
Abstract
Adenosine is an endogenous neuromodulator with anticonvulsive and neuroprotective activity. Adenosine levels are normally kept in the range of 20 to 200 nmol/L by low basal expression of its main metabolic enzyme, adenosine kinase (ADK). Dysfunction of the adenosinergic system has been demonstrated to contribute to epileptogenesis. To investigate whether upregulation of ADK may render the brain more susceptible to ischemic cell death, mutant mice overexpressing an Adk transgene in brain were subjected to middle cerebral artery occlusion (MCAO). One day after either 15 or 60 mins of MCAO, wild-type (WT) animals had infarct areas encompassing about 5% and 50% of their ischemic hemisphere, respectively. In marked contrast, the volume of the infarcts increased three-fold in Adk transgenic mutants after 15 mins of MCAO, and after 60 mins of MCAO all mutants died within 24 h. Pretreatment of the mutants with the ADK inhibitor 5-iodotubercidin led to lesions similar to those in WT mice. Thus, low levels of ADK are essential to maintain adenosine-mediated neuroprotection. We conclude that pathologic overexpression of ADK as in epilepsy may also render the brain more susceptible to injury from ischemia. Consequently, ADK emerges as a rational therapeutic target to enhance neuroprotection.
Introduction
Cerebral ischemia and transient periods of hypoxia are known to induce a dramatic increase in extracellular adenosine, which is thought to provide neuroprotection during cerebral ischemia and other neuronal insults (Berne et al, 1974). In fact, excessive adenosine release appears to be one of the mechanisms by which the brain attempts to protect itself from cell injury. Thus, adenosine is an endogenous neuroprotectant of the brain (Cunha, 2005; Fredholm, 1997; Ribeiro, 2005). These potentially beneficial effects of adenosine are limited by a rapid turnover of this purine ribonucleoside. Adenosine levels in brain are regulated primarily by adenosine kinase (ADK), which removes (protective) adenosine via phosphorylation to AMP (cyclic 3′,5′-adenosine monophosphate) (Arch and Newsholme, 1978). The adult brain is normally kept under tonic adenosine-mediated inhibitory tone, which can be relieved by adenosine receptor antagonists, such as caffeine (Fredholm et al, 1999). High levels of protective adenosine are maintained in the setting of low-level expression of ADK. Adult brain ADK is primarily expressed in astrocytes (Boison, 2005; Gouder et al, 2004). Thus, in pathologic conditions of brain, which involve astrogliosis, such as in partial epilepsy, ADK is upregulated, thus contributing to an enhancement of seizure susceptibility by a reduction of ambient adenosine (Boison, 2005; Gouder et al, 2004). Indeed, overexpression of ADK can be a direct cause for seizures (Fedele et al, 2005). Since overexpression of ADK can occur in pathologies with astrogliosis, such as in epilepsy, and likely also after other forms of brain injury, it is important to know how ADK levels might affect the brain's susceptibility to ischemic cell death.
To study ADK as a potential therapeutic target for the modulation of adenosine levels for neuroprotection in stroke, transient focal cerebral ischemia was induced in transgenic mice, which overexpress ADK in their brains (Adktm1−/−-Tg(UbiAdk)) (Fedele et al, 2005). Owing to a targeted disruption of the endogenous Adk gene, these mice lack the endogenous astrocytic enzyme, but contain an Adk transgene under the control of the human ubiquitin promoter, which leads (i) to an overexpression of ADK in brain with a 2.2-fold overall increase in enzymatic ADK-activity and (ii) to a novel expression of ADK in neurons (Fedele et al, 2005). As a result of the gene mutations, Adktm1−/− Tg(UbiAdk) mice are characterized by reduced adenosinergic tone, as was evidenced by a lack of an A1 receptor antagonist-mediated increase in EPSC responses in whole-cell recordings of CA3 hippocampal pyramidal neurons in acute slices from the mutant animals (Fedele et al, 2005). This finally manifests in behavioral hyperexcitability and enhanced seizure susceptibility (Fedele et al, 2005).
Materials and methods
Experimental Groups
Male C57BL/6 (Charles River) and Adktm1−/−-Tg(UbiAdk) mice of the same genetic background weighing 25 to 30 g were housed under diurnal lighting conditions (12 h darkness and 12 h light). Experiments were performed according to the international guidelines for animal research. All experiments were performed in accordance with the American animal protection legislation and approved by the Institutional Animal Care and Use Committee of Legacy Research.
Focal Ischemia
Transient focal ischemia was induced by suture occlusion of the middle cerebral artery (MCAO) in male mice anaesthetized using 1.5% isofluorane, 70% N2O and 28.5% O2. Ischemia was induced by introducing a coated filament (6.0; Doccol, Redlands, CA, USA) from the external carotid artery into the internal carotid artery, and advancing it into the proximal middle cerebral artery, thereby occluding the middle cerebral artery. Rectal and temporalis muscle temperature was maintained at 37°C ± 0.5°C with a thermostatically controlled heating pad and lamp. Cerebral blood flow was monitored by transcranial laser Doppler (Transonic System Inc., Ithaca, NY, USA). All surgical procedures were performed under an operating stereomicroscope.
Determination of the Infarct Volume
To evaluate the infarct volume, animals were killed with an overdose of isofluorane 24 h after ischemia. Brains were quickly removed, placed into a preformed mouse brain mould calibrated for removal of 1 mm slices with regular intervals, and sectioned coronally at 1 mm intervals using a razor blade, and stained by immersion in 2% (w/v) vital dye 2,3,5-triphenyltetrazolium hydrochloride (TTC). The infarction area was calculated by subtracting the TTC stained undamaged area of the ischemic hemisphere from the area of the non-ischemic hemisphere (Swanson and Sharp, 1994). The infarct volume was calculated by summing up infarction areas of all sections and multiplying these by slice thickness. The percentage of the infarct volume was calculated by dividing the infarct volume by the total ipsilateral hemispheric volume as described previously (Pignataro et al, 2004a, b). The individual who performed the image analysis was masked to the study groups.
Experimental Protocol
Adktm1−/−-Tg(UbiAdk) (= Adk-tg) and wild-type (= WT) mice were treated with either 15 or 60 mins of MCAO (n = 5 per genotype and treatment). An additional group of Adk-tg mice received intraperitoneal injections of 3.1 mg/kg 5-iodotubercidin (ITU) (dissolved in 20% DMSO in saline and diluted to 5% DMSO in saline) 15 mins before either 15 or 60 mins of MCAO (n = 5 per treatment). To control for potential vehicle mediated effect, each experimental animal was either injected with 3.1 mg/kg ITU (100 μL i.p.) or with vehicle alone (100 μL of 5% DMSO in saline, i.p.). All animals were killed and analyzed after 24 h of reperfusion. Individuals masked to the treatment groups performed all manipulations and analyses.
Statistical Analysis
Values are expressed as means ± s.e. Statistical analysis was performed with analysis of variance followed by Newmann-Keul's test. Statistical significance was accepted at the 95% confidence level (P < 0.05).
Results
Overexpression of Adenosine Kinase has Lethal Consequences after 60 mins of Middle Cerebral Artery Occlusion
To investigate whether the susceptibility to ischemic brain insults depends on ADK expression levels, we subjected WT and Adktm1−/−-Tg(UbiAdk) mice to an injurious ischemic insult, which consisted of 60 mins occlusion of the middle cerebral artery followed by 24 h of reperfusion. Achievement of ischemia was confirmed by monitoring regional cerebral blood flow in the area of the left middle cerebral artery. There were no significant differences in the reduction of cerebral blood flow during MCAO between mutant (18.4%±8.3% of normal), ITU-treated mutant (15.8%±5.9%), and control animals (21.1%+5.9%). The percentage of infarct volume in the WT animals was 50.57±2.35 (Figure 1). In contrast, all the Adktm1−/−-Tg(UbiAdk) mice subjected to 60′ of MCAO died within 12 to 20 h after MCAO. To verify the causal role of ADK overexpression for the death of the mutants after 60 mins of MCAO, a specific inhibitor of ADK, ITU, was injected (3.1 mg/kg, i.p.) in Adktm1−/−-Tg(U-biAdk) mice 15 mins before 60 mins of MCAO. Strikingly, the ITU treated animals were rescued from the lethal consequences of 60 mins of MCAO. The percentage of the ischemic volume in these animals was 59.07±3.43, and thus similar to WT animals after 60 mins of MCAO (Figure 1).
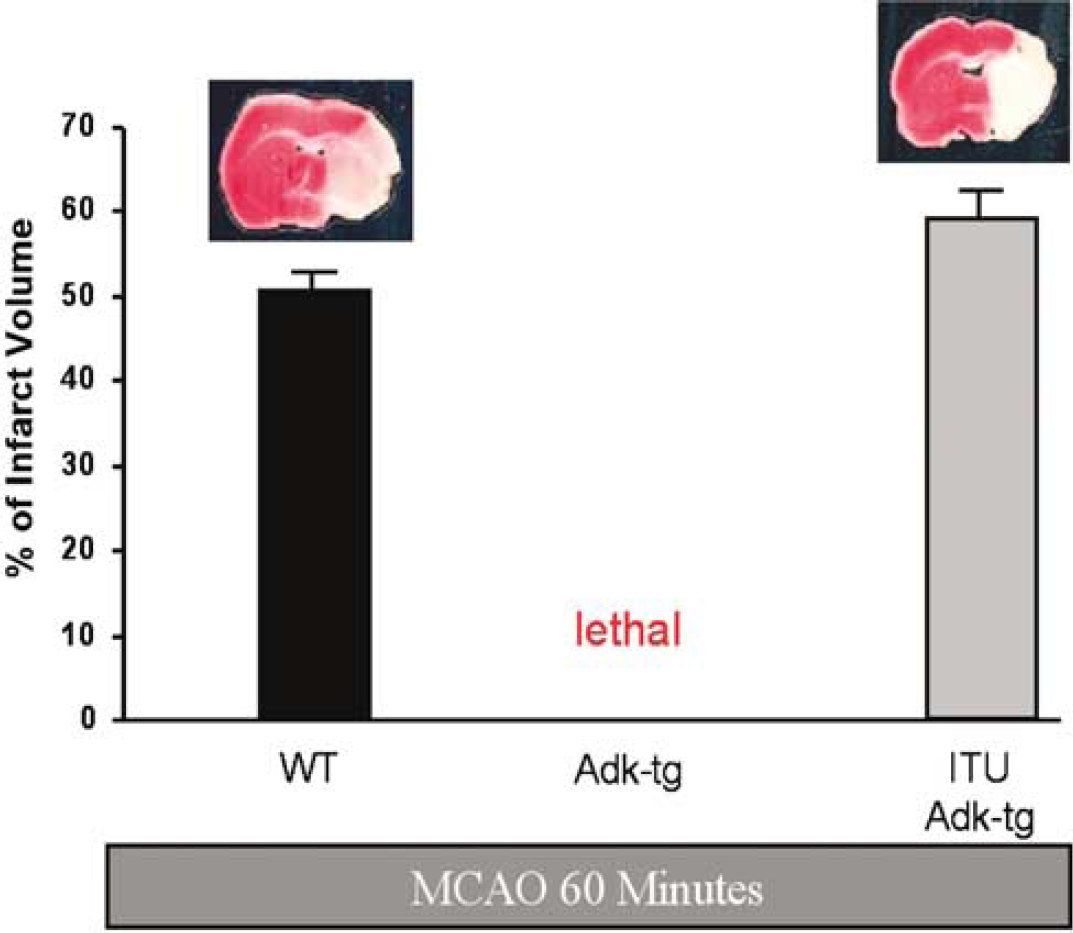
Effect of 60 mins of cerebral ischemia on wild type (WT) and Adk-transgenic (Adk-tg) mice and Adk-transgenic mice pretreated with 5′-ITU (ITU Adk-tg) (3.1 mg/kg, ip 15 mins before MCAO). All mice were euthanized 24 h after MCAO and analyzed by quantification of the infarct size after TTC staining. The percent of infarct volume per ischemic brain hemisphere is given and representative TTC-stained brain sections are shown. Each experimental group consisted of five animals.
Overexpression of Adenosine Kinase Leads to Augmentation of Infarct after 15 mins of Middle Cerebral Artery Occlusion
In WT mice 15 mins of MCAO is usually considered to be a noninjurious injury, associated only with a small infarct volume. To investigate whether overexpression of ADK in brain alters the susceptibility to noninjurious ischemic brain injury, we subjected Adktm1−/−-Tg(UbiAdk) and WT mice to mild ischemia, by occluding the left middle cerebral artery for 15 mins, followed by 24 h of reperfusion (n = 5, each). Achievement of ischemia during MCAO, as confirmed by monitoring regional cerebral blood flow by transcranial laser Doppler, was not significantly different in mutants (13.5%±4.2% of normal), ITU-treated mutants (14.7%±3.4%) or WT (16.6%± 4.6%). The infarct volume was determined 24 h after MCAO. The percentage of infarct volume in the WT animals was 6.28± 1.35. In contrast, Adktm1−/−-Tg(UbiAdk) mice displayed a significantly enlarged infarct volume of 19.81%± 7.23% (Figure 2), which was accompanied by corresponding motor deficits. To confirm that the observed effect was due to the transgenic overexpression of ADK, we injected a control group of Adktm1−/− Tg(UbiAdk) mice with the ADK inhibitor ITU(3.1 mg/kg, i.p.). As a result of ADK inhibition, the infarct volume after 15 mins of MCAO was 5.05 ± 0.76 5, and thus reduced to the lesion size of WT animals.
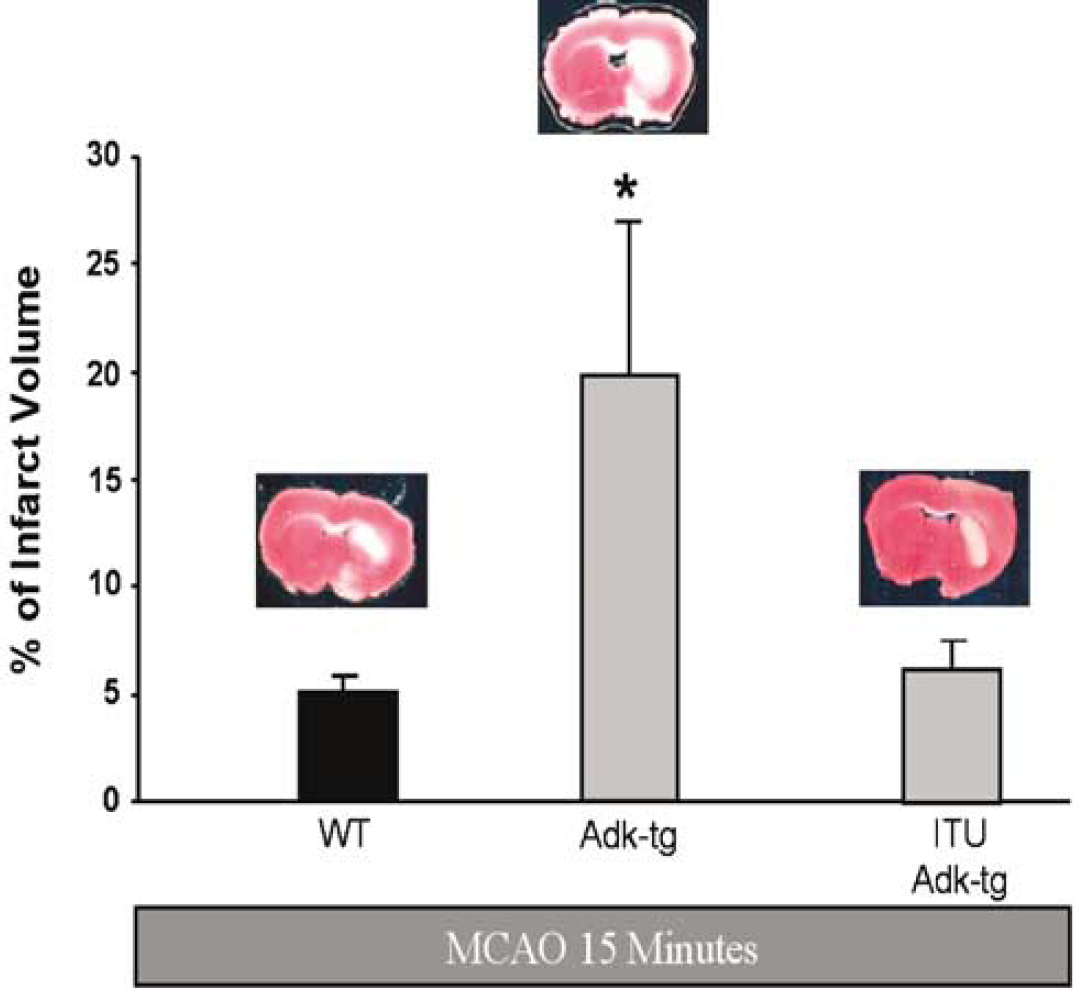
Effect of 15 mins of cerebral ischemia on wild type (WT) and Adk-transgenic (Adk-tg) mice and Adk-transgenic mice pretreated with 5′-ITU (ITU Adk-tg) (3.1 mg/kg, ip 15 mins before MCAO). All mice were euthanized 24 h after MCAO and analyzed by quantification of the infarct size after TTC staining. The percent of infarct volume per ischemic brain hemisphere is given and representative TTC-stained brain sections are shown. Each experimental group consisted of five animals. *P < 0.05 versus WT ischemic mice and ITU treated group.
Discussion
Purines, such as adenosine and adenosine 5′ triphosphate (ATP), are crucial cellular components used as a source of energy or as parts of nucleic acids. Besides these vital cellular functions, purines are able to act as signaling molecules that mediate physiologic effects by interacting with purine receptors. The effects of adenosine are mediated by activation of adenosine receptors (A1, A2A, A2B, and A3) that couple to heterotrimeric G proteins to access diverse intracellular signaling pathways. Thus, by specific interaction with different G proteins, by specific affinities for adenosine, and by specific spatial distribution within the brain, these receptors allow a high degree of complexity in the effects of adenosine (Fredholm et al, 2005). Adenosine exerts its potent inhibitory effects mainly by activation of the high-affinity A1 receptors that are linked to inhibitory G proteins, which (i) inhibit adenylyl cyclase, (ii) activate G-protein-dependent inwardly rectifying K+ (GIRK) channels, (iii) inhibit Ca2+ channels, and (iv) activate phospholipase C. As a consequence, the release of various neurotransmitters, in particular of glutamate, is inhibited via presynaptic A1 receptors (Fredholm et al, 2005). Pharmacologically, adenosine and its A1 selective analogues are effective in seizure suppression (Boison, 2005) and neuroprotection (Ribeiro, 2005). Furthermore, A1 receptor activation is a prerequisite to prevent the spread of epilepsy induced cell death (Fedele et al, 2006). Thus, adenosine has potent neuroprotective effects, which are mediated by A1 receptors.
In contrast, the tonic activation of A2A receptors may contribute to neuronal injury in several neurologic disease models (Fredholm et al, 2005). Thus, A2A receptor antagonists provide neuroprotection in models of neuronal cell loss (Cunha, 2005) and adult A2A receptor knockout mice display attenuated focal ischemic brain damage (Chen et al, 1999). However, neuroprotection by activation of the low affinity A3 receptors depends on the differential impact of these receptors on both neuronal and non-neuronal elements of the cerebral tissue (Von Lubitz et al, 2001).
Because the manifold actions of adenosine via its receptors, a tight regulation of adenosine levels is of crucial importance. Since intra- and extracellular adenosine is rapidly equilibrated by bidirectional, equilibrative nucleoside transporters, the extracellular concentrations of adenosine are regulated by the interplay of intra- and extracellular enzymes of adenosine metabolism. Based on its low KM for adenosine, ADK is the primary route of adenosine metabolism (Arch and Newsholme, 1978). A high flux rate in a substrate cycle between adenosine and AMP, involving 5′-nucleotidase and ADK, allows minor changes in the activity of ADK to be rapidly translated into major changes in adenosine (Arch and Newsholme, 1978). Consequently, under conditions of stress, for example during ischemia, in which ATP consumption exceeds its formation, ADK could rapidly be inhibited by an increase in ADP and subsequent aggregation (Sen et al, 2006). Thus, ADK is rapidly inactivated after oxygen glucose deprivation in cultured neurons (Lynch et al, 1998).
While inactivation of ADK may be beneficial, by increasing levels of protective adenosine, upregulation of ADK has been associated with astrogliosis (Gouder et al, 2004), a hallmark of several brain pathologies including epilepsy and ischemic brain injury. Remarkably, upregulation of ADK has been associated with epileptogenesis and can be the cause for seizures (Fedele et al, 2005; Gouder et al, 2004).
To address the question, whether upregulation of ADK may render the brain more susceptible to ischemic cell death, cerebral ischemia was induced in transgenic mice, which overexpress transgenic ADK in their brains (Adktm1−/−-TgUbiAdk)) (Fedele et al, 2005). Owing to a targeted disruption of the endogenous Adk gene, those mice lack the endogenous astrocytic enzyme, but contain an Adk transgene under the control of the human ubiquitin promoter, which leads to an overexpression of ADK in brain and to a novel expression of ADK in neurons (Fedele et al, 2005). As a consequence of the transgenic overexpression of ADK, the levels of ambient adenosine are reduced in the mutants (Fedele et al, 2005). It is important to have the ubiquitous transgenic ADK expression on top of a defined knockout of the endogenous gene to avoid compensatory mechanisms by regulatory mechanisms of the endogenous astrocytic enzyme (Gouder et al, 2004). In this model, we show for the first time that overexpression of ADK induced an increase in stroke volume after MCAO, thus demonstrating that adenosine plays a neuroprotective role in this pathology. In fact, a subthreshold MCAO-induced ischemic stimulus that caused minimal tissue damage in WT mice was here sufficient to induce extensive tissue damage of mice overexpressing ADK. To prove that the increased sensitivity to ischemia in the mutants is due to overexpression of ADK, the ADK inhibitor ITU was used to reverse the effect of ADK overexpression. The reversal of injury aggravation in the mutants after ITU application is consistent with earlier findings demonstrating neuroprotection in WT animals after 5′-deoxy-ITU administration (Jiang et al, 1997). The aggravation of MCAO-induced injury by overexpressed ADK is likely a central effect, since, in contrast to other organs analyzed, brain was found to be the only organ with overexpression of ADK (Fedele et al, 2005). For the same reasons, although not directly tested, we do not expect any major changes in cerebral blood flow in the mutants. Indeed, we were able to show that cerebral blood flow during MCAO was decreased to the same degree in mutant and WT animals.
This study also shows that the combined effect of ambient adenosine, mediated by the interplay of the different adenosine receptors, provides a neuroprotective tone in WT animals, which is abolished when ADK expression is increased. Thus, the reduced tone of adenosine in the mutants, as demonstrated previously in acute slice recordings (Fedele et al, 2005), is likely to lead to a reduction of A1 receptor activation, thus decreasing neuroprotection. At the same time, A2A receptor activation is expected to be reduced, a manipulation considered to be neuroprotective (Cunha, 2005). Owing to the overall significant decrease of neuroprotection in the Adktm1−/−-Tg(UbiAdk) mice observed in this study, we conclude that the A1 receptor mediated effect dominates over a potential A2A receptor mediated effect. Since A2B and A3 receptors are less frequent and require higher levels of adenosine to be activated (Fredholm et al, 2001), they are less likely to play a dominant role after overexpression of ADK.
In conclusion, low levels of ADK are essential to maintain adenosine-mediated endogenous neuroprotection and pathologic increases in ADK may render the brain more susceptible to injury. Consequently, ADK emerges as a rational therapeutic target to enhance neuroprotection in ischemia.
Footnotes
Acknowledgements
This work was supported by NIH Grant RO1NS024721 and the Good Samaritan Hospital Foundation.