
Abstract
Involvement of adenosine and adenosine triphosphate-sensitive potassium (KATP) channels in the development of ischemic tolerance has been suggested in global ischemia, but has not been studied extensively in focal cerebral ischemia. This study evaluated modulating effects of adenosine A1 receptor antagonist DPCPX (8-cyclopentyl-1,3-dipropylxanthine) and mitochondrial KATP channel blocker 5HD (5-hydroxydecanoate) on the development of tolerance to focal cerebral ischemia in rats. Preconditioning with 30-minute middle cerebral artery occlusion (MCAO) reduced cortical and subcortical infarct volume following 120-minute MCAO (test ischemia) given 72 hours later. The neuroprotective effect of preconditioning was attenuated by 0.1 mg/kg DPCPX given before conditioning ischemia (30-minute MCAO), but no influence was provoked when it was administered before test ischemia. DPCPX had no effect on infarct volume after conditioning or test ischemia when given alone. The preconditioning-induced neuroprotection disappeared when 30 mg/kg 5HD was administered before test ischemia. These results suggest a possible involvement of adenosine A1 receptors during conditioning ischemia and of mitochondrial KATP channels during subsequent severe ischemia in the development of tolerance to focal cerebral ischemia.
Keywords
A brief period of cerebral ischemia may limit damage from a subsequent severe ischemic insult. This phenomenon is known as ischemic tolerance and has been observed in various animal models of forebrain ischemia (Heurteaux et al., 1995; Kitagawa et al., 1990) and focal cerebral ischemia (Barone et al., 1998; Chen et al., 1996; Nakamura et al., 2002; Puisieux et al., 2000; Shimizu et al., 2001). Although stress responses including enhanced expression of heat shock proteins can play an important role in ischemic tolerance in the brain (Chen et al., 1996; Puisieux et al., 2000), several other mechanisms seem to be involved in tolerance induction (Dirnagl et al., 2003; Kirino, 2002). Elucidation of the mechanisms of ischemic tolerance is in the area of interest because the tolerance has been experimentally induced by clinically approved drugs (Dirnagl et al., 2003). Previously, we have shown that an adenosine A1 receptor antagonist attenuates rapid tolerance to focal cerebral ischemia and that the tolerance may be mediated through A1 receptor activation (Nakamura et al., 2002). Involvement of adenosine or adenosine triphosphate-sensitive potassium (KATP) channels in the development of ischemic tolerance has been suggested in global ischemia models (Heurteaux et al., 1995; Hiraide et al., 2001; Kawahara et al., 1998), cross-tolerance models (Plamondon et al., 1999; Xu et al., 2002), and in vitro studies (Pérez-Pinzón et al., 1996; Pérez-Pinzón and Born, 1999; Reshef et al., 1998, 2000). In addition, neuroprotective effects of adenosine receptor agonists (Lange-Asschenfeldt et al., 2003; von Lubitz, 1999) and mitochondrial KATP channel openers have been reported (Kis et al., 2003; Liu et al., 2002; Nakagawa et al., 2002; Rajapakse et al., 2002; Shake et al., 2001; Shimizu et al., 2002). However, no study has investigated involvement of these receptors in the delayed tolerance in focal cerebral ischemia models.
A recent human study showed that patients with repeated attacks of cerebral ischemia had a better neurologic outcome than patients without a previous ischemic attack (Moncayo et al., 2000). Another human study revealed that adenosine plasma levels were markedly increased and that the adenosine surge lasts days after transient ischemic attack and weeks after stroke (Pasini et al., 2000). These studies suggest existence of ischemic tolerance in human brains and possible roles of adenosine in the endogenous neuroprotective mechanisms.
There are two distinct KATP channels: one in the cell membrane (surface KATP channel) and the other in the inner mitochondrial membrane (mitochondrial KATP channel). Ischemic tolerance has been intensively studied in the heart and accumulating evidence supports that mitochondrial KATP channels play a pivotal role in the myocardial protection (O'Rourke, 2000). In the brain, however, the role of mitochondrial KATP channels in ischemic tolerance remains to be elucidated, although inhibition of preconditioning-induced protection by a nonspecific antagonist acting on both surface and mitochondrial KATP channels has been reported in global ischemia (Heurteaux et al., 1995). Recently, a mitochondrial KATP channel proteins were partially purified from rat brain mitochondria and exhibited ligand-binding properties similar to those of heart mitochondrial KATP channels (Bajgar et al., 2001). The amount of mitochondrial KATP channels in the brain was reported to be much higher than in the heart (Bajgar et al., 2001), suggesting the possibility that mitochondrial KATP channels play an important role in neural function.
The purpose of the present study was to determine whether activation of adenosine A1 receptors and mitochondrial KATP channels is involved in delayed tolerance to focal cerebral ischemia. We used a highly selective adenosine A1 receptor antagonist, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) and a selective antagonist of mitochondrial KATP channels, 5-hydroxydecanoate (5HD), and examined the effect of these drugs on the neuroprotection induced by ischemic preconditioning.
MATERIALS AND METHODS
Animals
This study was reviewed and approved by the Committee of the Ethics on Animal Experiment and carried out under the control of the Guidelines for Animal Experiment in Yamaguchi University School of Medicine. Male Wistar rats aged 8 to 9 weeks were used. Animals were given free access to food and water, and housed on a 12/12-hour light/dark cycle at constant temperature (~25°C).
General preparation and middle cerebral artery occlusion
Rats were anesthetized with inhalation of 3% isoflurane and 66% nitrous oxide. The tail artery was cannulated to monitor blood pressure, arterial blood gases, blood glucose, and hematocrit. After endotracheal intubation, rats were mechanically ventilated with 1.5% isoflurane and 66% nitrous oxide. A temperature probe was placed in the rectum and the temperature was monitored (Mono-a-therm 6510; Tyco Healthcare, Tokyo, Japan). Another temperature probe was placed on the skull under the right temporalis muscle and the cranial temperature was servo-controlled between 36.8 and 37.2°C by heating lamp (IFR-1000; Unique Medical, Japan) during surgical preparation and cerebral ischemia. Heparin 50 IU was administered through the tail artery.
Preparation for right middle cerebral artery occlusion (MCAO) was performed as previously described (Belayev et al., 1996; Nakamura et al., 2002). The right common carotid artery was exposed through a midline neck incision and gently dissected free of surrounding nerves and fascia, from its bifurcation to the base of the skull. The occipital artery and superior thyroid artery branches of the external carotid artery were dissected and coagulated. The external carotid artery was dissected further distally and coagulated along with the terminal lingual and maxillary artery branches, which were then divided. The internal carotid artery was isolated and carefully separated from the adjacent vagus nerve, and the pterygopalatine artery was ligated close to its origin. A 5–0 silk suture was tied loosely around the mobilized external carotid artery stump. Every surgical wound was infiltrated with 0.25% bupivacaine.
After 30-minute stabilization period with 1% (end-tidal) isoflurane and 66% nitrous oxide, a 3.5-cm length of 5–0 monofilament polypropylene suture (Prolene®) coated with silicone was inserted through the proximal external carotid artery stump into the internal carotid artery and advanced to occlude the origin of middle cerebral artery (MCA) 21 to 23 mm from the bifurcation of common carotid artery until a slight resistance was felt. The silk suture around the external carotid artery was tightened around the intraluminal monofilament suture to prevent bleeding.
Physiologic variables were measured 15 minutes before the start of ischemia.
Conditioning ischemia (30-minute MCAO)
Under anesthesia with isoflurane and nitrous oxide, 30-minute MCAO was induced. Thereafter, the intraluminal suture was carefully removed to avoid stripping silicone coating from the filament. The common carotid artery and internal carotid artery were inspected to ensure the return of good pulsation. The neck incision was closed, and the catheter in the tail artery and the thermistor placed on the skull were removed. The animals were then allowed to recover from anesthesia and were returned to a plastic box where the oxygen concentration was maintained at 85% to 90% with high flow of oxygen for 30 minutes.
Rats in the sham-operated groups underwent identical surgery but did not undergo conditioning ischemia.
Test ischemia (120-minute MCAO)
Three days after the sham operation or conditioning ischemia, rats were anesthetized with isoflurane and nitrous oxide and received recatheterization into the tail artery (proximal to the sites formerly catheterized) and mechanical ventilation. The rectal and cranial temperature probes were placed, the neck wound was reopened and the carotid arteries were exposed again. A new 5–0 silicon-coated monofilament suture was used to occlude the middle cerebral artery. After the intraluminal suture was placed, the neck incision was closed. The catheter in the tail artery was removed, the wound was closed, and rectal temperature probe was removed. The animals were then allowed to awaken from anesthesia and were moved to a plastic box at an environment with room air.
After 120 minutes of MCAO, rats were reanesthetized, the wound was reopened, and the intraluminal suture was removed. Good pulsation of the common and internal carotid arteries was ensured, the neck incision was closed, and the thermistor placed on the skull was removed. The animals were then allowed to recover from anesthesia.
Experimental groups
Experimental protocols are schematically presented in Fig. 1. The present study consisted of three experiments.
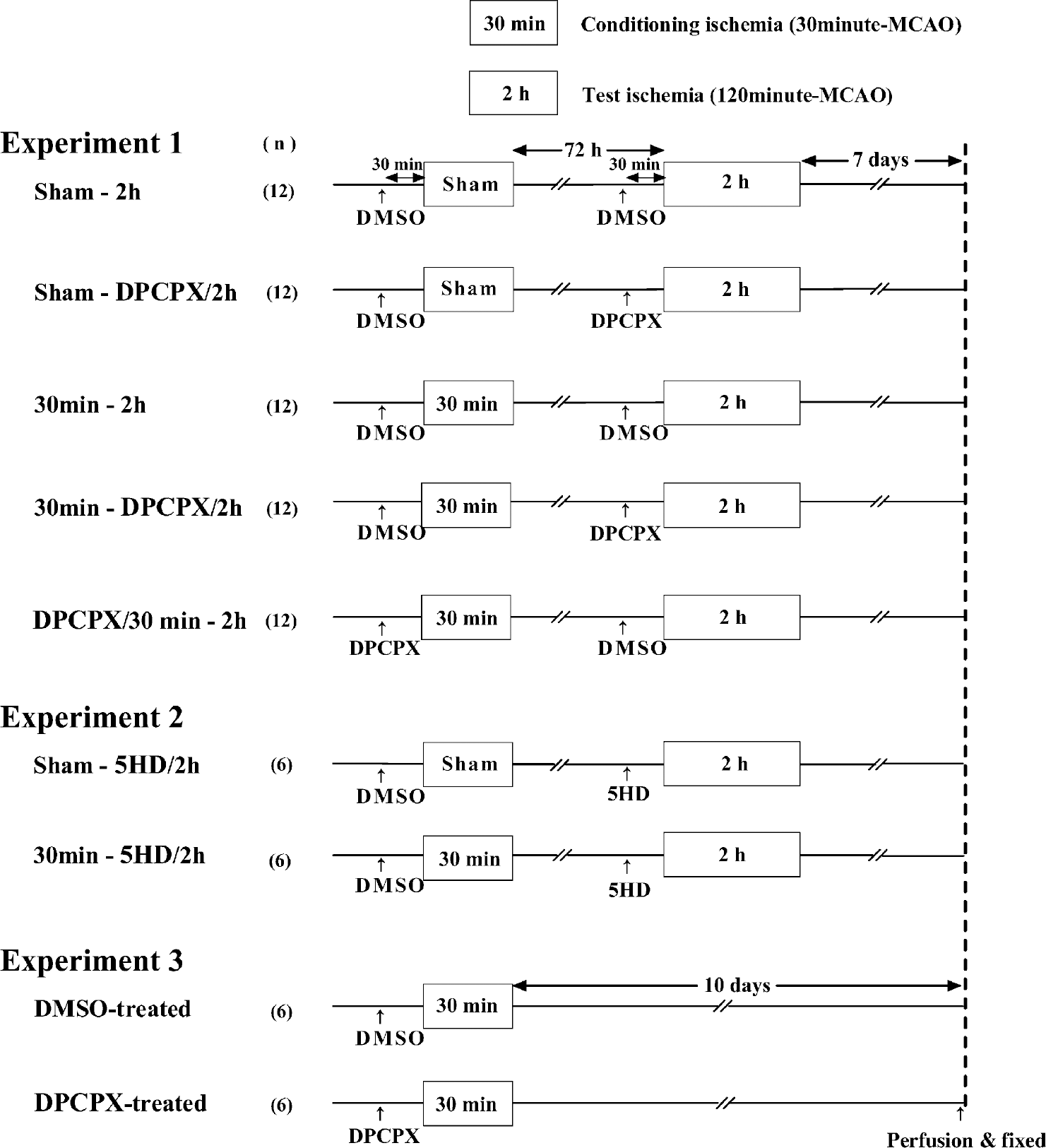
Experimental protocols. Experiment 1 was designed to determine whether activation of adenosine A1 receptors during conditioning ischemia (30-minute MCAO) and/or during test ischemia (120-minute MCAO) is required for preconditioning-induced neuroprotection to be developed. DPCPX (0.1 mg/kg; adenosine A1 receptor antagonist) or dimethyl sulfoxide (DMSO, vehicle control) was intraperitoneally administered 30 minutes before conditioning ischemia (or sham operation) or 30 minutes before test ischemia. Experiment 2 was designed to examine the role of mitochondrial KATP channels during test ischemia in development of tolerance. 5HD (30 mg/kg; mitochondrial KATP channel blocker) was intraperitoneally administered 30 minutes before test ischemia. The rats in experiments 1 and 2 were subjected to test ischemia 72 hours after conditioning ischemia or sham operation. Experiment 3 was designed to determine whether DPCPX influences the injury produced by conditioning ischemia. The rats were subjected to only 30-minute ischemia 30 minutes after intraperitoneal injection of DPCPX or DMSO. Rats were killed 7 days after test ischemia in experiments 1 and 2, or 10 days after 30-minute ischemia in experiment 3. Then, the brains were perfusion-fixed and stained with hematoxylin and eosin for determination of lesion volume (infarction and selective neuronal necrosis). MCAO, middle cerebral artery occlusion; DPCPX, 8-cyclopentyl-1,3-dipropylxanthine; 5HD, 5-hydroxydecanoate.
Experiment 1
Rats were randomly assigned to one of five groups (sham-2h, sham-DPCPX/2h, 30min-2h, 30min-DPCPX/2h, and DPCPX/30min-2h groups).
Thirty minutes before conditioning ischemia (30-minute MCAO), the DPCPX/30min-2h group received 0.1 mg/kg (0.5 mL/kg) intraperitoneal DPCPX dissolved in dimethyl sulfoxide (DMSO) and the other four groups received the same volume of DMSO. Three days later, the sham-DPCPX/2h and 30min-DPCPX/2h groups received 0.1 mg/kg DPCPX, and the other three groups received DMSO, 30 minutes before the start of test ischemia (120-minute MCAO).
The dose of DPCPX was chosen based on our previous study, in which we showed that the dose of DPCPX attenuated neuroprotective effects of rapid preconditioning (Nakamura et al., 2002).
Experiment 2
Rats were randomly assigned to the sham-5HD/2h or 30min-5HD/2h groups. Sham-5HD/2h and 30min-5HD/2h groups received sham operation and 30-minute ischemia, respectively. Three days later, both groups received 30 mg/kg 5HD 30 minutes before 120-minute MCAO. The dose of 5HD was selected based on two recent studies that showed abrogation of neuroprotective effects of pharmacologic preconditioning at the doses of 20 mg/kg and 40 mg/kg, respectively (Horiguchi et al., 2003; Liu et al., 2002).
In experiments 1 and 2, rats that died within 7 days after 120-minute MCAO were excluded from analysis and replaced in a blinded and randomized manner until each group in experiments 1 and 2 comprised 12 and 6 rats, respectively.
Experiment 3
Rats were randomly assigned to DMSO-treated or DPCPX-treated group (n = 6 in each group). In each group, rats received DMSO or 0.1 mg/kg DPCPX intraperitoneally, 30 minutes before the start of 30-minute MCAO.
Infarct volume measurement
Animals were anesthetized with isoflurane 7 days after 120-minute MCAO in experiments 1 and 2 and 10 days after 30-minute MCAO in experiment 3. The brains were perfusion-fixed with phosphate-buffered 4% paraformaldehyde following a flush of cold heparinized saline through a cannula inserted into the ascending aorta via the left ventricle. Brains were embedded in paraffin, coronally cut at 7 μm, and stained with hematoxylin and eosin. The areas of infarction and selective neuronal necrosis (SNN) were determined with the aid of a microscope and an image analyzer. The hemispheric infarcted area in each section was calculated by subtracting the noninfarcted area (including SNN) in the ipsilateral ischemic hemisphere from the contralateral nonischemic area. This technique minimizes the effect of edema on measurement of infarct size (Swanson et al., 1990). SNN areas with little, if any, swelling were directly measured. These measurements were taken from 25 standardized sections spaced 0.5 mm apart (4.2 to -7.8 mm from bregma), and the volume of infarction and SNN was calculated by summing the injured area of each section and multiplying by the interval (0.5 mm). For the measurements of SNN volume, only the area where the majority of neurons were affected was counted and occasional necrotic neurons were disregarded. An investigator blinded to the treatment groups determined the lesion volumes.
Statistical analysis
Comparison of the survival rates among groups was made with the χ2 test. Differences in the volume of infarction and SNN, and physiologic parameters were analyzed using analysis of variance. The PLSD method was used for post hoc testing. P < 0.05 was considered significant. All values are expressed as mean ± SD.
RESULTS
Survival rates and physiologic variables
In experiment 1, 11 rats died after neurologic deterioration within 7 days after 120-minute MCAO (2 in the sham-2h group, 5 in the sham-DPCPX/2h group, 1 in the 30min-2h group, and 3 in the DPCPX/30min-2h group). There were no significant differences in mortality rate among groups. In experiment 2, three rats in each group died within 7 days after 120-minute MCAO. All rats in experiment 3 survived for 10 days after 30-minute ischemia.
There were no significant differences in body weights at the start of experiments or on the day of brain-perfusion among groups in each experiment (291 ± 22 g and 220 ± 43 g in experiment 1, 284 ± 15 g and 196 ± 35 g in experiment 2, and 296 ± 17 g and 311 ± 22 g in experiment 3).
Physiologic variables in each experiment are presented in Table 1. There were no significant differences in any variables among groups in each experiment.
Physiologic variables
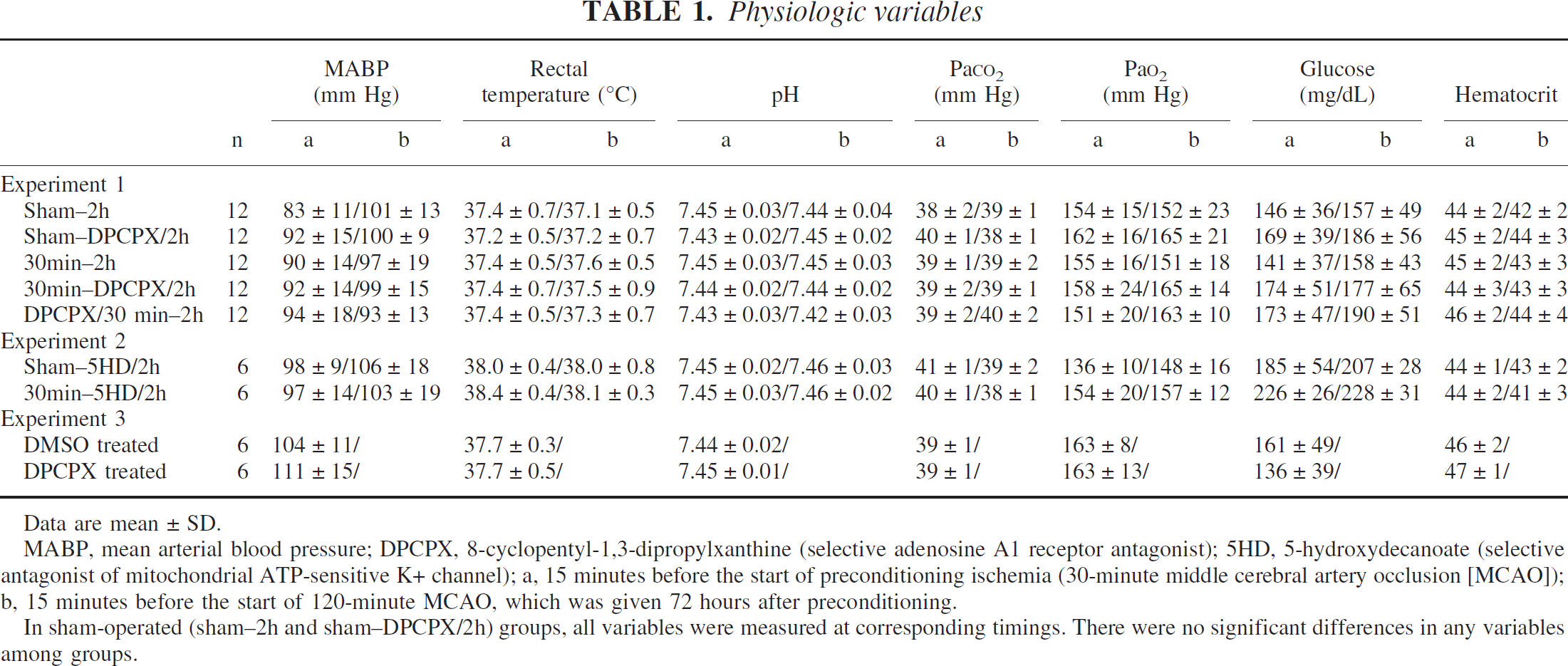
Data are mean ± SD.
MABP, mean arterial blood pressure; DPCPX, 8-cyclopentyl-1,3-dipropylxanthine (selective adenosine A1 receptor antagonist); 5HD, 5-hydroxydecanoate (selective antagonist of mitochondrial ATP-sensitive K+ channel); a, 15 minutes before the start of preconditioning ischemia (30-minute middle cerebral artery occlusion [MCAO]); b, 15 minutes before the start of 120-minute MCAO, which was given 72 hours after preconditioning.
In sham-operated (sham-2h and sham—DPCPX/2h) groups, all variables were measured at corresponding timings. There were no significant differences in any variables among groups.
Volume of infarction and selective neuronal necrosis
Figures 2 through 5 and Table 2 show the size of infarction and/or SNN in each experiment.
Volume of selective neuronal necrosis (mm3)
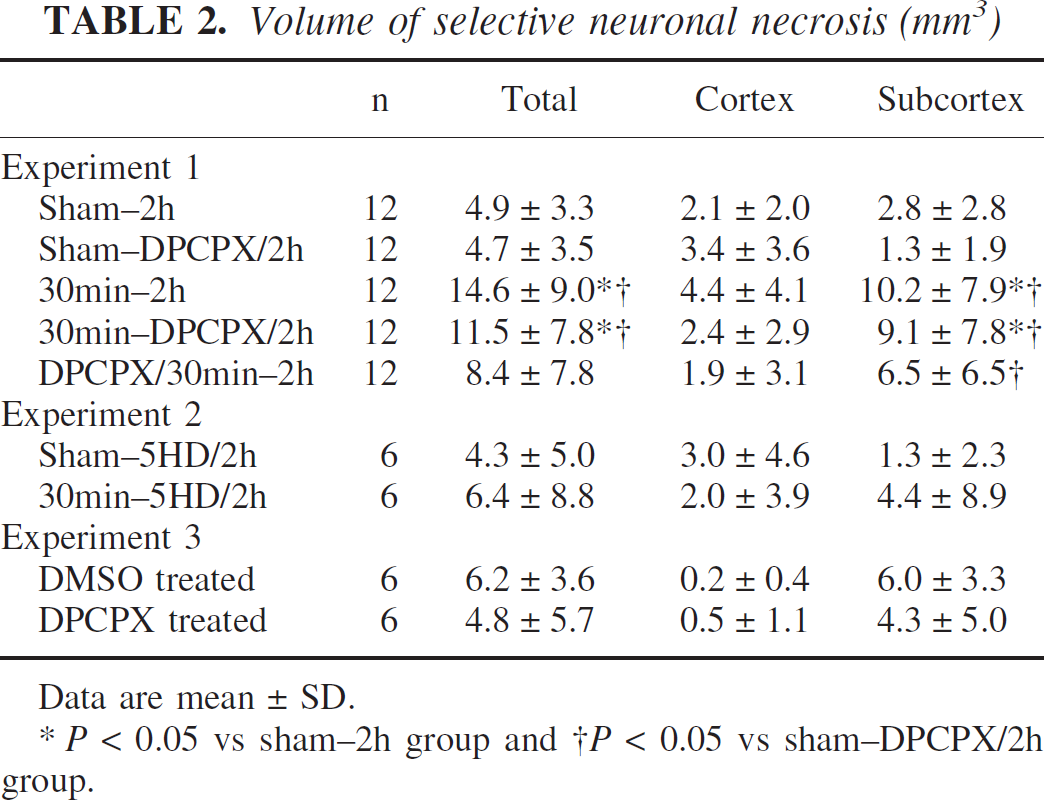
Data are mean ± SD.
* P < 0.05 vs sham-2h group and † P < 0.05 vs sham—DPCPX/2h group.
Experiment 1
Both cortical and subcortical infarct volume assessed 7 days after 120-minute MCAO was significantly smaller in rats preconditioned with 30-minute MCAO applied 72 hours before test ischemia, compared with that in the sham-operated group (Fig. 2). Significant reduction in infarcted areas induced by preconditioning was markedly seen in the forebrain cortex (Fig. 3).
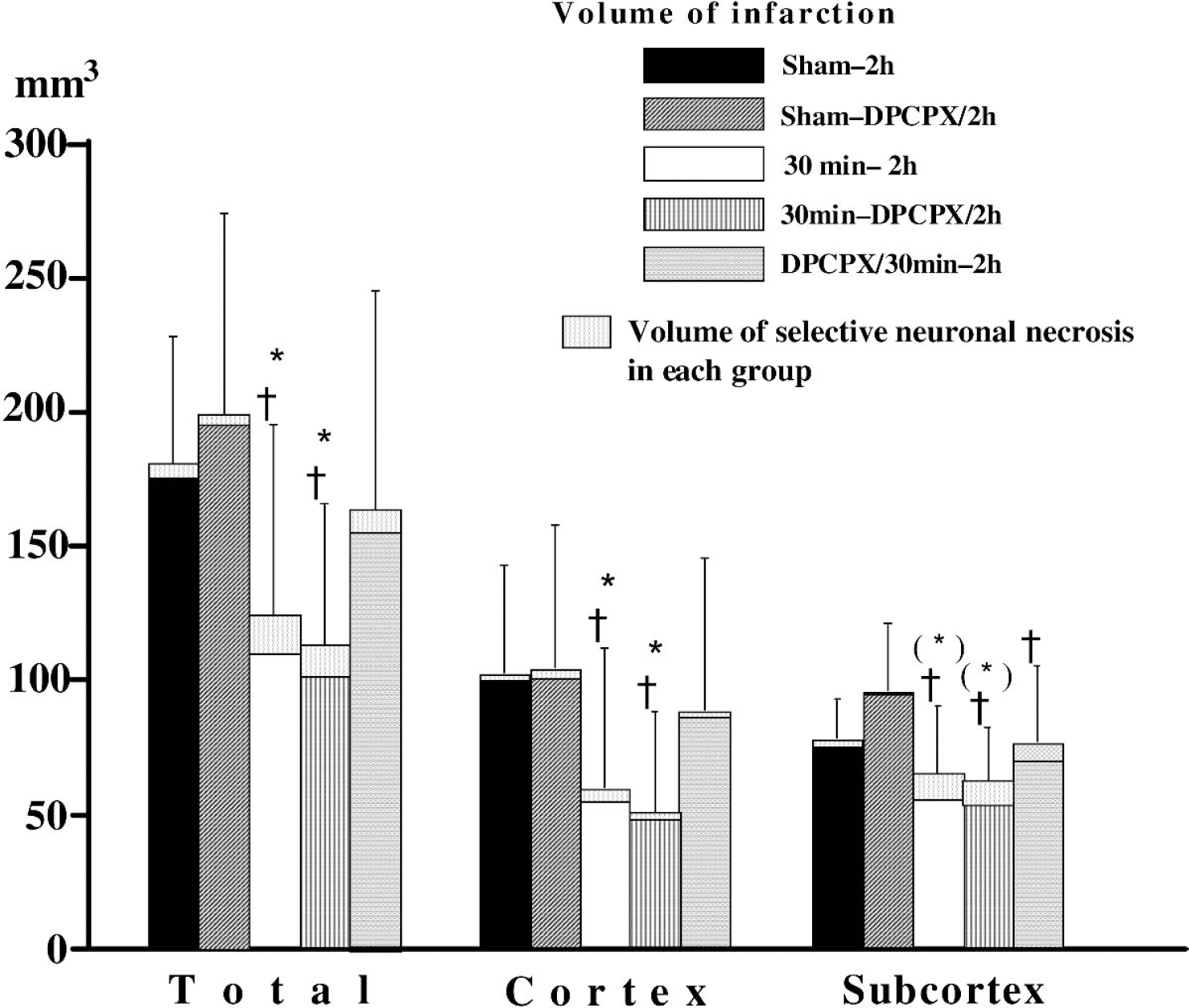
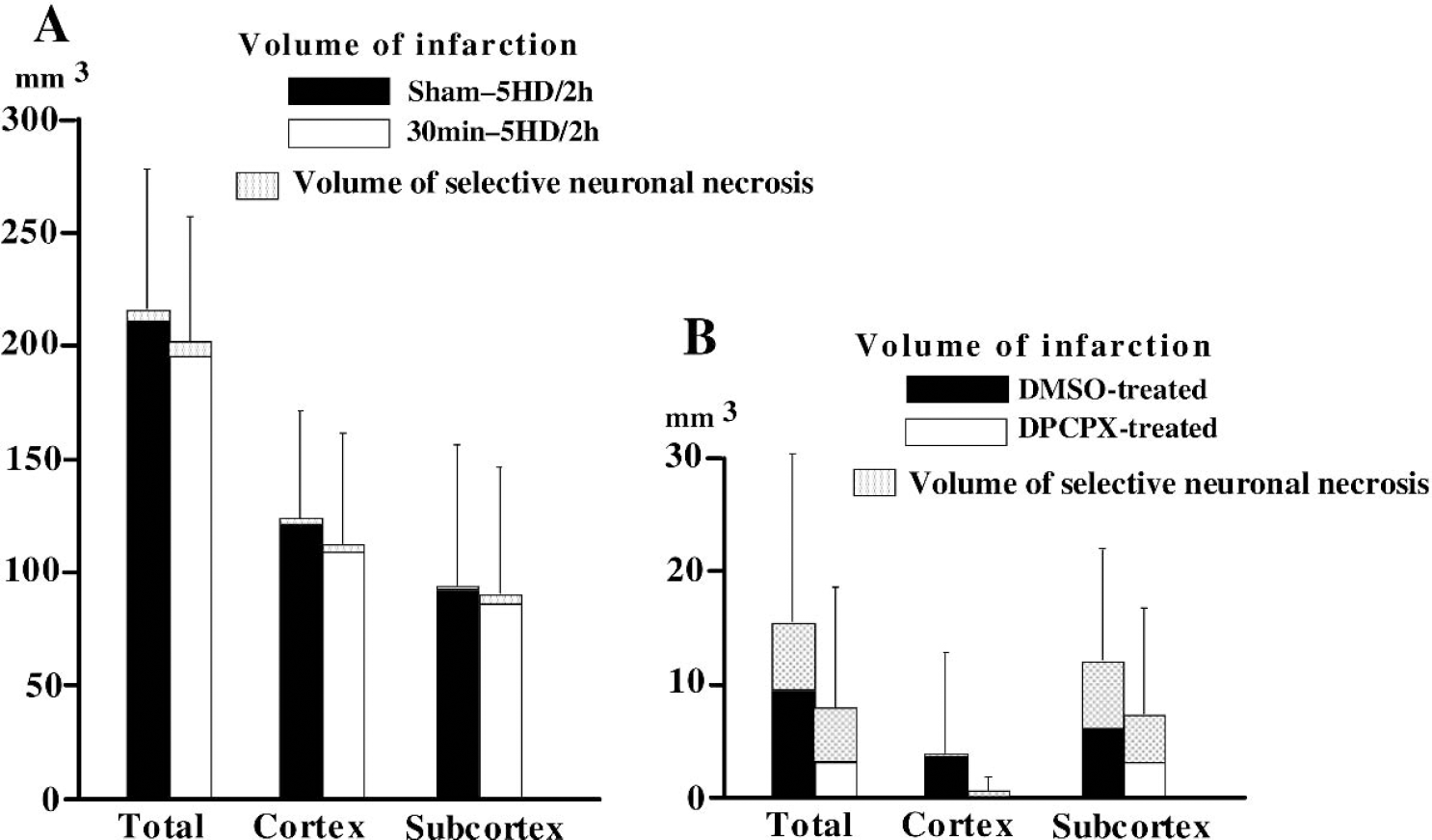
Effects of ischemic preconditioning (30-minute middle cerebral artery occlusion [MCAO]) on the volume of infarction and selective neuronal necrosis (SNN) after 120-minute MCAO given 72 hours later and modulating effects of selective adenosine A1 receptor antagonist on the preconditioning-induced neuroprotection (experiment 1). The sham-operated (sham-2h and sham-DPCPX/2h) groups and preconditioned (30min-2h, 30min-DPCPX/2h, and DPCPX/30min-2h) groups received 120-minute MCAO 72 hours after sham operation and conditioning ischemia (30-minute MCAO), respectively. The DPCPX/30min-2h group received DPCPX 30 minutes before the start of conditioning ischemia. The sham-DPCPX/2h and 30min-DPCPX/2h groups received DPCPX 30 minutes before the start of 120-minute MCAO. Preconditioning significantly reduced infarct volume both in the cortex and in the subcortex. The preconditioning-induced protection was attenuated by DPCPX given before conditioning ischemia, but was not influenced by DPCPX when administered before test ischemia (120-minute MCAO). Error bars indicate SD of total damaged volume (n = 12 per group). Total damage = infarct + SNN. *Infarct volume and total damaged volume were significantly different from sham-2h group. †Infarct volume and total damaged volume were significantly different from sham-DPCPX/2h group. (*)Compared with the sham-2h group, infarct volume was significantly different but total damaged volume was not. DPCPX, 8-cyclopentyl-1,3-dipropylxanthine (selective adenosine A1 receptor antagonist).
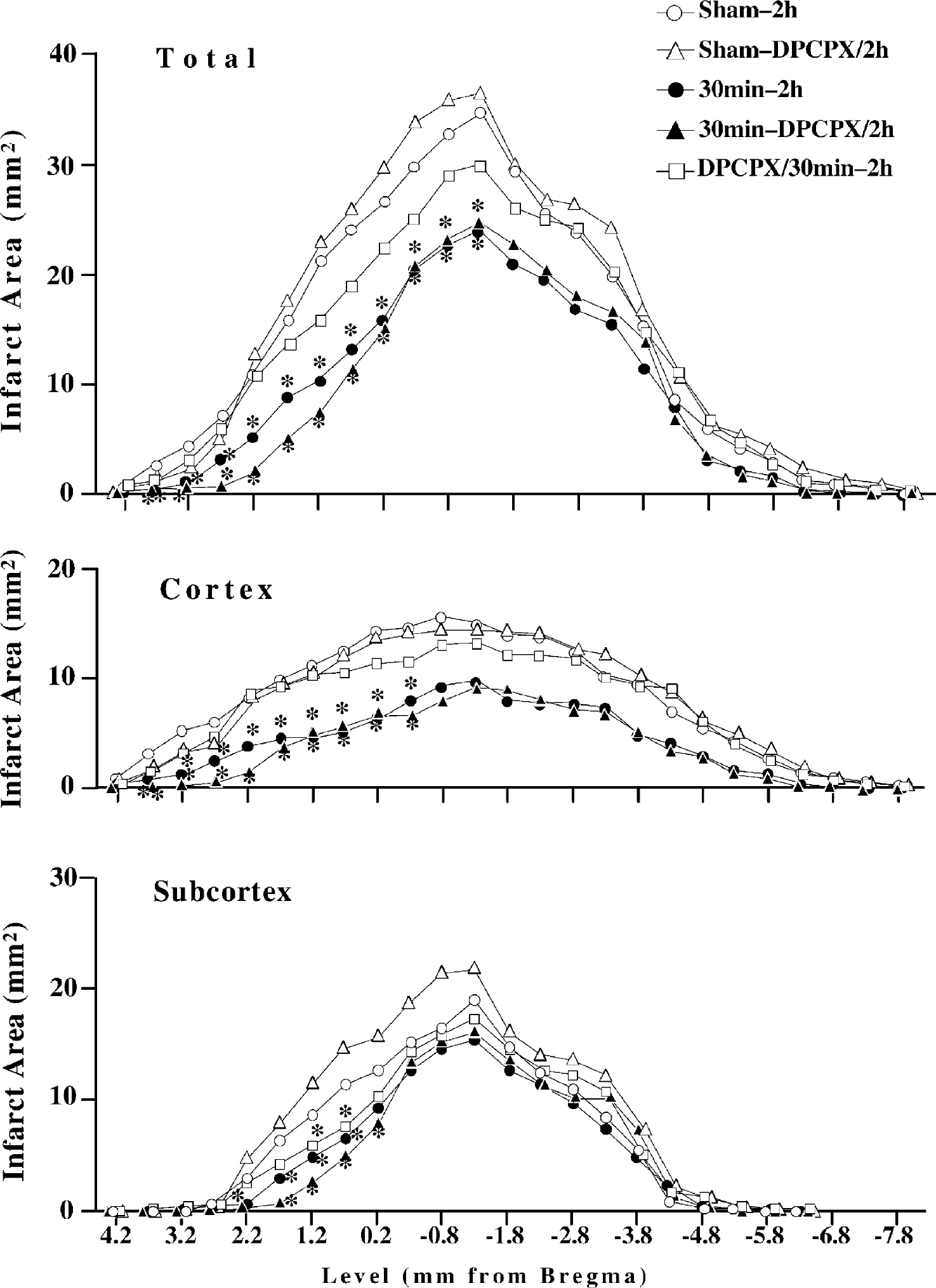
Infarcted area in each section after 120-minute middle cerebral artery occlusion (MCAO) with or without preconditioning (30-minute MCAO) and modulating effects of selective adenosine A1 receptor antagonist (experiment 1). The DPCPX/30min-2h group received DPCPX before conditioning ischemia. The sham-DPCPX/2h and 30min-DPCPX/2h groups received DPCPX before 120-minute MCAO. Data are means of each group (n = 12). *P < 0.05 versus sham-2h group. DPCPX, 8-cyclopentyl-1,3-dipropylxanthine (selective adenosine A1 receptor antagonist).
The adenosine A1 receptor antagonist DPCPX attenuated the neuroprotective effects of preconditioning when given before preconditioning, whereas DPCPX given before 120-minute MCAO produced no influence on the protection (Fig. 2 andFig. 3). There was no significant difference in infarct volume between the sham-2h and sham-DPCPX/2h groups, or between the 30min-2h and 30min-DPCPX/2h groups (Fig. 2).
The areas of SNN were observed around the infarcted regions both in the cortex and subcortex (Fig. 5). Table 2 shows the volume of SNN in each group. The SNN volume in the subcortex in two preconditioned groups (30min-2h and 30min-DPCPX/2h groups) was significantly larger than that in the groups without preconditioning (sham-2h and sham-DPCPX/2h groups). The SNN volume in the cortex did not differ among groups. Sum of infarct and SNN volumes in each group was also compared, and few changes were seen with the addition of SNN to infarct volume (Fig. 2).
Experiment 2
There were no significant differences in the volume of infarction or SNN between sham-5HD/2h and 30min-5HD/2h groups, indicating that 5HD administered before 120-minute MCAO blocked the neuroprotective effects of preconditioning (Fig. 4 and Table 2). This was in contrast to DPCPX, which had no influence on the preconditioning-induced protection when given before test ischemia (Fig. 2 and Fig. 3).
The volume of infarction and selective neuronal necrosis (SNN) after 120-minute middle cerebral artery occlusion (MCAO) with or without preconditioning (
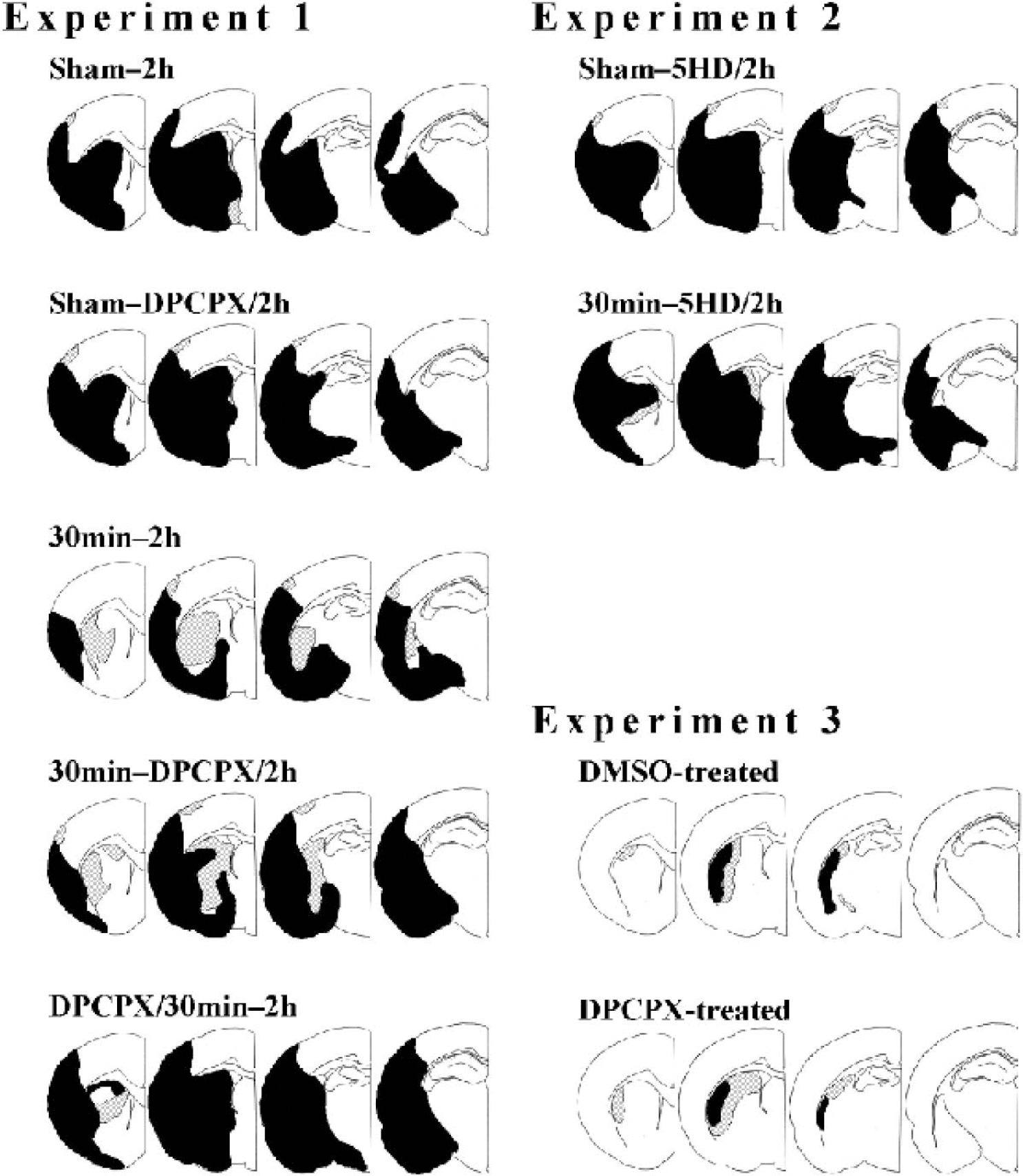
Schematic drawings of tissue damage 7 days after 120-minute middle cerebral artery occlusion (MCAO) or 10 days after 30-minute MCAO. Four out of 25 coronal sections from a representative animal in each group are shown (1.2, -0.3, -1.8, and -3.3 mm from bregma, respectively). Black areas represent infarct and hatched areas represent selective neuronal necrosis. DPCPX, 8-cyclopentyl-1,3-dipropylxanthine (selective adenosine A1 receptor antagonist); 5HD, 5-hydroxydecanoate (selective antagonists of mitochondrial ATP-sensitive K+ channels); DMSO, dimethyl sulfoxide (solvent of DPCPX).
Experiment 3
There were no significant differences in the volume of infarction or SNN after 30-minute MCAO between DMSO-treated and DPCPX-treated groups (Fig. 4 and Table 2).
DISCUSSION
The present study shows that, in focal cerebral ischemia, delayed neuroprotection induced by preconditioning is attenuated by a selective adenosine A1 receptor antagonist, DPCPX, given before preconditioning and by a selective mitochondrial KATP channel blocker, 5HD, given before subsequent severe ischemia. Neither antagonist at any dose studied had any influence on the infarct size after the conditioning insult or after the severe ischemic insult when given alone. The results indicate that exhibition of preconditioning-induced neuroprotection requires adenosine A1 receptor occupation during preconditioning and mitochondrial KATP channel opening during subsequent lethal ischemia.
The cascade underlying ischemic tolerance is of great interest but not fully understood. Recent studies indicate that liberation of adenosine and stimulation of A1 receptors play an important role as an initiator of ischemic preconditioning. We have previously shown that a selective A1 receptor antagonist, DPCPX, attenuated neuroprotective effects of rapid tolerance to focal cerebral ischemia (Nakamura et al., 2002). Similarly, the involvement of adenosine receptor activation as a trigger of delayed preconditioning has been suggested in global brain ischemia (Blondeau et al., 2000; Heurteaux et al., 1995). Concerning delayed ischemic tolerance to focal cerebral ischemia; this is the first study showing the involvement of adenosine A1 receptor activation during conditioning ischemia.
Propentofylline, an adenosine uptake inhibitor, administered 24 hours after conditioning ischemia potentiated tolerance to severe forebrain ischemia that was inflicted 24 hours thereafter (Kawahara et al., 1998). In addition, DPCPX given 3 hours after conditioning ischemia attenuated the beneficial effects of ischemic tolerance 21 hours later (Hiraide et al., 2001). These findings suggest that the activation of adenosine receptors is involved in mediating ischemic preconditioning. In the current study, DPCPX administered 72 hours after conditioning ischemia did not influence the neuroprotection induced by the preconditioning. The results suggest that adenosine A1 receptor stimulation is involved in triggering the preconditioning effects but is not necessary during subsequent lethal ischemia.
Adenosine, produced during cerebral ischemia, depresses the presynaptic release of glutamate and limits postsynaptic depolarization and Ca2+ influxes via A1 receptor activation (von Lubitz, 1999). An important step downstream of adenosine A1 receptor stimulation is KATP channel opening (Heurteaux et al., 1995; Reshef et al., 1998, 2000). In neuronal cultures, the opening of KATP channels was reported to be mandatory for the rapid and delayed ischemic tolerance by adenosine (Reshef et al., 2000).
Involvement of KATP channels in development of tolerance to forebrain ischemia has been reported, and the neuroprotective effect was abolished or mimicked by a nonselective KATP channel blocker (glibenclamide) (Heurteaux et al., 1995) or opener (cromakalim) (Blondeau et al., 2000). To our knowledge, the present study is the first to suggest the crucial role of mitochondrial KATP channel activation during focal cerebral ischemia in acquisition of tolerance. A few in vivo studies have shown neuroprotective effects of diazoxide, a selective mitochondrial KATP channel opener against cerebral ischemia and that the protective effects of diazoxide was abolished by 5HD, a selective antagonist of mitochondrial KATP channels (Liu et al., 2002; Rajapakse et al., 2002; Shake et al., 1999; Shimizu et al., 2002). These findings correspond well to the current results. Neuroprotection induced by mitochondrial KATP channel opening is possibly explained by optimizing energy state and suppression of mitochondrial Ca2+ overload during ischemia (Shimizu et al., 2002). Diazoxide is suggested to induce mild oxidative stress and preconditioning-like neuroprotection (Samavati et al., 2002). A recent report (Liu et al., 2003) shows that 5HD inhibits the preconditioning effect of diazoxide, while 5HD given alone produce neuroprotection through suppression of oxidative responses to ischemia.
A potassium conductance-independent pathway has recently been postulated for the effects of diazoxide and 5HD. Diazoxide attenuates succinate dehydrogenase activity and consequently inhibits mitochondrial respiration, and 5HD is converted to its coenzyme A derivative (5HD-Co A), which may affect fatty acid β-oxidation of mitochondrial metabolism (Hanley et al., 2002; Lim et al., 2002). Thus, these effects may account, in part, for the modification of ischemic tolerance by the agents. Nevertheless, the current study suggests the pivotal role of mitochondrial function for acquisition of ischemic tolerance and provides a valuable insight into understanding of pathways underlying preconditioning-induced neuroprotection.
This study indicates that both adenosine A1 receptor activation during conditioning ischemia and mitochondrial KATP channel opening during test ischemia are required for the induction of brain ischemic tolerance. Signal transduction pathways relating the two events may include protein kinase C, nuclear factor kB, and nitric oxide synthase (Baxter, 2002; Blondeau et al., 2001; Bolli, 2000; Centeno et al., 1999; Dirnagl et al., 2003; Kirino, 2002; Lange-Asschenfeldt et al., 2003; Pérez-Pinzón and Born, 1999; Raval et al., 2003; Reshef et al., 1998, 2000), though the cascade is speculative and remains to be elucidated.
We found that the volume of SNN in the subcortex (mainly in the caudoputamen) increased with preconditioning, despite a significant reduction in the infarct volume, indicating that the caudoputamen was salvaged from infarction but resulted in SNN. This may be attributable to the vulnerability of caudoputamen to MCAO in rats (Memezawa et al., 1992).
Although cerebral blood flow was not measured in the present study, no differences in blood flow have been reported in the tolerant regions between sham-operated and preconditioned animals before and during test focal ischemia (Barone et al., 1998; Chen et al., 1996; Shimizu et al., 2001). Similarly, a dose of diazoxide that showed neuroprotection against focal ischemia elicited no or minimal effects on cerebral blood flow during the ischemia (Shimizu et al., 2002), and 5HD pretreatment did not influence the blood flow and abolished the protective effects of diazoxide (Liu et al., 2002). DPCPX was also reported to have no significant effects on basal cerebral blood flow or carbon dioxide-evoked hyperemia (Phillis and O'Regan, 2003). Therefore, induced tolerance observed is not flow-related phenomenon.
In summary, the present study shows an involvement of adenosine A1 receptors during conditioning ischemia and mitochondrial KATP channels during subsequent lethal ischemic period in exerting the tolerance. A recent clinical study suggested that an endogenous neuroprotective mechanism similar to ischemic tolerance might play a role in patients with repeated attacks of cerebral ischemia (Moncayo et al., 2000). Although the preconditioning-induced protection is moderate in the current study and further investigation is needed to elucidate the detailed cascades of ischemic tolerance, the present findings may help to provide novel therapeutic strategies for cerebral protection against ischemic injury.