
Abstract
We studied the effect of decreased glucose concentration on cerebrovascular tone in vitro. Segments of rat middle cerebral arteries (MCA) were isolated, cannulated at both ends with glass micropipettes, and pressurized to 85 mm Hg. Decreasing the glucose in the extraluminal bath and luminal perfusate from 5.5 mmol/L to 1.0 or 0.5 mmol/L for 1.5 hours each had no significant effect on the diameter of the arteries. When all the glucose was removed from the extraluminal bath and luminal perfusate for 1.5 hours, the MCA dilated by 23% [252 ± 24 (SD) μm to 311 ± 7 μm (P < .5, n = 7)]. This dilation was 80% of the maximum dilation produced by removal of Ca+2 from the bathing solutions. Neither removal of the endothelium nor inhibition of the ATP-sensitive K channels with 10−5 mol/L glibenclamide altered the response of the isolated MCA to the removal of glucose. We conclude that rat MCA are relatively more resistant to substrate limitation compared to the brain as a whole.
Keywords
Inhibition of the glycolytic pathway in the brain, by either substrate limitation (hypoglycemia) or pharmacologic agents that block the pathway enzymes is accompanied by an increase in CBF (Horinaka et al., 1997a; Tanaka et al., 1985; Breier et al., 1993; Bryan, Jr. et al., 1987). Although the increase in CBF during glycolytic inhibition, mostly hypoglycemia, has been reported for a number of species including man, the most consistent findings have been in the rat (Horinaka et al., 1997a; Tanaka et al., 1985; Mujsce et al., 1989; Neil et al., 1987; Breier et al., 1993; Bryan, Jr. et al., 1987; Pelligrino and Albrecht, 1991; Abdul-Rahman et al., 1980).
The mechanism evoking the increase in CBF (decrease in cerebrovascular resistance) is not well understood. Several mechanisms including (1) stimulation of beta adrenoceptors by an unknown agonist (Bryan et al., 1994; Hollinger and Bryan, 1987), (2) adenosine (Horinaka et al., 1997b; Ruth et al. 1993), (3) nitric oxide (Ichord et al., 1994), and (4) an increase in extracellular potassium (during coma only) (Astrup and Norberg, 1976) have been implicated with the flow increase accompanying substrate limitation (Ichord et al., 1994; Ruth et al., 1993; Bryan et al., 1994; Hollinger and Bryan, 1987). A recent study has suggested that stimulation of ATP-sensitive potassium channels, linked to the stimulation of P1 purinoceptors by adenosine, are an important factor for the flow regulation during hypoglycemia (Horinaka et al., 1997b).
One possible mechanism not previously considered is a direct effect of decreased glucose concentrations on the tissues of the cerebral vasculature (smooth muscle or endothelium). The decrease in the concentration of glucose could dilate the cerebrovascular smooth muscle directly or through the endothelium by stimulating the release of an endothelium-derived relaxing factor.
The purpose of this study was to determine the effects of decreased glucose on cerebrovascular tissues uncomplicated by influences from parenchymal tissues of the brain. For this reason, we used the isolated rat middle cerebral artery. Initially, we tested the hypothesis that a decrease in the glucose concentration dilates the rat middle cerebral artery (MCA).
If the above hypothesis proved to be true, we wished to test a second and third hypothesis, both dealing with the mechanism(s) for the dilation. The second hypothesis was: the dilation produced by a decreased glucose concentration involves ATP-dependent K channels (KATPs). KATPs are found in a variety of tissues including the basilar and MCA of the rat (Fredricks et al., 1994; Faraci and Heistad, 1993; De Weille, 1992; Edwards and Weston, 1993). These channels, which are regulated by ATP, have been shown to dilate cerebral arteries during hypoxia or after treatment with KATP channel openers (Faraci and Heistad, 1993; Fredricks et al., 1994). During glucose deprivation, KATPs might open in the cerebrovascular tissue due to a decrease in ATP. The result would be hyperpolarization of the smooth muscle, closure of voltage-gated calcium channels, and ultimately dilation of the artery (Edwards and Weston, 1993). Reports of KATP channel activation due to decreased glucose concentrations have been reported for pancreatic β-cells and neurons in the brain (Edwards and Weston, 1993).
Third, we wished to test the hypothesis that the release of a relaxing factor(s) from the endothelium dilates the rat MCA when the glucose concentration is decreased. The endothelium can release a variety of relaxing factors (nitric oxide, prostacyclin, endothelium-derived hyper-polarizing factor, and others) after receptor stimulation, mechanical stimulation, or hypoxia (Rubanyi, 1991; Zakhary et al., 1996; Fredricks et al., 1994).
METHODS
In vitro studies
The experimental protocol was approved by the Animal Protocol Review Committee at Baylor College of Medicine. Male Long Evans rats were anesthetized with isoflurane and decapitated. The brain from each rat was removed from the cranium and placed in cold physiologic saline solution (PSS, 4°C). The left and right MCA (MCA) were carefully removed beginning at the circle of Willis and continuing 5 to 6 mm distally.
Arteries were placed in an arteriograph (Living Systems Inc., Burlington, VT) where micropipettes were inserted into both ends of each artery. A segment of each artery approximately 1 mm in length and lying between branch points was positioned between the tips of the two micropipettes. The artery was secured to the micropipettes with nylon sutures (9-0). Each artery was bathed in PSS equilibrated with 20% O2, 5% CO2, and balance N2. The PSS in the bath was maintained at 37°C.
Luminal or transmural pressure was maintained at 85 mm Hg by raising reservoirs, connected to the micropipettes by tygon tubing, to the appropriate height above each artery. Luminal perfusion was adjusted to 100 μL/min by setting the two reservoirs at different heights. Pressure transducers between the micropipettes and the reservoirs provided a measure of perfusion pressure. A flow meter (#11, Gilmont Instruments, Barrington, IL) measured luminal flow (Bryan, Jr. et al., 1996; Bryan, Jr. et al., 1995). The luminal perfusate was heated to 37°C and gassed before perfusing the lumen of each artery. Samples of PSS from the bath were analyzed for PO2, PCO2, and pH using a Corning model 280 analyzer (Medfield, MA).
The vessels were magnified using an inverted microscope equipped with a video camera and monitor. Outside diameters of the arteries were measured directly from the video screen.
After mounting, MCA were allowed to equilibrate for 1 hour before beginning any experiments. During this period, the diameter decreased by approximately 20% indicating spontaneously developed tone in the arteries. Glucose was decreased from 5.5 mmol/L to 0 mmol/L (glucose free) in several steps or in one step. Glucose was decreased in both the luminal PSS and extraluminal bath. At the end of each experiment Ca+2 free PSS containing 1 mmol/L egtazic acid (EGTA) was used to obtain the maximal dilation of the MCA.
In one study, the endothelium was removed by passing 8 mL of air through the lumen of the vessels (Fredricks et al., 1994). Care was taken to ensure that pressure did not exceed 85 mm Hg during this process. Removal of the endothelium was confirmed by the absence of a dilation to the addition of 10−5 mol/L UK14, 304, an α2 agonist (Bryan, Jr. et al., 1996; Bryan, Jr. et al., 1995).
In another study, we tested the ability of glibenclamide, a blocker of the ATP-dependent K channels (KATP), to inhibit the dilation produced by removal of the glucose. The efficacy of glibenclamide was determined by its ability to inhibit dilations produced by Iloprost, a stable prostacyclin analogue that dilates by activating the KATP channels (Fredricks et al., 1994).
Reagents and drugs
Serotonin was obtained from Sigma, St. Louis, MO. Glibenclamide was obtained from Research Biochemicals Incorporated, Natick, MA. Iloprost was a gift from Berlex Laboratories, Cedar Knolls, NJ. Glibenclamide was dissolved in dimethyl sulfoxide (DMSO); other compounds were dissolved in PSS or distilled water.
The PSS consisted of the following (Bryan, Jr. et al., 1996; Bryan, Jr. et al., 1995): NaCl 119 mmol/1, NaHCO3 24 mmol/L, KCl 4.7 mmol/L, KH2PO4 1.18 mmol/L, MgSO4 1.17 mmol/L, CaCl2 1.6 mmol/L, and EDTA 0.026 mmol/L.
Statistics
Data are expressed as the mean ± the SD. For comparison of responses, the Student's t test was used with a Bonferroni correction for multiple comparisons (where appropriate). The acceptable level of significance was defined as P < .05.
RESULTS
The mean diameter of MCA with endothelium [endothelium (+)] bathed in PSS with 5.5 mmol/L glucose was 252 ± 24 μm (n = 7). In a second group of MCA where the endothelium had been removed [endothelium (–)], the mean diameter was 212 ± 41 μm [(n = 7), P = .049 compared to endothelium (+)]. Removal of the endothelium was confirmed by the absence of a dilation to the addition of 10−5 mol/L UK14.304, an α2 agonist that requires intact endothelium to produce a dilation [21 ± 7% before removal versus 2 ± 2% after removal (n = 7); P = .0001] (Bryan, Jr. et al., 1996; Bryan, Jr. et al., 1995). The reduction of glucose to 1 mmol/L for 1.5 hours in the luminal PSS and extraluminal bath did not significantly change the diameters in either group when compared to the corresponding diameters at 5.5 mmol/L glucose (Fig. 1). At 0.5 mmol/L glucose, three of seven MCA in each group showed an increase in diameter greater than 10% compared to the diameter at 5.5 mmol/L glucose; however, the diameters did not show a statistically significant change when either group was taken as a whole. When all glucose was removed (0 mmol/L) for 1.5 hours, MCA dilations of 23% for endothelium (+) and 37% for the endothelium (–) were statistically significant when compared to the corresponding baseline diameters at 5.5 mmol/L glucose. On restoration of glucose to 5.5 mmol/L, the MCA with endothelium returned to the original diameter, whereas the diameters in the endothelium denuded group remained 15% larger than the original diameter (Fig. 1).
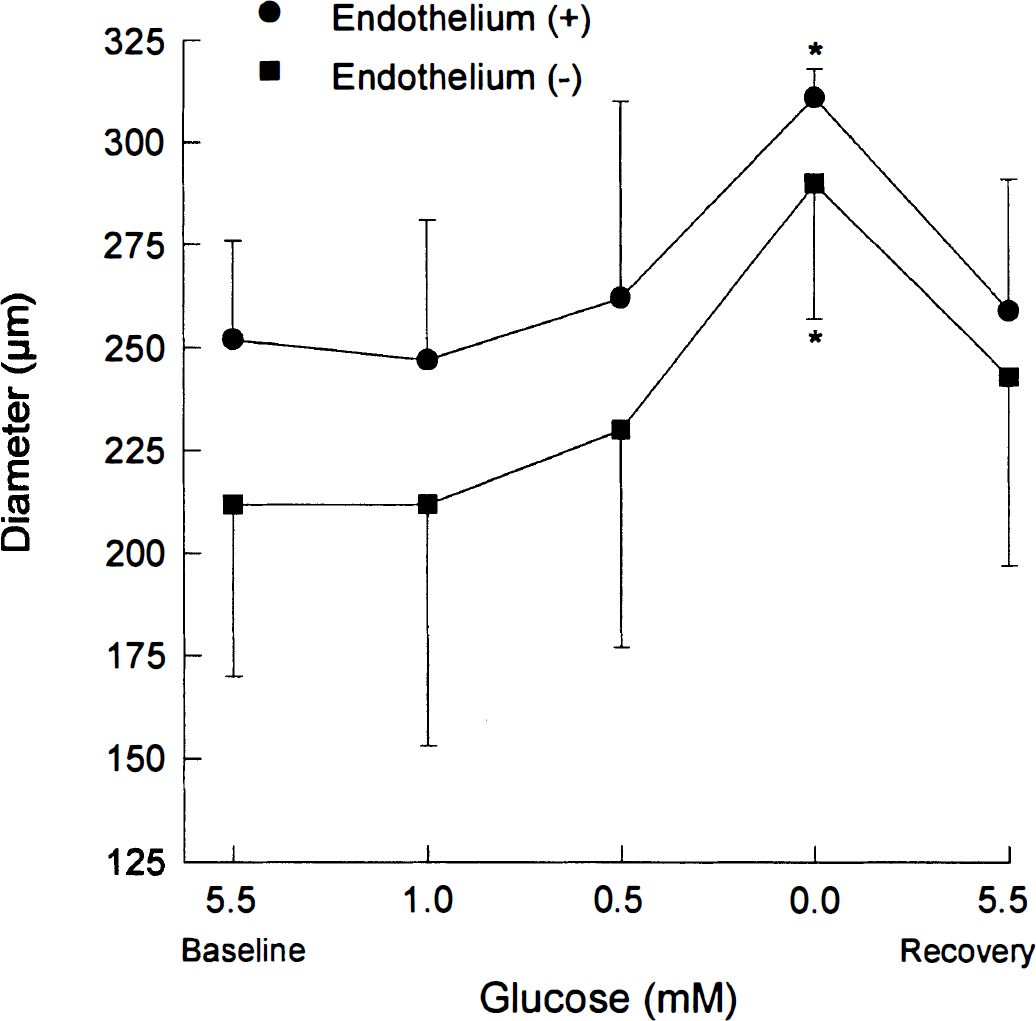
The effects of changing glucose concentration on the diameter (mean ± SD) of rat middle cerebral arteries with (circles) and without (squares) endothelium, n = 7 for each group, *P < .05 compared to the corresponding baseline at 5.5 mmol/L glucose.
In another group of MCA with intact endothelium, the diameter increased 56 ± 17 μm (n = 6) above the resting baseline (251 ± 34 μm) when the glucose concentration was changed directly to 0 mmol/L from 5.5 mmol/L (Fig. 2). Bathing the MCA in Ca+2-free PSS to obtain a maximum dilation further increased the diameters by 14 ± 7 μm (Fig 2).
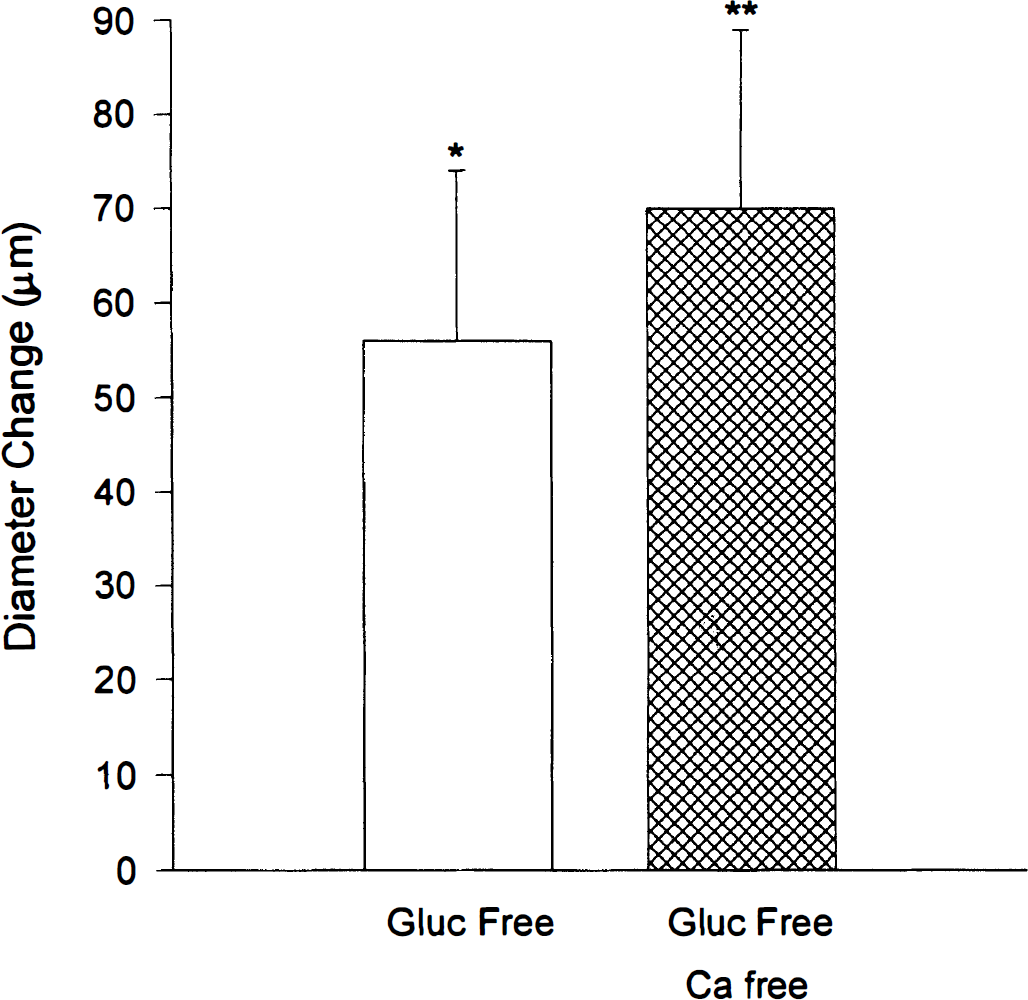
Change in diameter (μm, mean ± SD) of rat middle cerebral arteries (MCA) when the glucose concentration was changed from 5.5 mmol/L to 0 mmol/L and after removal of Ca+2. The resting diameter of the MCA was 251 ± 14 μm (n = 6). *P < .05 compared to diameter at 5.5 mmol/L glucose, **P < .05 compared to the diameter change in 0 mmol/L glucose (glucose free).
After 1.5 hours in PSS containing 0 mmol/L glucose, the MCA transiently constricted by 11 ± 5 μm (n = 7) when 10−6 mol/L serotonin was added in the extraluminal PSS (data not shown). This constriction was significantly different when compared to the 46 ± 7 μm (n = 6) decrease in diameter of MCA at 5.5 mmol/L glucose.
We tested the hypothesis that activation of ATP-dependent K channels (KATP) was responsible for the dilation in the MCA when the glucose was reduced to 0 mmol/L. Figure 3 shows the change in mean diameter produced by changing the glucose concentration in the PSS from 5.5 mmol/L to 0 mmol/L in two groups of MCA: one exposed to 10−5 mol/L glibenclamide, an inhibitor of the KATP channels, and the other, a control group, receiving only the DMSO vehicle. The diameters before addition of any drug or change in glucose concentration were 245 ± 39 μm (n = 6) for the DMSO controls and 253 ± 40 μm (n = 7) for the glibenclamide group. The presence of glibenclamide did not appear to affect either the rate of dilation or the maximum dilation.
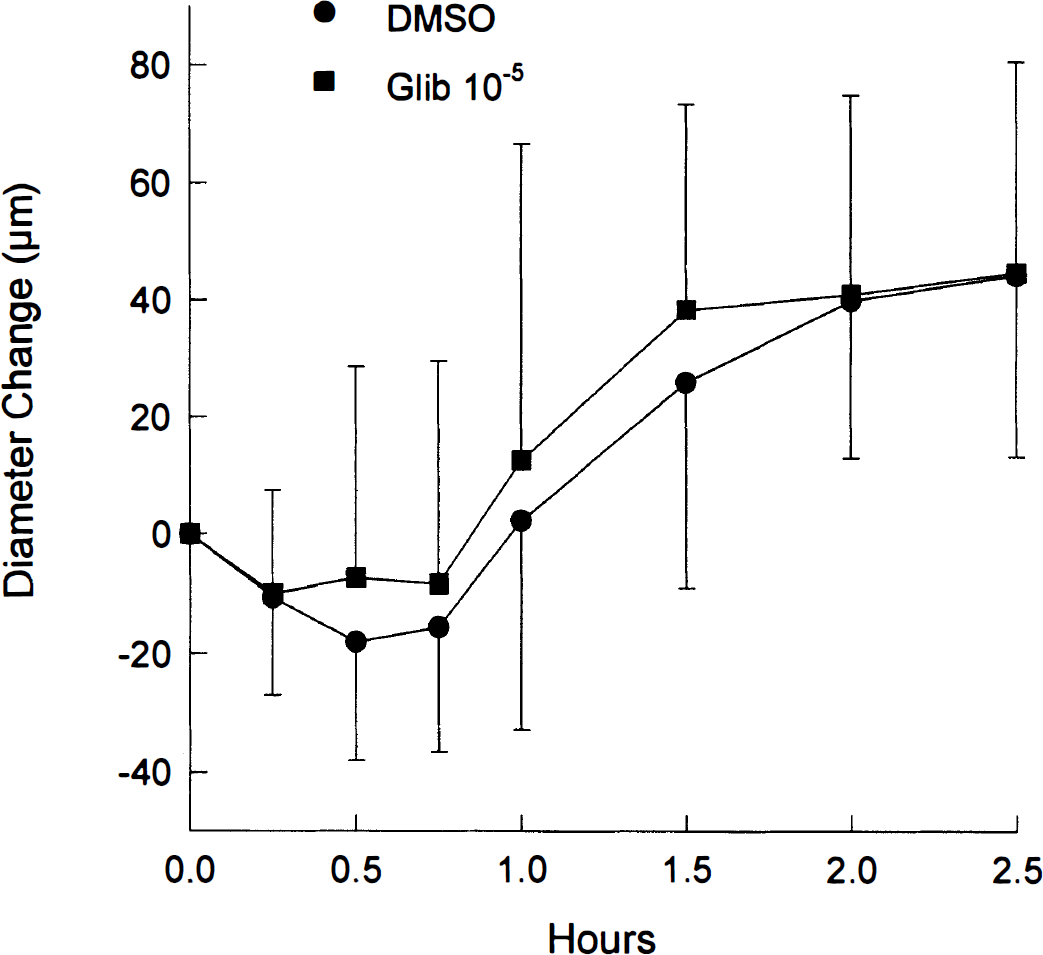
The change in diameter (mean ± SD) over time after the glucose concentration was changed to 0 mmol/L from 5.5 mmol/L in middle cerebral arteries treated with glibenclamide (glib, 10−5 mol/L) or vehicle (dimethyl sulfoxide).
To determine the efficacy of glibenclamide, we tested its ability to block a dilation produced by 20 pg/mL Iloprost, a stable prostacyclin analogue that dilates by activating the KATP channels (Fredricks et al., 1994). The increase in diameter produced by Iloprost (21 ± 6 μm, n = 4) was significantly reduced in the presence of 10−5 mol/L glibenclamide (4 ± 2, P < .001). This study indicates that we effectively blocked the KATP channels with glibenclamide.
DISCUSSION
Our results support the idea that the rat MCA dilates in response to a decrease in the glucose concentration; however, it is relatively resistant to glucose limitation compared to the brain as a whole. During hypoglycemia, glucose becomes limiting as a substrate in the brain and cerebral blood flow increases at plasma glucose concentrations between 2 and 2.5 mmol/L (Horinaka et al., 1997a; Bryan, Jr. et al., 1986; Bryan, Jr. et al., 1987). In the isolated MCA, we had to completely remove all glucose from the bathing solutions for approximately 1 hour before the vessel dilated. Because our incremental decrease of glucose in the bathing solution was from 0.5 mmol/L to 0 mmol/L, we conclude that a glucose concentration somewhere less than 0.5 mmol/L must be achieved before MCA tone is affected.
It is tempting to speculate that direct effects of glucose deprivation on the cerebral vasculature are not responsible for the increase in CBF during hypoglycemia because increases in CBF in the rat occurs at a much greater glucose concentration (2–2.5 mmol/L) than is required to dilate MCA (0.0 to 0.5 mmol/L). However, it is possible that other arteries and/or arterioles are not as resistant to the glucose reduction as found in the MCA. CBF is regulated by the resistances of all segments in the vascular tree. To definitively conclude the above, the direct effects of glucose deprivation in smaller arteries and arterioles in the brain must be determined.
The dilation to the reduction in glucose was similar in MCA with and without endothelium (Fig. 1). Therefore, the dilation must have been strictly a smooth muscle response without an involvement of the endothelium or endothelium-derived relaxing factors.
On dilation and plateau, the MCA were near but had not reached their maximal dilation (defined as the dilation produced by removal of calcium; Fig. 2). After the vasodilatory plateau, the constrictor response to serotonin was diminished, but nevertheless present, indicating that the mechanism for constriction was at least partially intact.
Our results indicate that the dilation was not due to activation of ATP-sensitive potassium channels since the presence of glibenclamide, an inhibitor of the channel, had no effect on the magnitude or rate of dilation during glucose deprivation (Fig. 3). We were initially surprised by this finding because it might be expected that ATP would decrease in the MCA over the duration of glucose deprivation in our study. However, it is possible that the maintenance of smooth muscle ATP after substrate depletion in the MCA is a major contributing factor for the lack of involvement of the KATP channels.
A recent study showed that KATP channels were involved with the increase in CBF when plasma glucose decreased to around 2 mmol/L during insulin-induced hypoglycemia in the rat; activation of these channels was likely due to the stimulation of purinoceptors by adenosine (Horinaka et al., 1997b). At first glance, this may seem to contradict our results; however, it must be remembered that our isolated vessel preparation did not include the parenchymal tissues. We speculate that adenosine released from nonvascular cells (neurons or glia) during insulin-induced hypoglycemia stimulated P1 purinoceptors on vascular smooth muscle to open the KATP channels. Without the parenchymal tissue, as is the case for the isolated MCA, there is no source for adenosine and, thus, no dilation due to adenosine.
Cerebral arteries and blood vessels in general are surprisingly resistant to substrate depletion. Data supporting this contention are as follows: (1) At least an hour of total substrate depletion was necessary for a dilation to occur (present study). (2) Even after 30 minutes of substrate depletion in bovine MCA, there was no significant change in the constrictor response to serotonin (Vinall and Simeone, 1986). (3) Suffusion of glucose-free artificial CSF over the pial surface did not alter the diameter of the pial vessels or affect the response to several dilators (Mayhan and Patel, 1995). (4) In the rabbit aorta, the response to a number of constrictor agents was diminished by 30% or less after 2 hours of substrate deprivation (Altura and Altura, 1970). At 10 hours of substrate deprivation, the constrictor responses to epinephrine and potassium were still present. In another study, 3 hours of glucose deprivation in rabbit aorta did not affect the constrictor response to physiological concentrations of epinephrine (Coe et al., 1968). (5) In the rat portal vein, substrate deprivation had minimal effects on energy stores; phosphocreatine and ATP were maintained at control levels while glycogen was decreased by 50% after 2 hours of substrate deprivation (Hellstrand et al., 1977).
The fact that viability of the blood vessels can be maintained for relatively long periods after substrate depletion implies that the balance between ATP generation and use is favorable for long-term substrate deprivation. This balance can be accomplished by efficient energy use and/or use of free fatty acids, endogenous substrates, or energy stores for the production of ATP.
In summary we report that rat MCA dilated after 1 hour of total substrate depletion. This dilation involved neither the endothelium nor KATP channels. The rat MCA is relatively resistant to substrate limitation.