage. Parametric data were evaluated by ANOVA, followed by Tukey-Kramer post hoc tests when appropriate. P-values <0.05 were considered statistically significant. Brain damage scores were expressed as median and other values expressed as mean ± standard error of the mean (s.e.m.). Statistical analysis was performed with StatView (Version 5.0.1, SAS Institute Inc., Cary, NC, USA).
Results
Overexpression of Hsp70 Reduces H/I-Induced Brain Injury
Brain injury was determined in H&E-stained sections by using a 0 to 24 brain damage scoring system described previously (Sheldon et al, 1998). The brain damage score was significantly lower in Hsp70 Tg as compared with Wt mice at both 5 and 14 days after H/I insult (P<0.001). The median brain damage scores on P12 and P21 were 7 (n = 39) and 8 (n = 15) in Hsp70 Tg versus 21 (n = 32) and 22 (n = 18) in Wt mice, respectively (Figure 1). The mortality rate was 3.9% in Hsp70 Tg and 7.1% in Wt mice, 4 out of 102 Tg and 7 out of 98 Wt mice died during the period of hypoxia exposure and 2 additional mice each in the Tg and Wt groups died after restoration of normoxia.
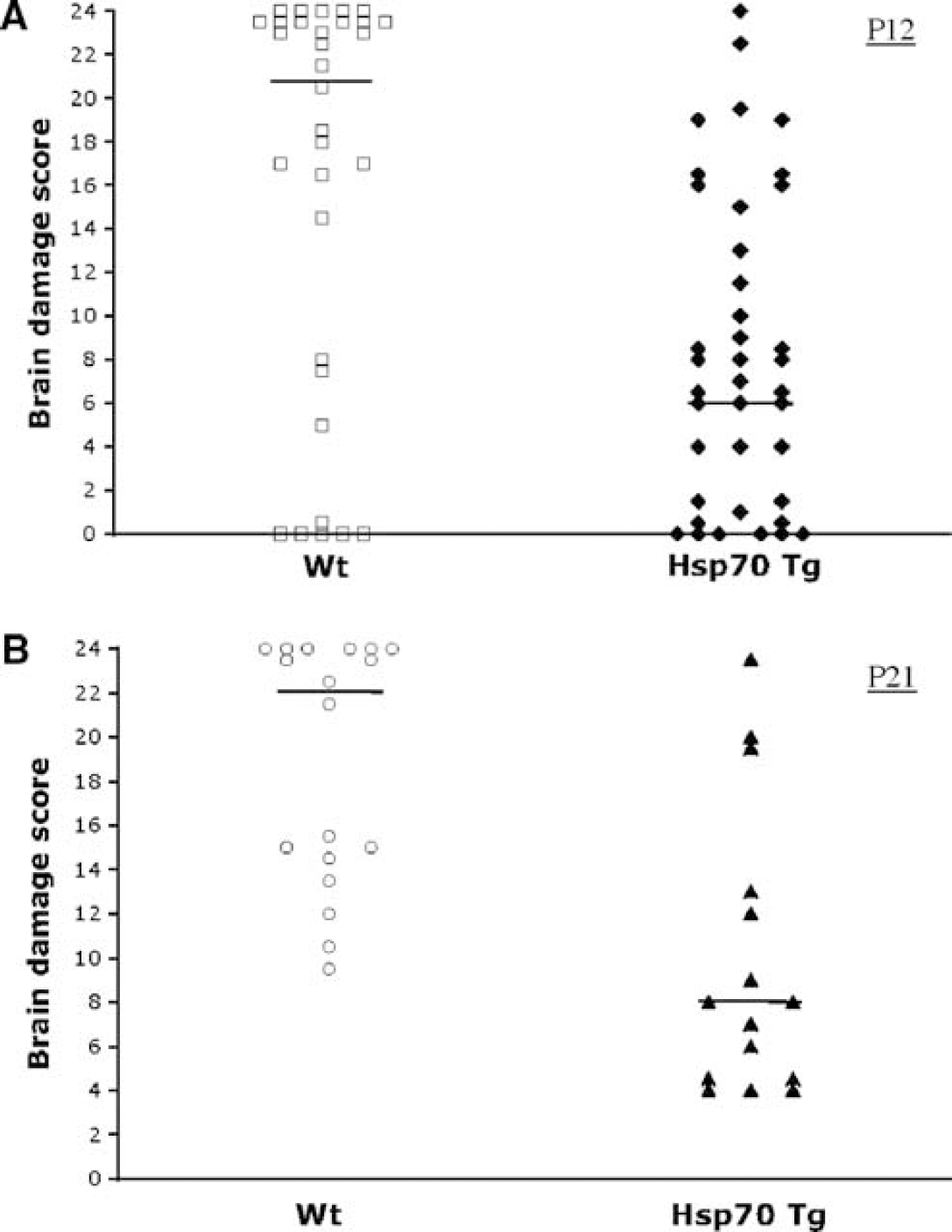
Reduced brain damage in Hsp70 transgenic (Tg) mice as compared with wild-type littermates (Wt) 5 and 14 days after hypoxic/ischemic (H/I) injury. Significantly less brain damage was detected in Hsp70 Tg than in Wt mice on both P12 (
The neuroprotective effect seen in Hsp70 Tg mice was not attributed to variance in CBF because there was no significant difference in cortical CBF between Tg and Wt mice at any given time during the investigation (Figure 2A). Cortical CBF in the injured hemisphere was reduced to approximately 50% of that in the uninjured hemisphere within 5 mins after the induction of hypoxia and returned to baseline level soon after restoration of normoxia (Figure 2A). Body temperature also did not appear to be the contributing factor for Hsp70-related neuroprotection, as there was no significant difference in rectal temperature during hypoxia and up to 30 mins after restoration of normoxia, between the Tg and Wt mice (Figure 2B).
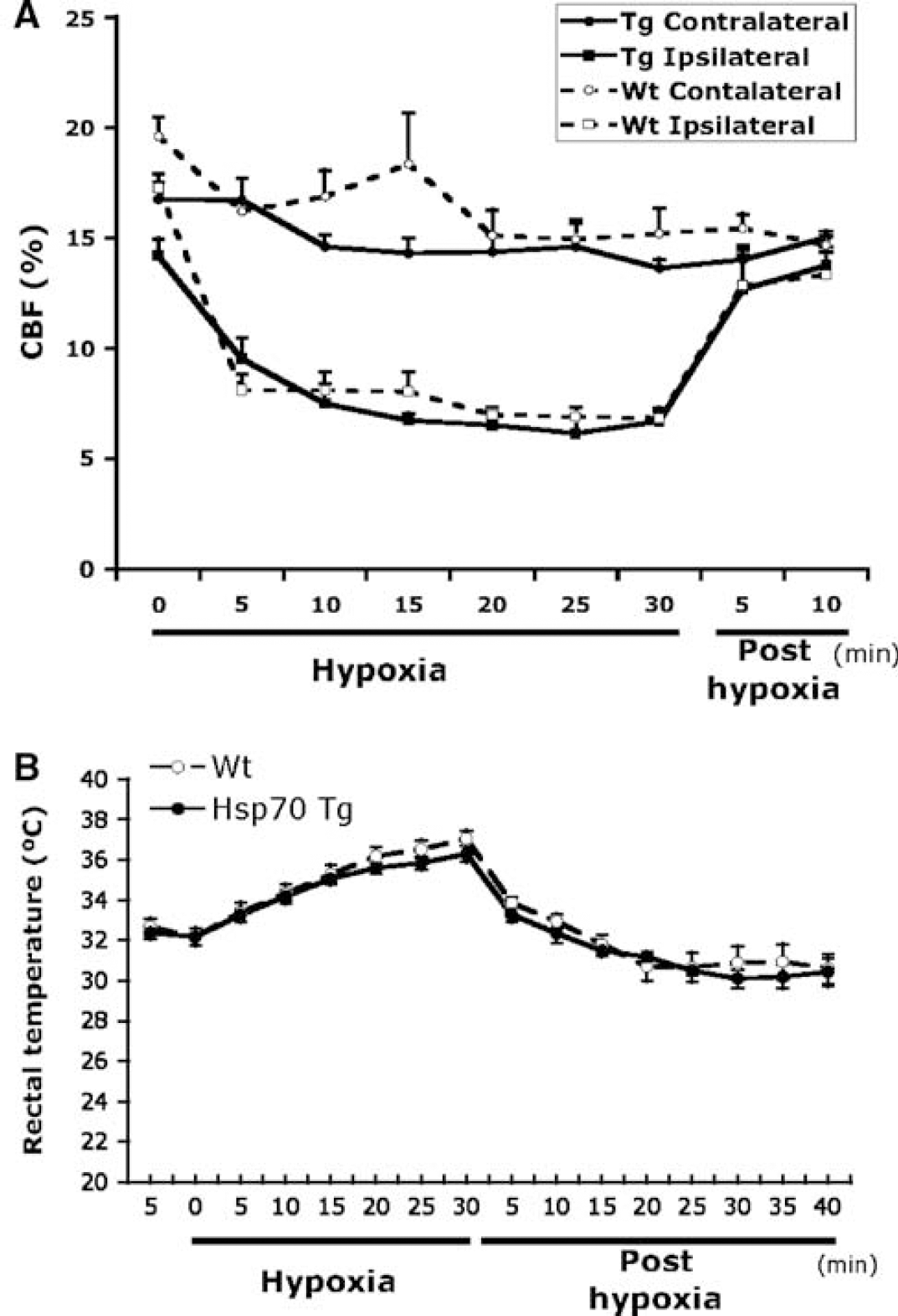
Lack of significant differences in cortical cerebral blood flow (CBF) and body temperature between Hsp70 transgenic (Tg) and wild-type (Wt) mice during and after hypoxic/ischemia (H/I) insult. (
Continuous Injury Evolution in the Gray and White Matter after P12
As delayed injury often occurs after neonatal brain injury (Northington et al, 2001a, b), we compared the areas of cortical mantle in mice between P12 and P21 to determine if injury progressed beyond 5 days after H/I. The ratio of ipsilateral versus contralateral cortical mantle area was obtained to eliminate individual variance. The mean ratios of cortical mantle were significantly smaller in the brains on P21 than P12 in both Tg (P<0.05) and Wt (P<0.05) mice (Figure 3A), suggesting a progressive loss or shrinkage of cortex between these time points. The mean ratios of cortical mantle were also significantly smaller in Tg compared with Wt mice, both on P12 (P<0.05) and P21 (P< 0.005), confirming a smaller cortical injury observed in the Tg. MBP immunohistochemistry showed significantly more extensive damage to white matter on P21 than P12 in both Tg and Wt mice (Figure 3B), corroborating the continuous evolution of injury seen in the gray matter. Hsp70 overexpression did not affect the morphology of the MBP staining pattern (data not shown).
High Constitutive Level of Hsp70 in Tg Mice Versus Robust Induction of Hsp70 in Wt Mice
To determine the level of constitutive and inducible Hsp70 protein at the time of H/I injury, Western blot analysis was performed in both Hsp70 Tg and Wt brains. In brain samples from naïve mice, a 9- to 10-fold higher constitutive Hsp70 level was found in Hsp70 Tg as compared with Wt mice (P< 0.0001) (Figure 4B), consistent with our previous observation in adult Hsp70 Tg mice (Rajdev et al, 2000). Interestingly, the ratio of Hsp70 in the injured versus uninjured hemisphere was higher in Wt compared with that in Tg at 6, 12, and 24 h after H/I, suggesting that more inducible Hsp70 was produced in the Wt mice responding to the H/I insult. However, despite a more robust Hsp70 induction in the Wt mice after H/I injury that is not associated with protection, there was still a greater amount of total Hsp70 in Tg than in Wt mice (Figures 4A and 4C).
Immunohistochemical analysis of Hsp70 revealed the spatial distribution of constitutive (Figure 4D) and total Hsp70 expression at 6 h after H/I insult (Figure 4E) on P7. In the uninjured hemisphere, strong immunoreactivity was observed in cerebral cortical, striatal, and hippocampal cells with neuronal morphologic features in Hsp70 Tg brains. On the contrary, little or no immunoreactivity was observed in Wt brains (Figure 4D). This finding is consistent with our previous results showing strong constitutive Hsp70 overexpression in adult Hsp70 Tg mice (Rajdev et al, 2000). In the injured hemisphere, an upregulation of Hsp70 expression was observed in cerebral cortical, striatal, and hippocampal cells in both Hsp70 Tg and Wt mice, also consistent with previous reports in neonatal rat H/I (Ferriero et al, 1990). Specifically, Hsp70 immunoreactive cells were found throughout the hemisphere in Tg mice with increased density in the peri-infarct areas. By comparison, Hsp70 immunoreactive cells were observed only in the cortex and striatum, and throughout most of the hippocampus of Wt mice subjected to H/I insult (Figure 4E).
Reduction of Cytosolic Cytochrome c Translocation in Hsp70 Tg After H/I
To investigate how Hsp70 overexpression affected mitochondrial proapoptotic protein activation and translocation after H/I, differential fractionation and Western blotting were performed on lysates from 26 Hsp70 Tg mice and 22 Wt mice at 6, 12, and 24 h after H/I. Robust Cox signals were detected in the mitochondrial fractions (Figure 5) from either Hsp70 or Wt mice but not from the cytosolic fractions (data not shown), indicating proper subcellular fractionations without cross-contamination. Cytosolic cytochrome c translocation from mitochondria occurred as early as 6 h after H/I in both Hsp70 Tg and Wt mice (Figure 5A). Significantly more cytosolic cytochrome c was detected in Wt mice (P<0.05) in a time-dependent manner after H/I, whereas a substantially smaller increase in cytosolic cytochrome c translocation was observed in Hsp70 Tg mice at 12 and 24 h after H/I.
Lack of Smac/DIABLO Cytosolic Translocation During the First 24 h After H/I
In contrast to the significantly increased levels of cytochrome c found in the cytosol, little or no Smac/DIABLO translocation to the cytosol was observed in either Hsp70 Tg or Wt mice within the first 24 h after H/I (Figure 5B).
AIF Translocation into the Cytosol
AIF translocation to the cytosol began as early as 6 h after H/I with a significant amount of AIF found in the cytosol in both genotypes. The amount of AIF in the cytosol was significantly increased at 12 and 24 h after H/I in both Tg and Wt mice as compared with the uninjured controls (Figure 5C). There was no significant difference in the amount of cytosolic AIF between the two genotypes at any time point investigated. These results suggest that over-expression of Hsp70 does not affect AIF translocation from mitochondria into cytosol.
Protein-Protein Interaction between Hsp70 and AIF
Coimmunoprecipitation was performed to investigate the physical interaction between Hsp70 and AIF after H/I insult by using whole-cell lysates from the injured and uninjured hemispheres of both Hsp70 Tg (n = 5) and Wt (n = 4) mice. Immunoprecipitation using an antibody against AIF brought down significantly more Hsp70 protein from the injured hemispheres of Tg than Wt mice at 24 h after H/I (P<0.05) (Figure 6).
Reduction of AIF Nuclear Translocation in Hsp70 Tg Mice
To determine whether interaction between AIF and Hsp70 affected further downstream apoptotic events, lysates from cytosolic, and nuclear fractions of 5 Tg and 11 Wt mice were used in Western blotting. Significantly less AIF was detected in the nuclear extracts from the injured hemispheres of Tg than Wt mice (P<0.05) (Figure 7).
Discussion
Hsp70 protein is a potent cell death-inhibitory factor that protects against an array of stressful stimuli. Members of the HSP family are in general activated by oxidative stress, and likely help maintain viability under this stress condition (Barrett et al, 2004). The principal finding in this study is that baseline Hsp70 overexpression protects neonatal brains from H/I injury and its protective effect is associated with the reduction of AIF translocation to the nucleus due to an increased binding of Hsp70 to AIF. Overexpression of Hsp70 also correlated with a reduction of cytosolic translocation of cytochrome c after H/I injury in the neonatal mice. To our knowledge, this is the first report to show a neuroprotective effect of Hsp70 in neonatal brains against H/I injury and its specific interaction with apoptosis pathways involving cytochrome c and AIF.
The neuroprotective effect of Hsp70 against a variety of insults is best exemplified in both in vitro (Kelly et al, 2001; Lee et al, 2001; Lowenstein et al, 1991; Papadopoulos et al, 1996; Rordorf et al, 1991) and in vivo (Hoehn et al, 2001; Rajdev et al, 2000; Tsuchiya et al, 2003a; Yenari et al, 1998) models of cerebral ischemia. Although there are few inconsistent observations with regard to its neuroprotective effect (Lee et al, 2001; Plumier et al, 1997), levels of Hsp70 expression prior to the induction of injury might hold the key in determining these results. Similar to the adult Hsp70 overexpressors (Rajdev et al, 2000), our neonatal Hsp70 Tg mice have about a 10-fold higher constitutive Hsp70 expression than the Wt mice (Figure 4), supporting our previous speculation that higher constitutive Hsp70 expression may be required to protect brains against ischemic injury (Tsuchiya et al, 2003b). At 6 h after H/I insult, the ratio of Hsp70 protein detected by Western blotting in the injured versus uninjured hemisphere was actually higher in Wt than Tg (Figure 4), suggesting that the ratio of inducible versus total Hsp70 after H/I injury is higher in the Wt than Tg mice. This is not surprising because the Hsp70 transgene is produced by using a chimeric transgene that is under the control of human cytomegalovirus enhancer and chicken-actin promotor, which are not the optimal elements for Hsp70 gene transcription (Abravaya et al, 1992; Giaccia et al, 1992; Liu et al, 1993; Park and Craig 1991; Perry et al, 1994). However, despite the increased level of inducible Hsp70 elicited in the Wt mice, the total amount of Hsp70 is not sufficient to protect the neonatal brain from H/I damage. This suggests that the presence of Hsp70 before the onset of injury is a prerequisite to the protection conferred. It is also possible that lesser protection observed in the Wt mice was because of either an insufficient quantity of, or delayed timing in, Hsp70 induction. In addition, biosynthesis of Hsp70 after H/I injury might be detrimental to the brain at a time when energy metabolism is at a critical state. Lastly, in keeping with the previous observation that H/I injury evolves over time (Northington et al, 2001a), our data showing that the degree of injury was greater on P21 relative to P12 (Figure 3) further indicate that delayed injury outlasts the early and transient induction of Hsp70 protein. This might also explain the lack of significant neuroprotection seen in the Wt mice despite robust induction of Hsp70.
Cytochrome c is the first apoptogenic intermembrane protein identified as being released from mitochondria during apoptosis (Liu et al, 1996b). We previously showed that Hsp70 overexpression protected adult murine brains from permanent focal ischemic injury presumably by reducing cytochrome c release and inhibiting apoptosis (Tsuchiya et al, 2003b). Consistent with our report showing reduced cytosolic cytochrome c release in adult Hsp70 overexpressors, we also noticed a reduction in cytochrome c translocation to the cytosol in the neonatal Hsp70 Tg mice after H/I injury (Figure 5A). Recent studies showed that Smac/DIABLO translocation from mitochondria into cytosol occurs 24 h after focal cerebral ischemia in adult mice (Saito et al, 2003) and 12 h after global ischemia in adult rats (Tanaka et al, 2004). However, in the present study, there was no significant difference in the amount of cytosolic Smac/DIABLO in either the Tg or Wt mice within 24 h after H/I injury (Figure 5B). This implies that the regulation of Smac/DIABLO translocation after H/I in the neonatal brain may differ from that after focal ischemia in the adult brain. In addition, our results also suggest that prevention of cytochrome c release from mitochondria may be specific, since Hsp70 overexpression had very little effect on Smac/DIABLO cytosolic translocation within 24 h after H/I insult.
AIF, a mitochondrial intermembrane protein, has been found to translocate from mitochondria to cytosol and nuclei in a caspase-independent fashion. Recent studies showed that AIF transolocates from mitochondria into the cytosol and nuclei after global and focal ischemia in the adult rodent (Cao et al, 2003; Plesnila et al, 2004), and this translocation was blocked by Bcl-2 transfection after focal ischemia in rat (Zhao et al, 2004). A decrease of AIF in the mitochondrial fraction was also observed 24 h after neonatal HI (Zhu et al, 2003), consistent with our finding that AIF might be translocated from the mitochondrial to nuclear fractions (Figure 7). The fact that there was less nuclear AIF in Tg compared with Wt mice 24 h after H/I suggests that over-expression of Hsp70 may prevent the nuclear import of AIF in Tg mice (Figure 7), leading to reduced apoptosis. In addition, more Hsp70 was coimmunoprecipitated with AIF in Hsp70 Tg than Wt mice (Figure 6), whereas similar amounts of cytosolic AIF were detected in both Hsp70 Tg and Wt mice during the first 24 h after H/I (Figure 5C). Moreover, preincubation of AIF with Hsp70 yielded a more efficient inhibition of apoptosis in vitro than preincubation of nuclei with Hsp70 (Ravagnan et al, 2001), suggesting that Hsp70 is required to antagonize AIF before the import of AIF into the nuclear compartment to prevent apoptosis. These findings support our results showing that robust neuroprotection by Hsp70 against neonatal H/I in Hsp70 Tg mice was due to high constitutive Hsp70 expression before the onset of injury.
Hsp70 did not substantially prevent AIF release from mitochondria to cytosol, but did prevent cytochrome release 24 h after H/I insult in Hsp70 Tg mice. Although both proapoptotic proteins are confined to mitochondrial intermembrane space and released in a similar fashion, the mechanisms of release and regulation by Hsp70 are quite different between cytochrome c and AIF. Inhibition of AIF by microinjection of a neutralizing antibody or direct inactivation of the AIF gene can prevent mitochondrial membrane permeabilization (MMP), whereas microinjection of AIF itself or transfection with AIF gene constructs can trigger the release of cytochrome c or release of AIF itself from mitochondria (Ferri et al, 2000; Joza et al, 2001; Loeffler et al, 2001). The ability of Hsp70 in preventing MMP might be attributed to its effect in antagonizing AIF directly (Polla et al, 1996; Ravagnan et al, 2001). Because AIF release might occur after cytochrome c release (Arnoult et al, 2002), it is also possible that Hsp70 might act both upstream of mitochondrial release of cytochrome c to reduce translocation into cytosol and downstream in reducing apoptotic events in Hsp70 Tg mice. Although there is no feasible method with which to measure the amount of AIF binding to Hsp70 in the cytosol while the cells are alive, our data showing an enhanced AIF-Hsp70 interaction leading to reduced nuclear import of AIF and brain injury are in keeping with our current knowledge about the role of AIF and apoptosis.