
Abstract
Neuronal injury may be dependent upon the generation of the free radical nitric oxide (NO) and the subsequent induction of programed cell death (PCD). Although the nature of this injury may be both preventable and reversible, the underlying mechanisms that mediate PCD are not well understood. Using the agent nicotinamide as an investigative tool in primary rat hippocampal neurons, the authors examined the ability to modulate two independent components of PCD, namely the degradation of genomic DNA and the early exposure of membrane phosphatidylserine (PS) residues. Neuronal injury was determined through trypan blue dye exclusion, DNA fragmentation, externalization of membrane PS residues, cysteine protease activation, and the measurement of intracellular pH (pHi). Exposure to the NO donors SIN-1 and NOC-9 (300 μmol/L) alone rapidly increased genomic DNA fragmentation from 20 ± 4% to 71 ± 5% and membrane PS exposure from 14 ± 3% to 76 ± 9% over a 24-hour period. Administration of a neuroprotective concentration of nicotinamide (12.5 mmol/L) consistently maintained DNA integrity and prevented the progression of membrane PS exposure. Posttreatment paradigms with nicotinamide at 2, 4, and 6 hours after NO exposure further demonstrated the ability of this agent to prevent and reverse neuronal PCD. Although not dependent upon pHi, neuroprotection by nicotinamide was linked to the modulation of two independent components of neuronal PCD through the regulation of caspase 1 and caspase 3-like activities and the DNA repair enzyme poly(ADP-ribose) polymerase. The current work lays the foundation for the development of therapeutic strategies that may not only prevent the course of PCD, but may also offer the ability for the repair of neurons that have been identified through the loss of membrane asymmetry for subsequent destruction.
Keywords
Neuronal injury can be initiated by several different stimuli that ultimately lead to either apoptosis or necrosis. Neuronal apoptosis or programed cell death (PCD) is an active, controlled, and deliberate process of cell destruction that is present during cell development and injury (Kerr et al., 1972). In contrast, neurons that undergo necrosis suffer diffuse organelle damage with the subsequent discharge of toxic products to neighboring cells. Programmed cell death without effective modulation may precipitate neurodegenerative diseases, such as Alzheimer's disease, Huntington's disease, and Parkinson's disease (Thomas et al., 1995). Although the mechanisms that result in PCD are multiple in nature, elucidating the underlying pathways that mediate PCD may serve to develop effective therapeutic strategies against neurodegeneration.
Experimental models have illustrated that ischemic generation of the free radical nitric oxide (NO) can be a trigger for the subsequent induction of neuronal PCD in a variety of cell types, such as cortical neurons (Brune et al., 1999), hippocampal neurons (Maiese et al., 1999; Maiese and Vincent, 2000), and mesencephalic neurons (Hunot et al., 1996). Cellular pathways generated by NO that result in PCD are varied, but include constitutive and inducible neuronal endonucleases (Vincent and Maiese, 1999b; Vincent et al., 1999a), intracellular acidification (Ito et al., 1997; Vincent et al., 1999b), and cysteine proteases (Maiese and Vincent, 1999; Uehara et al., 1999). As a result of its close link to the molecular pathways that lead to PCD, NO functions not only as a potential therapeutic target, but also as a valuable investigational agent.
Neuronal PCD is believed to proceed through two dynamic, but distinct, pathways that involve DNA fragmentation and the loss of membrane asymmetry with the exposure of membrane phosphatidylserine (PS) residues (Vincent and Maiese, 1999a; Maiese and Vincent, 2000). These processes are functionally independent determinants of neuronal PCD. The internucleosomal cleavage of genomic DNA into fragments may be a late event during PCD and ultimately commit a cell to its death. In contrast, the redistribution of membrane PS residues can be an early event during PCD that usually precedes DNA fragmentation (Rimon et al., 1997; Vincent and Maiese, 1999a) and may serve to “tag” injured cells for phagocytosis (Verhoven et al., 1999). Recent work that uses the ability to follow the progressive externalization of membrane PS residues in adherent monolayer living neurons over time has provided evidence that neuronal PCD may also be reversible in nature (Vincent and Maiese, 1999a; Maiese and Vincent, 2000).
Studies using neuroprotective regiments, such as the application of either trophic factors (Kiprianova et al., 1999) or metabotropic glutamate receptor agonists (Maiese et al., 1999; Vincent et al., 1999a; Maiese and Vincent, 2000), have further assisted in demonstrating the concept of reversible neuronal injury. Previous work has shown that the initial stages of PCD can be significantly prevented with appropriate therapeutic intervention, such as during neuronal ischemia (Schulz et al., 1998) and free radical exposure (Brune et al., 1999; Maiese and Vincent, 1999; Vincent et al., 1999a). Further support for the reversible nature of neuronal PCD has been suggested with the agent nicotinamide, an essential precursor of nicotinamide adenine dinucleotide. Nicotinamide was observed to be neuroprotective at least two hours after the onset of permanent focal cerebral ischemia in rats (Ayoub et al., 1999). Although nicotinamide appears to be an attractive therapeutic agent against neuronal injury and is membrane diffusable to directly modulate hippocampal action potentials (Wallis et al., 1996), the underlying mechanisms that determine this neuroprotection have not been elucidated.
In the current study, the authors used nicotinamide as an investigational tool to demonstrate that NO-induced neuronal PCD is not only reversible, but preventable, through the preservation of genomic DNA integrity and through the extended maintenance of cellular membrane asymmetry. The current studies illustrate that the neuroprotective ability of nicotinamide is robust and novel. Nicotinamide, through its ability to regulate caspase 1 (interleukin converting enzyme, ICE) and caspase 3 (CPP32), can modulate independent components of PCD by maintaining DNA integrity and cellular membrane asymmetry. Nicotinamide also offers enhanced protection through the modulation of the DNA repair enzyme poly(ADP-ribose) polymerase (PARP) and the prevention of membrane PS exposure that can protect neurons from being “tagged” for destruction.
MATERIALS AND METHODS
Primary hippocampal neuronal cultures
Hippocampi were obtained from E-19 Sprague-Dawley rat pups and incubated in dissociation medium (90 mmol/L Na2SO4, 30 mmol/L K2SO4, 5.8 mmol/L MgCl2, 0.25 mmol/L CaCl2, 10 mmol/L kynurenic acid, and 1 mmol/L HEPES with the pH adjusted to 7.4) containing papain (10 U/mL) and cysteine (3 mmol/L) for two 20-minute periods. The hippocampi were then rinsed in dissociation medium and incubated in dissociation medium containing trypsin inhibitor (10 to 20 U/mL) for three 5-minute periods. Cells were washed in growth medium (Leibovitz's L-15 medium; GibcoBRL, Gaithersburg, MD, U.S.A.) containing 6% sterile rat serum (Bioproducts for Science, Indianapolis, IN, U.S.A.), 150 mmol/L NaHCO3, 2.25 mg/mL transferrin, 2.5 μg/mL insulin, 10 nmol/L progesterone, 90 μmol/L putrescine, 15 nmol/L selenium, 35 mmol/L glucose, 1 mmol/L L-glutamine, penicillin and streptomycin (50 μg/mL), and vitamins. Dissociated cells were plated at a density of ∼1.5 × 103 cells/mm2 in 35 mm polylysine/laminin-coated plates (Falcon Labware, Lincoln Park, NJ, U.S.A.). Neurons were maintained in growth medium at 37°C in a humidified atmosphere of 5% CO2 and 95% room air. All experiments were performed with neurons that had been in culture for 2 weeks. Nonneuronal cells accounted for 10% to 20% of the total cell population.
Experimental treatments
Nitric oxide administration was performed by replacing the culture media with media containing 3-morpholinosydnonimine (SIN-1) (300 μmol/L) (Alexis Corporation, San Diego, CA, U.S.A.) or 6-(2-hydroxy-1-methyl-2-nitrosohydrazino) -N-methyl-1-hexanamine (NOC-9) (300 μmol/L) (Calbiochem, San Diego, CA, U.S.A.) for 5 minutes. The authors have shown previously that SIN-1 and NOC-9 are directly toxic to hippocampal neurons through a mechanism that involves the release of NO with a 5-minute application of 300 μmol/L resulting in the death of approximately 70% to 80% of neurons over a 24-hour period (Maiese and Boccone, 1995; Vincent and Maiese, 1999b). More than one NO generator was used as a control to demonstrate that the neurons were responding to NO rather than to other by-products of these agents. After treatment with the NO donors, the culture medium was replaced with fresh growth medium and the cultures were placed in a normoxic, humidified incubator at 37°C with 5% CO2 for periods determined by the specific experimental paradigm. In both pre-and postparadigm applications, nicotinamide exposure was continuous.
Neuronal survival assays
Hippocampal neuronal injury was determined by bright field microscopy using a 0.4% trypan blue dye exclusion method 24 hours after treatment with the NO donors. Neurons were identified by morphology and the mean survival was determined by counting 8 randomly selected nonoverlapping fields with each containing approximately 10 to 20 neurons (viable + nonviable) in each 35-mm Petri dish. Mean survival from each culture dish represents an n = 1 determination. Each experiment was replicated 4 to 6 times independently with different cultures.
Assessment of DNA fragmentation
Genomic DNA fragmentation was determined by the terminal deoxynucleotidyl transferase nick end labeling (TUNEL) assay. Briefly, neurons were fixed in 4% paraformaldehyde/0.2% picric acid/0.05% glutaraldehyde in phosphate buffered saline solution (PBS: 10 mmol/L KH2PO4, 37 mmol/L Na2HPO4, 87 mmol/L NaCl, 53 mmol/L KCl, pH 7.4). Neurons were permeabilized using 0.1% Triton X-100, then endogenous peroxidase was blocked using 0.3% H2O2 in methanol. The 3′-hydroxy ends of cut DNA were labeled with biotinylated dUTP (Promega, Madison, WI, U.S.A.) using the enzyme terminal deoxytransferase (Promega). Incorporation of the label was detected using streptavidin-peroxidase (Sigma, St. Louis, MO, U.S.A.) and visualized with 3,3′-diaminobenzidine (Vector Laboratories, Burlingame, CA, U.S.A.).
Reversible assessment of phosphatidylserine residues externalization
Externalization of membrane PS residues was determined by annexin V labeling as described in previous studies (Vincent and Maiese, 1999a; Maiese and Vincent, 2000). The growth medium was removed from culture plates, annexin V conjugate was applied in a final concentration of 3 μg/mL and incubated at 37°C in a humidified atmosphere in the dark for 10 minutes. Plates were then rinsed twice using fresh binding buffer. Neurons were examined using Leitz DMIRB microscope (Leica, McHenry, IL, U.S.A.) and Oncor Image 2.0 imaging software (Oncor, Gaithersburg, MD, U.S.A.). Images were acquired using a cooled charge-coupled device with transmitted light and fluorescent single excitation light at 490 nm and detected emission at 585 nm. After examination, the annexin V label was detached by washing three times in dissociation buffer (10 mmol/L HEPES, pH 7.5, 150 mmol/L NaCl, 5 mmol/L KCl, 2.8 mmol/L MgCl2), which differed from binding buffer in that the calcium was replaced with magnesium. Plates could then be reexamined to confirm that the annexin V was completely removed and returned to the incubator for a further specified period. Plates were restained using the same method. By drawing a grid on the bottom of the culture dish, the same fields of neurons could be relocated for sequential imaging. The proportion of stained neurons in each field was counted with 20 to 100 neurons per field and 6 to 10 fields per plate giving an n = 1 determination.
Assessment of ICE and CPP32-like activity
At specific times after NO exposure, caspase 1 (ICE) and caspase 3 (CPP32) activity were determined as previously described (Maiese and Vincent, 1999). Neurons were harvested and resuspended in 100 μL lysis buffer (25 mmol/L HEPES, pH 7.5, 5 mmol/L MgCl2, 1 mmol/L EGTA, 0.5% Triton X-100, 10 μg/mL PMSF, 1 μg/mL aprotinin, 1 μg/mL leupeptin). The neuronal suspensions were homogenized using a sonic dismembrator (Model 550; Fisher Scientific, Pittsburgh, PA, U.S.A.) pulse mode (0.5 seconds on, 0.5 seconds off, for 30 seconds) in an eppendorf tube on ice. An aliquot of supernatant containing 30 μg protein was incubated with 250 μmol/L colorimetric substrates, Ac-YVAD-pNA for ICE or Ac-DEVD-pNA for CPP32 (Calbiochem, San Diego, CA, U.S.A.). The total reaction mixture volume was 100 μL in reaction buffer (50 mmol/L HEPES, pH 7.5, 10 mmol/L DTT, 1% sucrose, 0.1% CHAPS). Reaction mixtures were incubated at 37°C and the absorbance was measured at 405 nm for 1 hour. The mean rate of substrate cleavage in micromoles per minute (μmol/min·g) was calibrated with standard p-nitroaniline solutions.
Western blot analysis for PARP cleavage
Cells were washed once with PBS and harvested from the Petri dishes with gentle scraping using ice-cold 1.4% EDTA/PBS. Neurons were resuspended in PBS containing protease inhibitors (10 μg/mL PMSF, 1 μg/mL aprotinin, 1 μg/mL leupeptin) and lysed by sonication on ice for 30 seconds. After protein determination, equal protein concentration of cell extracts were added to a 2× sample buffer (62.5 mmol/L Tris-HCl, pH 6.8, 6 mol/L urea, 10% glycerol, 2% SDS, 0.00125% bromophenol blue, 5% β-mercaptoethanol) and incubated at 65°C for 15 minutes. Each sample (50 μg/lane) was then subjected to 7.5% SDS-polyacrylamide gel electrophoresis, followed by a transfer onto a nitrocellulose membrane with plate electrodes (BioRad, Hercules, CA, U.S.A.) at high-field position in a buffer containing 25 mmol/L Tris, 192 mmol/L glycine, and 20% methanol for 2.5 hours at 4°C at 400 mA. Membranes were blocked overnight at 4°C in 5% nonfat dry milk in Tris-buffered saline (50 mmol/L Tris-HCl pH 7.4, 150 mmol/L NaCl) with 0.1% Tween 20 (TBST) and incubated with rabbit anti-PARP polyclonal antibody (1:500) for 2.5 hours at room temperature. Blots were washed with TBST, incubated for 2 hours with goat anti-rabbit IgG (Pierce, Rockford, IL, U.S.A.) coupled to horseradish peroxidase, and then washed again with TBST. The antibody-reactive bands were revealed by enhanced chemiluminescence (Pierce) using a phosphorimager with chemiluminescence capability (BioRad GS-525).
Measurement of intracellular pH
Similar to the authors' previous work (Vincent et al., 1999b), intracellular pH (pHi) was measured using the fluorescent probe BCECF (2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxy-fluorescein) (Molecular Probe, Eugene, OR, U.S.A.). The acetoxymethyl derivative of BCECF was dissolved in anhydrous dimethylsulfoxide to 10 mmol/L. Directly before use, BCECF was diluted in a serum-free medium to 1.25 μmol/L. A volume of 750 μL of the BCECF solution was added to dishes and incubated at 37°C for 8 minutes. The plates were rinsed three times, and the medium was replaced with 1 mL HEPES-buffered balanced saline solution (HBBSS: 118 mmol/L NaCl, 3 mmol/L KCl, 1 mmol/L MgCl2, 1.5 mmol/L CaCl2, 10 mmol/L glucose, 25 mmol/L Hepes, pH 7.2). Using a Leitz DMIRB microscope (Leica, McHenry, IL, U.S.A.) and Oncor Image 2.0 calcium ratio imaging software (Oncor), the ratio of fluorescence at 490 nm over 440 nm was determined with a fixed emission wavelength of 535 nm. This provided a linear response with increasing pH over the range of 6.5 to 7.5. To produce a standard curve, neurons were loaded with BCECF then imaged in HBBSS with the pHi adjusted to a range of values also containing 125 mmol/L KCl and the proton ionophore nigericin (25 μmol/L; Sigma). The pHi was allowed to equilibrate with the extracellular medium for 10 minutes before imaging. Values for pHi were calculated from the equation of the linear portion of the graph of fluorescence ratio versus pHi. To transiently modulate pHi of culture neurons, ammonium chloride (15 mmol/L) in growth medium was applied to cultures, the medium was then replaced with fresh growth medium after 5 minutes. This procedure has been shown to cause temporary intracellular acidification without altering the extracellular pH (Thomas, 1984).
Statistical analysis
For each experiment involving assessment of neuronal survival, neuronal PCD and caspase activity differences between groups were statistically analyzed by means of analysis of variance and the Student's paired t-test.
RESULTS
Nicotinamide alters neuronal survival at elevated concentrations
As compared with neuronal survival in untreated control cultures (86 ± 4%, mean ± SD), no significant toxicity was present in the cultures containing nicotinamide in the concentrations of 1 mmol/L to 25 mmol/L over a 24-hour period (Fig. 1). Concentrations of nicotinamide greater than 50 mmol/L were toxic and yielded a significant decrease in neuronal survival.
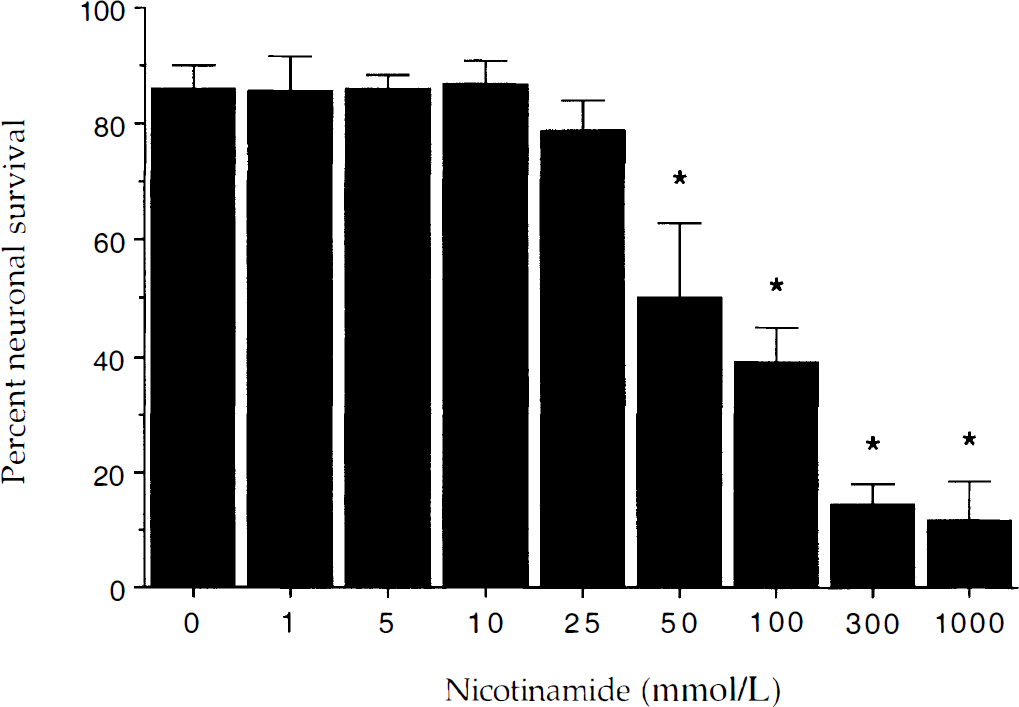
Nicotinamide alters neuronal survival at elevated concentrations. To examine the effect of nicotinamide alone on neuronal viability, increasing concentrations of nicotinamide (1 to 1000 mmol/L) were applied to neuronal cultures for a 24-hour period. Neuronal survival in the presence of nicotinamide at the concentrations of 1 mmol/L, 5 mmol/L, 10 mmol/L, and 25 mmol/L was not significantly different from the cultures in the absence of nicotinamide. Neuronal toxicity of nicotinamide was seen in the cultures with high concentration of nicotinamide (50 mmol/L, 100 mmol/L, 300 mmol/L, and 1000 mmol/L; * P < 0.01, analysis of variance). Neuronal survival was based on the percentage of the total number of neurons (viable plus nonviable) and determined by using a 0.4% trypan blue dye exclusion assay. Mean survival was determined by counting eight randomly selected nonoverlapping fields containing 10 to 20 neurons. Each culture dish represents an n = 1 determination with each experiment replicated independently 4 to 6 times using different cultures. Data for each concentration of nicotinamide represent the mean and SD.
Nicotinamide increases neuronal survival during NO toxicity
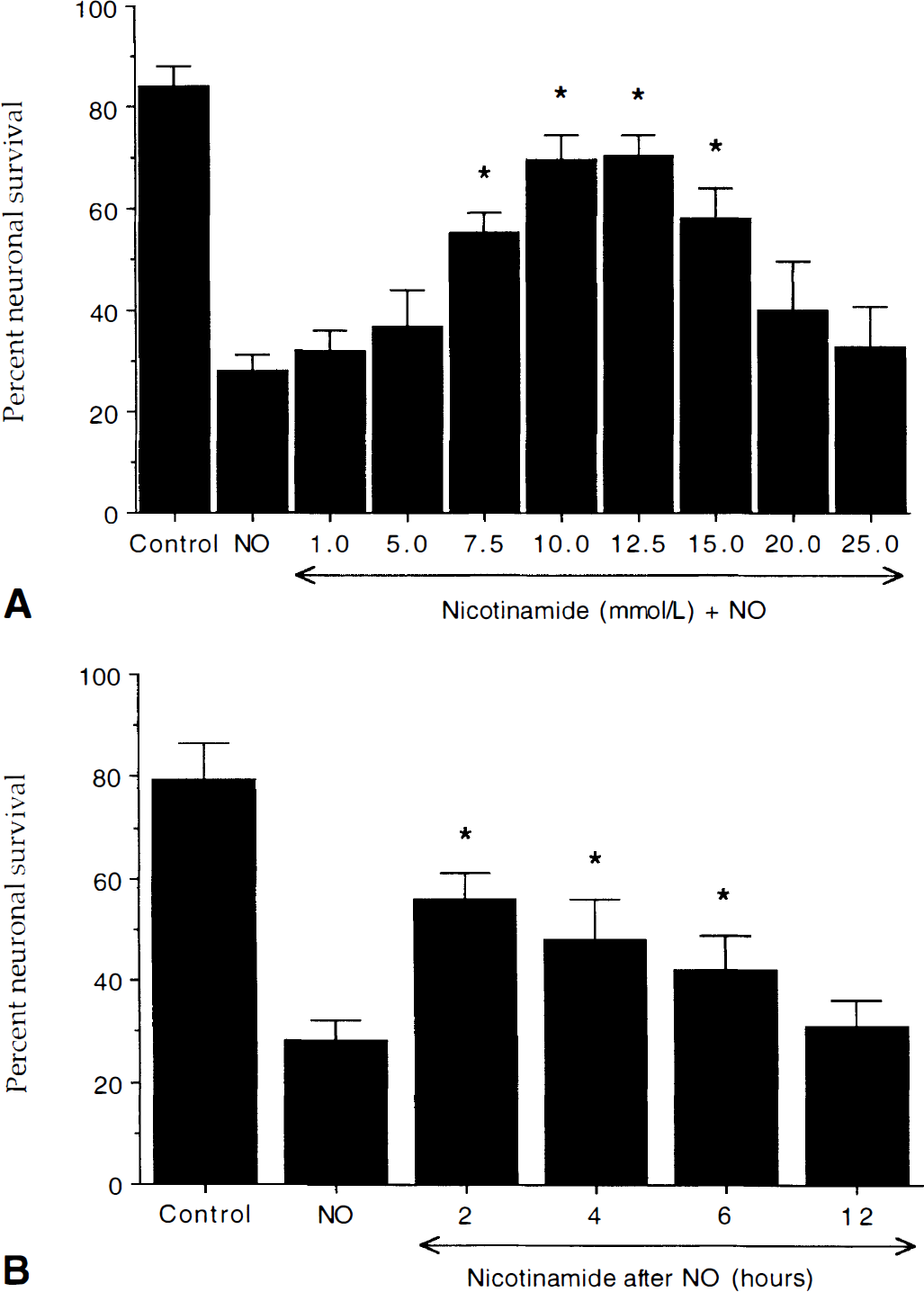
Nicotinamide increases neuronal survival during nitric oxide (NO) exposure in concentration-specific manner.
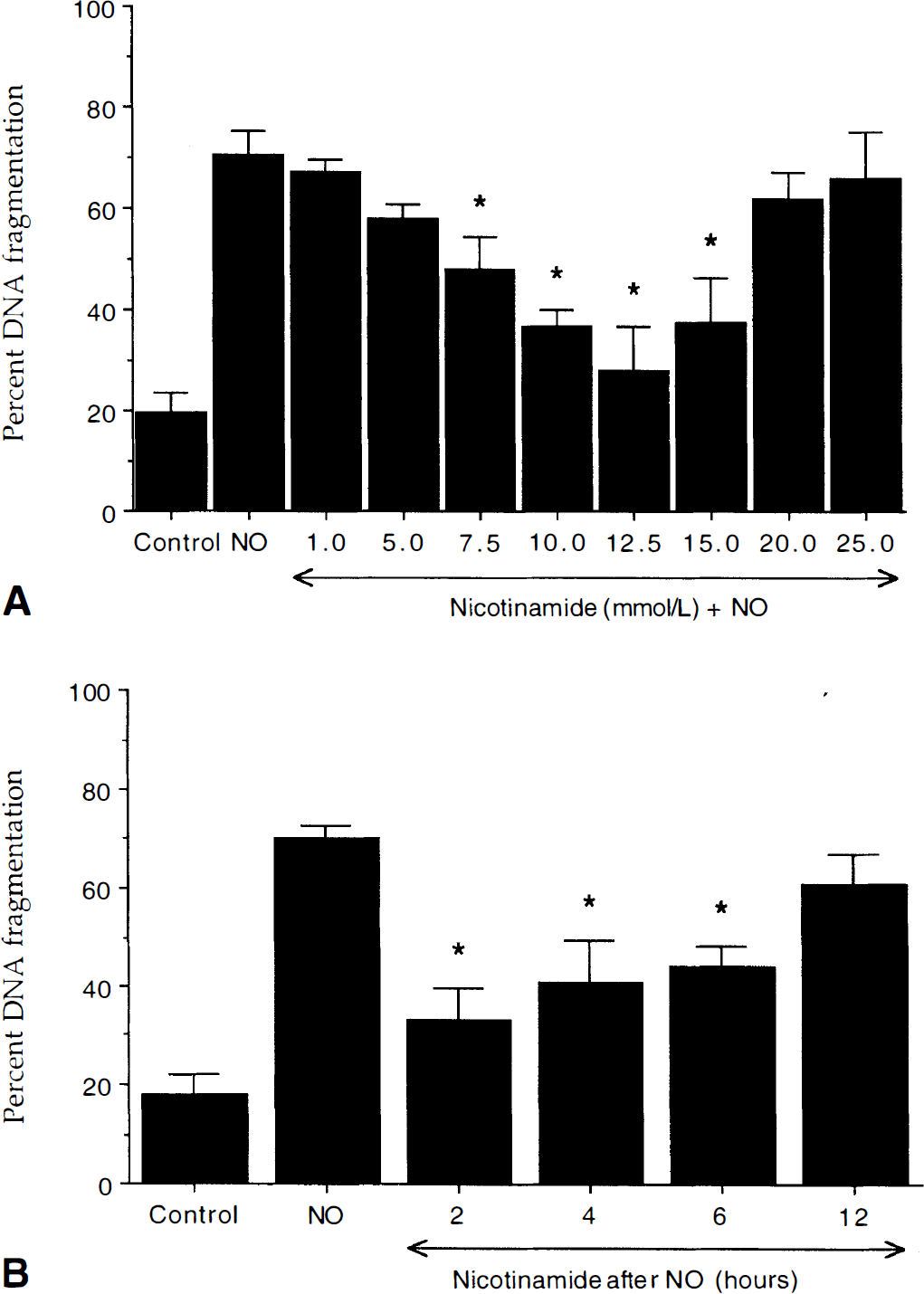
Nicotinamide prevents neuronal DNA fragmentation during nitric oxide exposure.
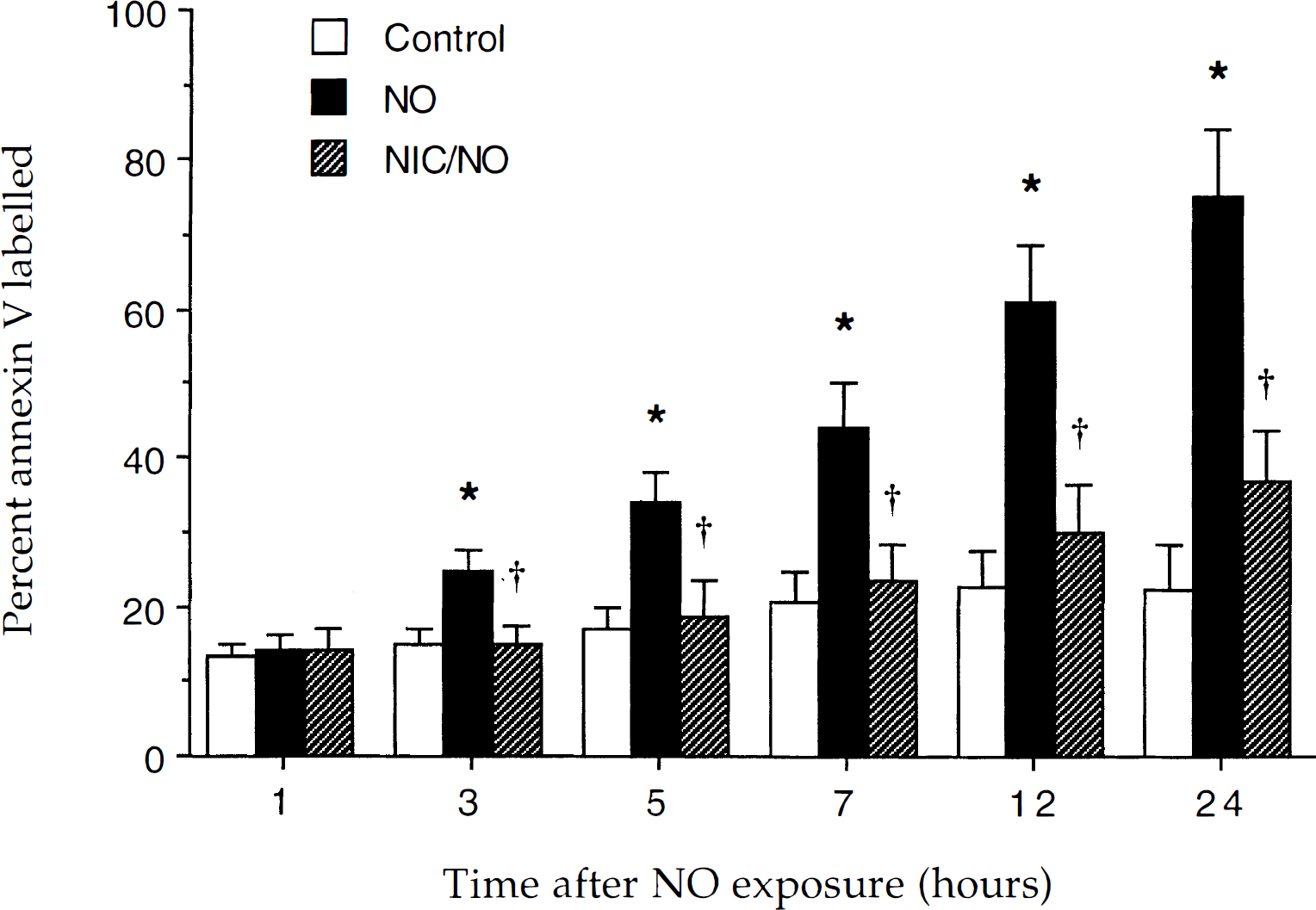
Nicotinamide (NIC) prevents the externalization of membrane phosphatidylserine residue induced by nitric oxide (NO) exposure. The percentage of neurons labeled with annexin V was determined at each indicated time point after exposure to NO (SIN-1 or NOC-9, 300 μmol/L). To simplify the figure, the results for the two NO donors were combined. Over a 24-hour period after NO exposure, annexin V staining progressively increased to approximately 76% (* P < 0.01, analysis of variance). Pretreatment with 12.5 mmol/L nicotinamide 1 hour before NO exposure significantly prevented the progression of annexin V positive neurons during NO exposure (†P < 0.05, analysis of variance). Data represent the mean and SD from eight to nine individual experimental cultures. In each experiment, the percentage of labeled neurons was counted in 3 to 7 discrete fields with 5 to 25 neurons in each field.
In posttreatment paradigms, the authors further assessed the ability of nicotinamide to prevent membrane PS exposure after NO exposure. A representative sequence of transmitted (T) and fluorescent (F) light images of the same microscope field for a neuron is illustrated in Fig. 5. In this treatment paradigm, nicotinamide (12.5 mmol/L) was applied 6 hours after exposure to NOC-9 (300 μmol/L). A neuron is labeled for surface PS at 3 hours after NO exposure as indicated by a white arrow. After application of nicotinamide at the 6-hour period, the neuron rapidly reversed the externalization of membrane PS residues by the 7-hour period. This neuron that reversed PS externalization remained negative for surface PS over a 24-hour period.
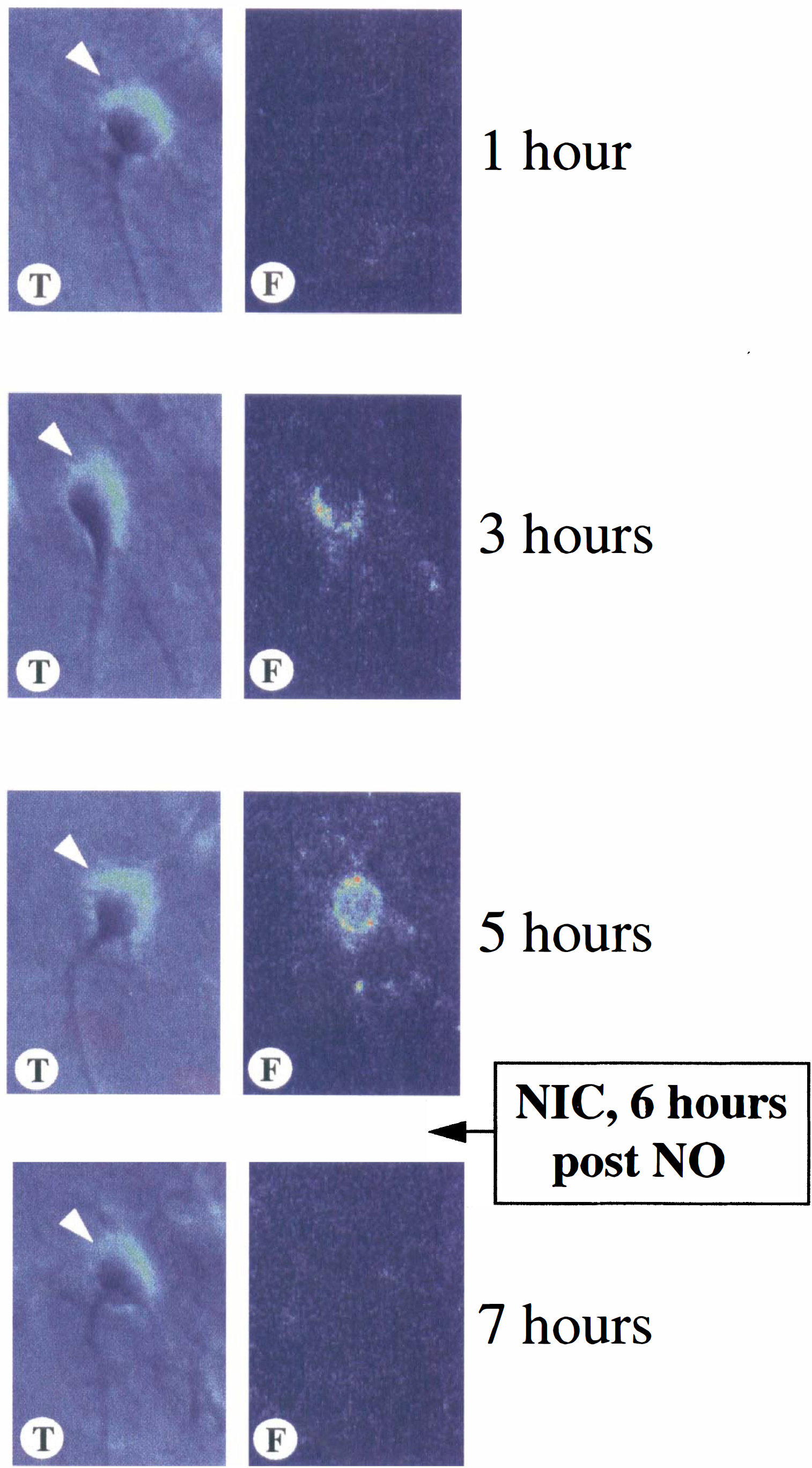
Nicotinamide rapidly reverses nitric oxide- (NO) induced phosphatidylserine externalization. Neurons were repeatedly labeled with annexin V phycoerythrin at 1, 3, 5, and 7 hours after exposure to NO. The NO donors SIN-1 (300 μmol/L) and NOC-9 (300 μmol/L) were used. NOC-9 was the NO donor in the representative figure. At each time-point indicated, the same field of neurons was imaged using transmitted light (T) microscopy. Corresponding fluorescent light (F) images were obtained using 490 nm excitation and 585 nm emission wavelengths to locate the annexin V-phycoerythrin label. The annexin V label was completely removed in calcium-free buffer after each time-point. Nicotinamide (NIC) (12.5 mmol/L) was applied one time at the specific period indicated after NO exposure. A 6-hour post-NO treatment is illustrated. One neuron is illustrated and labeled for PS externalization at the 3-hour period as indicated by the white arrow. At the 7-hour period, 1 hour after NIC application, membrane PS exposure is reversed in the neuron. This neuron that reversed PS externalization remained negative for surface PS over a 24-hour period.
Quantitation of surface PS after posttreatment regiments was performed in the same manner as the pretreatment experiments above. As shown in Fig. 6A, administration of NO alone resulted in the exposure of membrane PS residues in approximately 76% of the neuronal population over a 24-hour period and this increase was prevented by nicotinamide in each posttreatment group. This attenuation of membrane PS exposure by nicotinamide occurred within 1 hour of application of nicotinamide (Fig. 6B to Fig 6D). In addition, these posttreatment paradigms were able to reverse some of the initial expression of membrane PS exposure after NO administration.
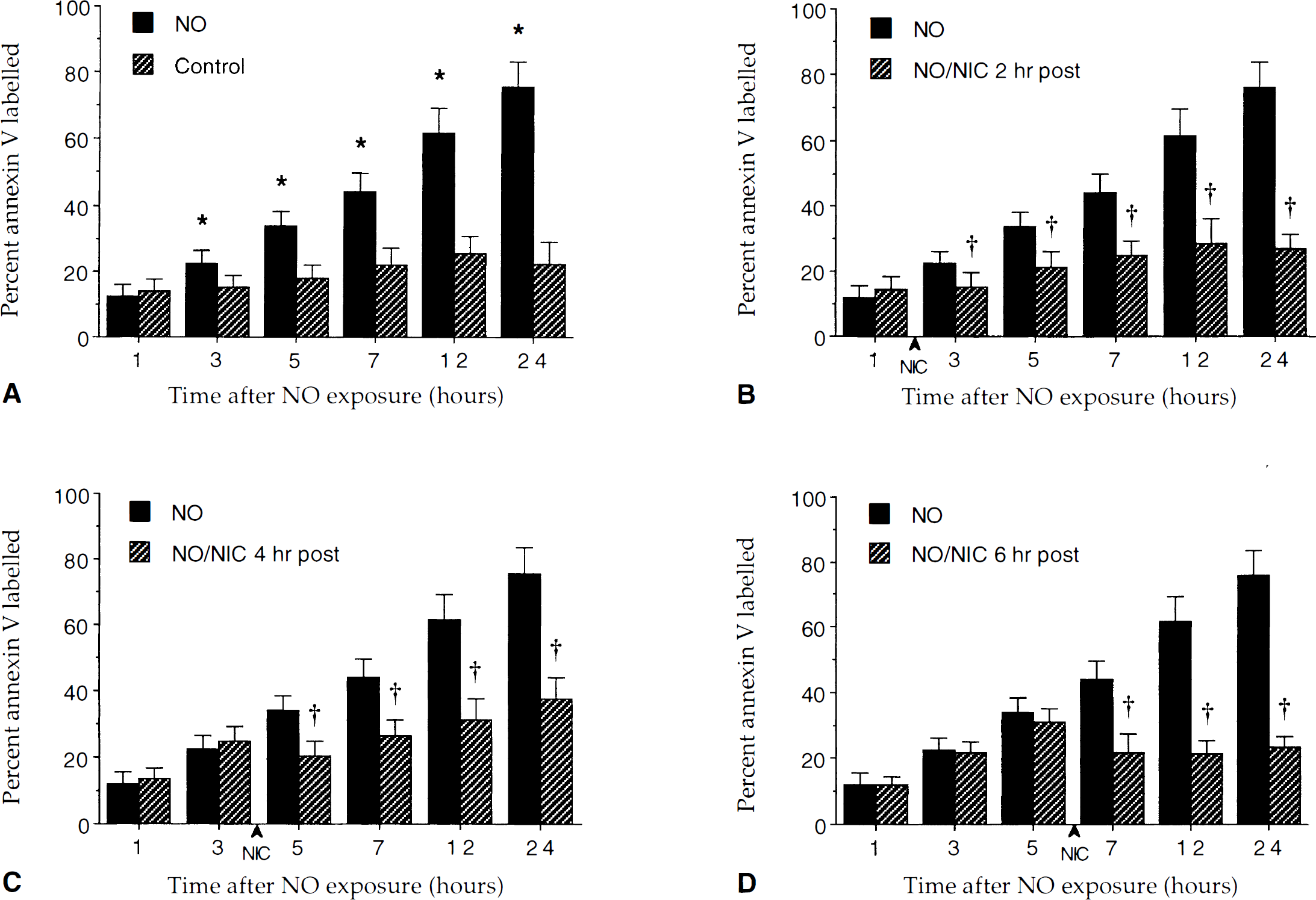
Posttreatment with nicotinamide (NIC) prevents the increase in the externalization of membrane phosphatidylserine residues induced by NO exposure. The percentage of neurons labeled with annexin V was determined at each indicated time point after exposure to NO (SIN-1 or NOC-9, 300 μmol/L). In contrast to untreated control cultures, NO exposure induced a progressive increase in the labeling of membrane PS residues over a 24-hour period (
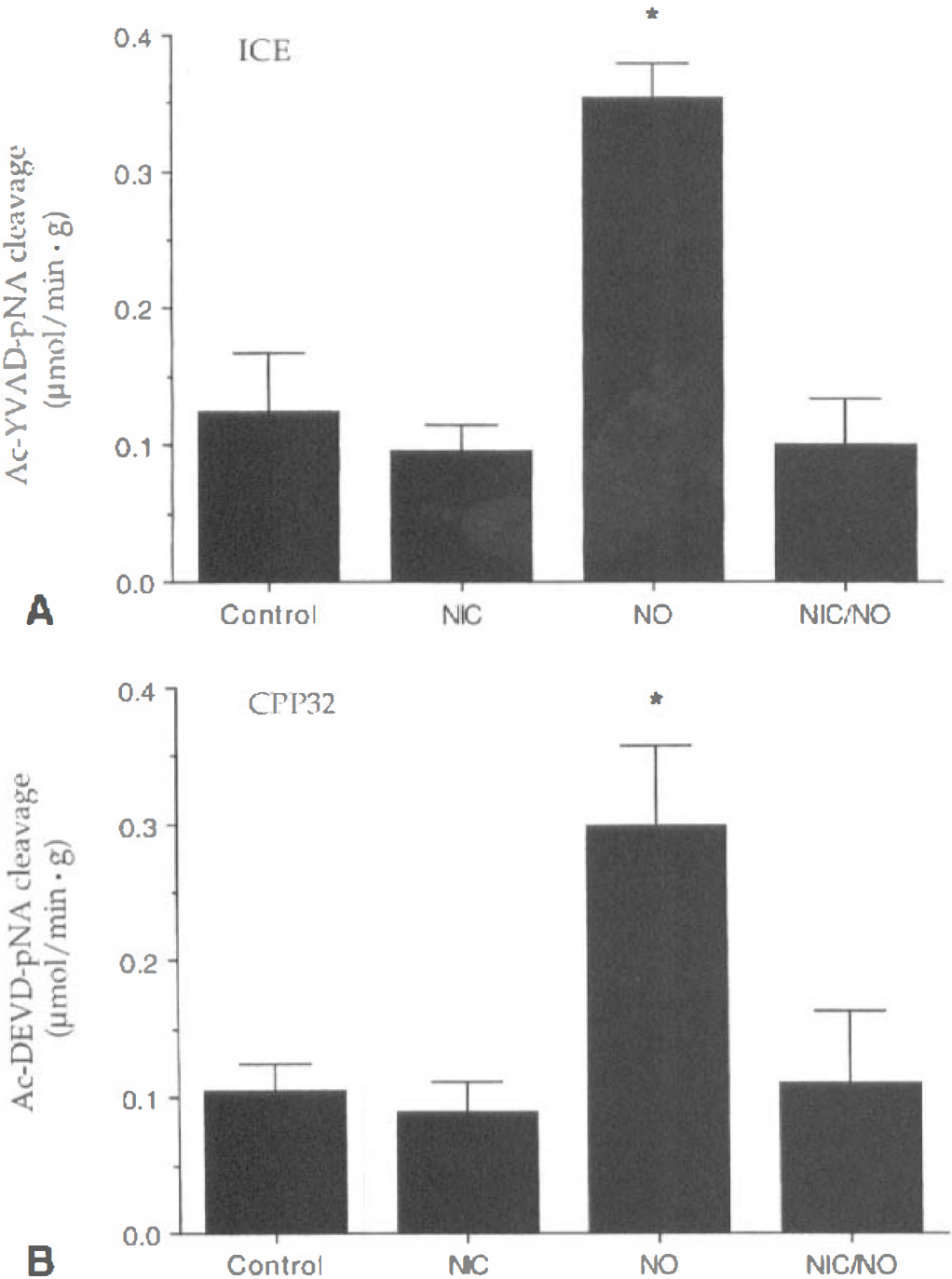
Nicotinamide (NIC) prevents the induction of ICE and CPP32 activities after NO exposure. ICE
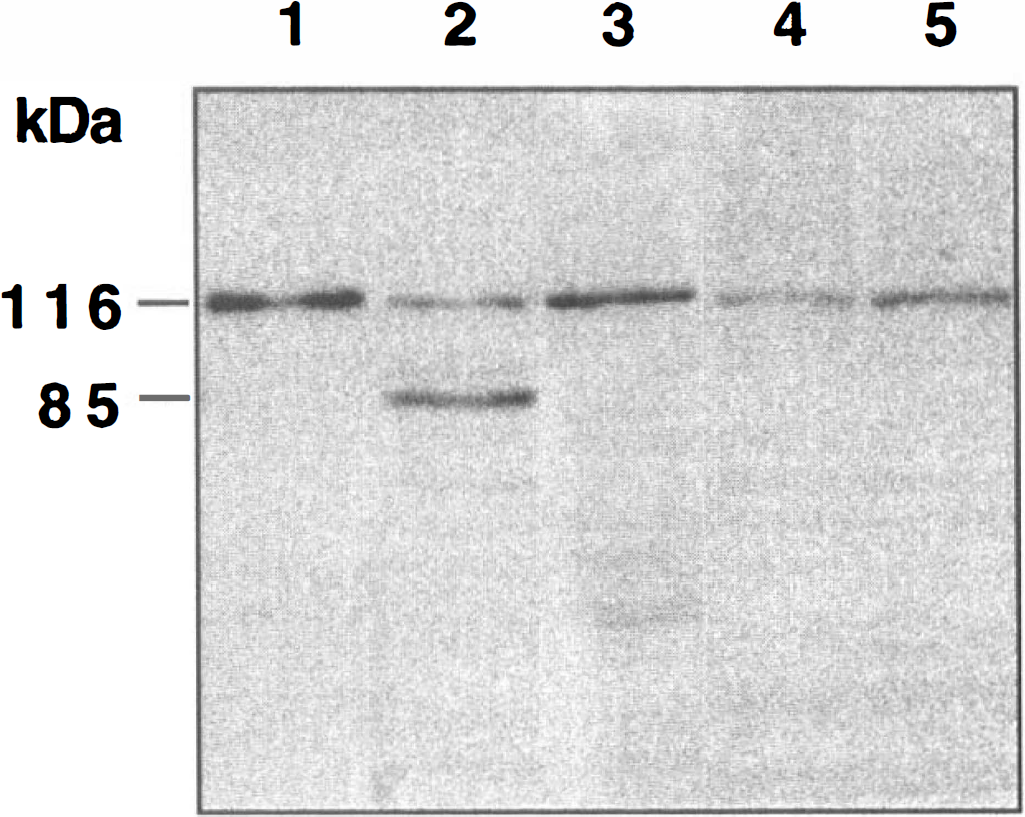
Nicotinamide inhibits PARP proteolysis during NO toxicity. A representative Western blot is illustrated. Total protein extracts were prepared from either untreated control cultures or from extracts prepared 12 hours after a 5-minute NO exposure with or without pretreatment of nicotinamide (12.5 mmol/L, 1 hour before NO exposure). The NO donors used were SIN-1 (300 μmol/L) or NOC-9 (300 μmol/L). To simplify the figure, the results for the two NO donors were combined. For a positive control, lanes 1 and 2 used 20 μL of protein extract from ∼75,000 human HL60 leukemia cells uninduced (lane 1) or induced (lane 2) to undergo apoptosis by the agent etoposide. In lanes 3, 4, and 5, equal amounts of neuronal protein extracts (25 μg/lane) were separated by 7.5% SDS-PAGE and were then immunoblotted with polyclonal anti-PARP antibody. Detection was by enhanced chemiluminescence. In lanes 1 and 2, extracts of the human HL60 cells were analyzed to compare migration of PARP and its 85 kDa proteolytic fragment. Lane 3 contained an extract from untreated rat hippocampal cultures. In lane 4, a decrease in the amount of 116 kDa PARP occurred within 12 hours after NO exposure. In lane 5, pretreatment with nicotinamide (12.5 mmol/L) 1 hour before NO exposure inhibited PARP cleavage during NO exposure. Molecular weight markers are indicated on the left of the Western blot.
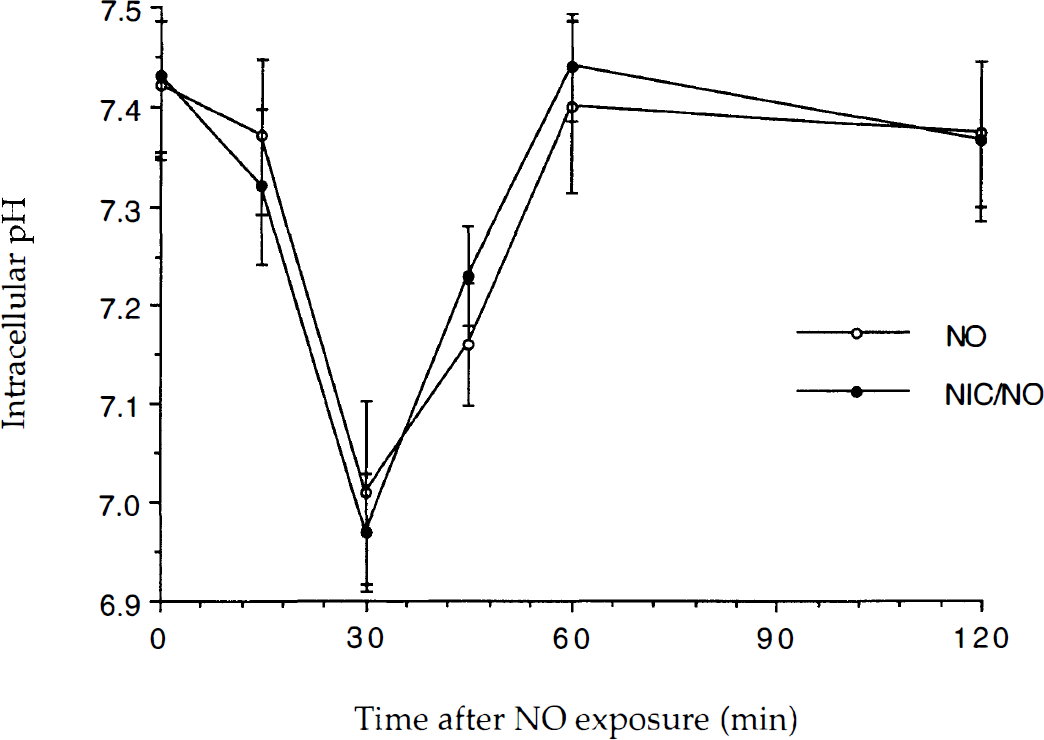
Neuroprotection by nicotinamide does not involve pHi modulation. To examine whether nicotinamide protects neurons from nitric oxide (NO) toxicity through pHi modulation, the effect of nicotinamide (NIC) on pHi over time was determined using the fluorescent probe BCECF. The NO donors, SIN-1 or NOC-9 (300 μmol/L), were applied for a 5 minute period and pHi was determined at the times indicated. To simplify the figure, data for the two NO donors were combined. Experiments were performed on at least three separate occasions with different cultures. Figure illustrates cumulative data for ≥ 42 neurons at each time point. The pHi in untreated control cultures was 7.42 ± 0.07. No significant difference between the pHi generated by NO and NO with NIC administration was found.
To further investigate whether nicotinamide prevented acidification-induced neuronal injury, the authors used an experimental model for the induction of intracellular acidification that could replicate the changes in pHi produced during NO exposure (Thomas, 1984; Vincent et al., 1999b). As shown in Fig. 10, application of NH4+(−) alone significantly reduced neuronal survival over a 24-hour period. Administration of nicotinamide did not alter neuronal survival during intracellular acidification as compared with the untreated control group.
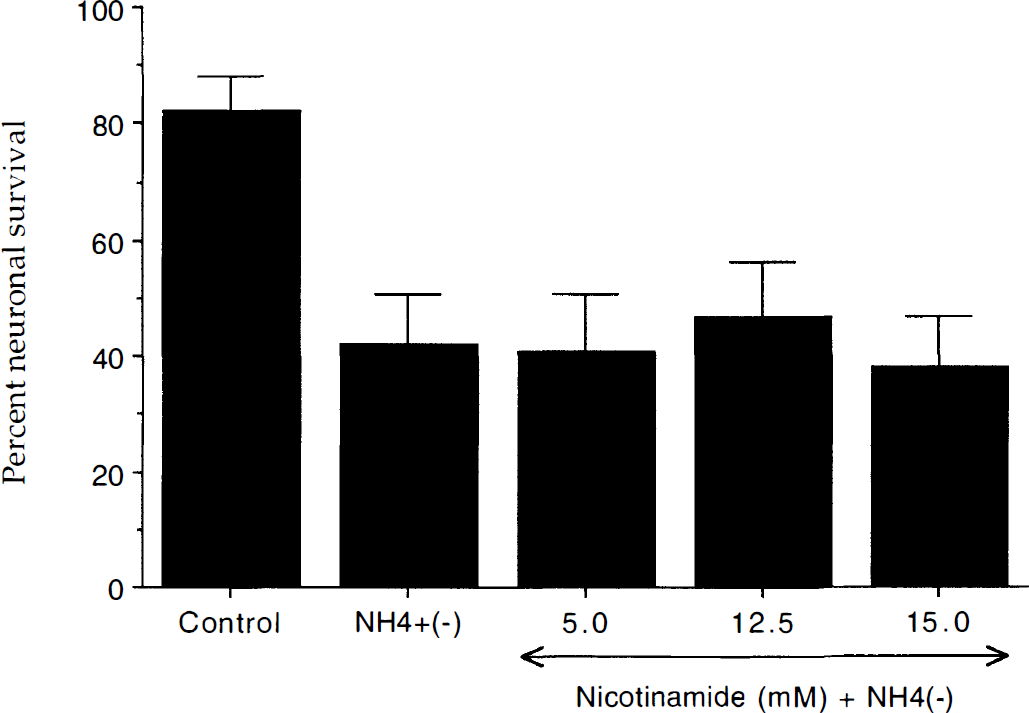
Nicotinamide (NIC) does not protect neurons from intracellular acidification. Intracellular acidification was performed by the addition of ammonium ions (NH4+ (+)) and the subsequent removal of the ammonium ions (NH4+ (−)) for transient acid-loading. Neuronal survival was quantitated at 24 hours after the addition of ammonium ions using a trypan blue dye exclusion assay. In the absence of ammonium ions untreated control neuronal survival was 82 ± 6%. Intracellular acidification decreased neuronal survival from approximately 82% to 42% over a 24-hour period. Pretreatment with nicotinamide (5 mmol/L, 12.5 mmol/L, and 15.0 mmol/L) 1 hour before the addition of ammonium ions did not increase neuron survival compared with NH4+ (−) only treated neurons. Mean proportion of stained neurons was calculated by counting 8 randomly selected nonoverlapping fields containing 10 to 20 neurons. Each culture dish represents an n = 1 determination with each experiment replicated independently 4 to 6 times using different cultures.
DISCUSSION
The cellular pathways that determine PCD are broad in nature but can be responsive to modulation. However, approaches to the prevention of neuronal injury first require an understanding of the mechanisms of PCD during neurodegeneration. In this respect, we have used nicotinamide as an investigative tool to examine the ability of this agent to protect or rescue neurons in culture from NO-induced PCD.
We demonstrate that nicotinamide can reduce NO-induced neuronal PCD and increase neuronal viability in a concentration-specific manner. Treatment with nicotinamide in a range of 7.5 to 15.0 mmol/L can significantly protect neurons from NO toxicity. This concentration range is similar to other injury paradigms of neuroprotection with nicotinamide (Wallis et al., 1996) and parallels the concentrations used in clinical studies that have demonstrated no detrimental effects on the cardiovascular system (Stratford et al., 1992). Concentrations of nicotinamide that were greater than 20 mmol/L were not protective and became toxic at 50 mmol/L when present during a 24-hour incubation period in neuronal cultures. Interestingly, the neuroprotective effects of nicotinamide in animal models of cerebral ischemia were also concentration-specific with toxicity illustrated at elevated concentrations. Only intraperitoneal injections of nicotinamide that reached 500 mg/kg, but not greater, were able to significantly reduce focal cerebral ischemia (Ayoub et al., 1999). Although unclear at this time, there may be several reasons for a limited concentration range of nicotinamide that offers protection from injury. For example, exposure to high concentrations of nicotinamide has been found to inhibit the function of rat beta-cells, decrease DNA content of adult rat islet cells, and induce cell death in fetal rat islet cells (Reddy et al., 1995). Nicotinamide also can induce the release of choline in hippocampal slices (Erb and Klein, 1998). Although an increased release of choline may sometimes be neuroprotective against ischemic injury (D'Orlando and Sandage, 1995), high concentrations of nicotinamide can trigger excessive release of choline that may precipitate neuronal injury (Koppen et al., 1993).
Programmed cell death involves at least two distinct pathways that may each mediate neuronal injury. Nuclear DNA fragmentation and loss of membrane asymmetry are separate processes that can lead to neuronal PCD (Vincent and Maiese, 1999a; Maiese and Vincent, 2000). Our studies with nicotinamide have indicated a separate biologic role for DNA degradation that is distinct from the inversion of membrane PS residues. Similar in its ability to prevent neuronal injury after NO exposure, our initial work with nicotinamide prevented the induction DNA degradation after NO exposure. Protection against DNA degradation was concentration dependent and most effective in the range of 7.5 mol/L to 15.0 mmol/L. Concentrations greater than 20.0 mmol/L were ineffective against NO-induced PCD.
The current work suggests that nicotinamide provides a unique protective ability that can maintain intact membrane asymmetry for extended periods after neuronal injury that is not seen with other neuroprotective regiments. Administration of nicotinamide during NO exposure prevented and maintained the induction of membrane PS inversion over a 24-hour period. The externalization of membrane PS residues can mediate cell injury independently from DNA fragmentation during PCD (Maiese and Vincent, 2000). It is conceivable that nicotinamide maintains membrane PS asymmetry through the modulation of random “flip-flop” membrane phospholipids (Bratton et al., 1997). Alternatively, nicotinamide may maintain cellular energy metabolism because exposure of membrane PS residues on the membrane surface is an active process facilitated by an ATP-dependent membrane translocase (Verhoven et al., 1999). With the prolonged maintenance of membrane PS asymmetry, nicotinamide also provides an enhanced level of neuroprotection. Membrane PS externalization is believed to function as a cellular “marker” or “tag” for neurons that are to be identified for subsequent removal (Verhoven et al., 1999; Maiese and Vincent, 2000). The removal of neurons that have been “tagged” with membrane PS exposure may ultimately be deleterious to an organism through the elimination of neurons that may otherwise be functional and irreplaceable. Prevention of membrane PS exposure by neuroprotective strategies, such as with nicotinamide, may prevent or lessen the severity of a neuronal injury.
The use of posttreatment strategies with nicotinamide illustrated that PCD appears to be reversible in nature rather than being a series of fixed, committed cellular pathways that result in neuronal injury. During the 2-, 4-, and 6-hour posttreatment regiments, nicotinamide was able to reverse an initial progression of membrane PS inversion and maintain the suppression of PS exposure over the next 24-hour period after NO exposure. These results suggest that PCD, at least along the pathway that involves membrane PS exposure, is dynamic and reversible in nature. In addition, similar to the pretreatment regiments, the posttreatment paradigms with nicotinamide not only reverse PS exposure, but also maintain membrane asymmetry at significantly lower levels than injured untreated neurons. These results suggest that the rapid reversal and restoration of membrane asymmetry during a critical period of repair by nicotinamide may impart an additional advantage for prolonged neuronal survival.
The ability of nicotinamide to prevent genomic DNA degradation and maintain membrane PS asymmetry may be closely linked to the modulation of cysteine protease activity. Nitric oxide is thought to be one of the signal transduction systems that can elicit cysteine protease activity and can directly stimulate caspase 1 and caspase 3-like activities (Brune et al., 1999; Maiese and Vincent, 1999). Caspase 1 has been linked to the modulation of membrane PS residues through cytoskeletal proteins such as fodrin (Cryns et al., 1996). Caspase 3 can lead to the direct degradation of DNA through the enhancement of DNase activity (Enari et al., 1998). Application of nicotinamide directly prevented the activation of caspase 1 and caspase 3-like activities after NO exposure, suggesting that nicotinamide may maintain genomic DNA integrity and membrane PS asymmetry through the modulation of cysteine protease activity.
In addition to directly downregulating cysteine protease activity, nicotinamide also prevented the cleavage of the specific CPP32 substrate poly(ADP-ribose) polymerase (PARP) that is required for DNA-repair (Berger, 1985). Our work illustrates that nicotinamide prevented the degradation of PARP during NO-induced PCD. After NO exposure, nicotinamide maintained the intact form of 116kDa PARP. Without the application of nicotinamide during NO exposure, a decrease in the amount of 116 kDa PARP was evident. Proteolytic fragments of PARP, such as the 85 kDa form, were not evident suggesting subsequent PARP cleavage during NO administration similar to other ischemic injury paradigms (Taylor et al., 1997). In addition, because nicotinamide alone can lead to the ribosylation of PARP (Bredehorst et al., 1980), the absence of the 85 kDa form of PARP is not a result of possible ribosylation of epitope binding sites. It is conceivable that nicotinamide maintains DNA integrity through at least two possible pathways that involve PARP. First, nicotinamide may prevent PARP degradation and allow for DNA repair through the direct inhibition of CPP32 activity. Second, nicotinamide also may modulate the activity of PARP to prevent excessive energy depletion that may be detrimental. Excessive activation of PARP has been shown to deplete NAD and ATP and subsequently lead to cell death through energy depletion (Endres et al., 1997). Nicotinamide concentrations of at least 1 mmol/L have been shown to provide sufficient stores of NAD during PARP activation (Smets et al., 1990).
Although nicotinamide may offer protection against PCD through a series of cellular pathways, one attractive mechanism to enhance neuronal survival is through the regulation of intracellular acidification. Nitric oxide can lead to a rapid and robust intracellular acidification that directly enhances neuronal endonuclease activity and leads to subsequent PCD (Vincent and Maiese, 1999b; Vincent et al., 1999b). However, our work demonstrated that nicotinamide does not directly prevent the induction of intracellular acidification by NO and does not prevent neuronal injury during intracellular acidification paradigms. These results suggest that nicotinamide maintains genomic DNA integrity through mechanisms that are independent of intracellular pH.
In summary, we have used the agent nicotinamide to illustrate that neuronal PCD can be a reversible process during its initial stages and is composed of two independent components. The first component is the degradation of genomic DNA which may impact on immediate neuronal survival and is dependent upon cysteine protease activity. The second component in NO-induced PCD involves the loss of membrane asymmetry and the subsequent externalization of PS residues. The loss of membrane asymmetry may subsequently “tag” neurons for subsequent destruction and also may be linked to enhanced cysteine protease activity during NO exposure. Nicotinamide is a unique and attractive neuroprotectant in its ability to maintain prolonged genomic DNA integrity and membrane PS asymmetry during pretreatment and posttreatment regiments. The current work initiates the development of more efficacious therapeutic strategies against neurodegeneration that may not only reverse the course of PCD, but also prevent the “tagging” of neurons for subsequent destruction.