
Abstract
After 24-hour middle cerebral artery occlusion (MCAO) in spontaneously hypertensive rats, brain ceramide level increased from baseline reached 595% (ischemic core) and 460% (perifocal/penumbral areas); brain glucosylceramide synthase (GCS) activities in these areas simultaneously decreased by 70% and 50%, respectively. Ten-minute MCAO preconditioning significantly attenuated 24-hour MCAO-induced ceramide accumulation by 40% to 60% in ischemic core and perifocal areas, and GCS activities improved by 60% to 70% in both areas. Thus, potentially toxic levels of brain ceramide induced by MCAO were attenuated to intermediate levels in preconditioned animals; brain GCS activity was relatively preserved. In ischemic tolerance, GCS appears to modulate otherwise high levels of brain ceramide.
Keywords
Brief ischemia (ischemic preconditioning) provides significant protection against subsequent severe ischemia in brain (Barone et al., 1998; Chen and Simon, 1997; Dirnagl et al., 2003; Kirino, 2002; Kitagawa et al., 1990) and heart (Baxter, 1997; Murry et al., 1986).
We have induced tolerance to brain ischemia by preconditioning with proinflammatory cytokines (Nawashiro et al., 1997; Ohtsuki et al., 1996) or bacterial endotoxin (LPS) (Tasaki et al., 1997) and have identified the sphingolipid, ceramide, as a downstream mediator of ischemic tolerance. Although ceramide participates in various cellular stress responses including apoptosis (for review see Ballou et al., 1996; Hannun 1996; Luberto et al., 2002; Pettus et al., 2002) and inflammation (Horton, 1999), our studies show that ceramide has a protective effect in the middle cerebral artery occlusion (MCAO) model in spontaneously hypertensive rats and in the neonatal asphyxia model in rat pups (Chen et al., 2001; Furuya et al., 2001). De novo synthesized ceramide increases after preconditioning of cultured brain cells with hypoxia, oxygen/glucose deprivation, or tumor necrosis factor (Ginis et al., 1999; Liu et al., 2000) and after LPS preconditioning in rats (Zimmermann et al., 2001). In contrast, sphingomyelin hydrolysis during severe ischemia results in very high levels of ceramide that promote brain cell death (Kubota et al., 1996; Nakane et al., 2000).
Ceramide can be converted to glycosphingolipids (major pathway) or to sphingomyelin (via sphingomyelin synthase), or degraded to sphingosine (via deacylation by ceramidases) (Hannun and Luberto 2000; Merrill et al., 1997) to control intracellular levels. Glucosylceramide synthase (GCS), which is inducible by an increase in endogenous ceramide (Abe et al., 1996; Komori et al., 2000), catalyzes glucosylation of ceramide by transfer of uridine diphosphate (UDP)-glucose and produces glucosylceramide (Ichikawa and Hirabayashi, 1998; Radin, 1994). Glucosylation of ceramide by GCS augments cellular resistance to tumor necrosis factor-induced (Liu et al., 1999) and ceramide-induced cell damage (Komori et al., 1999; Liu et al., 2001).
We hypothesized that tolerance induced by ischemic preconditioning is associated with modulation of the ceramide elevation in brain during a later, more severe brain ischemia and that GCS activity plays a role in ischemic preconditioning.
MATERIALS AND METHODS
Animals
Male spontaneously hypertensive rats (Taconic Farms, Germantown, NJ, U.S.A.), weighing 300 to 350 g, were used for their increased sensitivity and decreased variability during focal ischemia (Barone et al., 1992). All animals had food and water ad libitum. Body temperature was maintained at 37°C by heating pad throughout surgery and postsurgery recovery. Procedures on animals and their housing and care followed the Guide for the Care and Use of Laboratory Animals 1996, National Research Council. The Institutional Animal Care and Use Committee of GlaxoSmithKline Pharmaceuticals approved all animal procedures.
Focal ischemia and brain tissue sampling
Animals were anesthetized with pentobarbital (65 mg/kg, intraperitoneal) and underwent right MCAO as described previously (Barone et al., 1992, 1998). A 10-minute temporary MCAO was performed 24 hours before pMCAO on the basis of previous data in many studies (Barone et al., 1998; Currie et al., 2000; McLaughlin et al., 2003; Purcell et al., 2003; Wang et al., 1998) showing that 10 minutes of temporary MCAO produced no brain injury, but reduced the response to ischemic injury 24 hours later (e.g., from approximately 115 mm3 to approximately 48 mm3 infarct size).
Ceramide studies
Lipids were extracted from brain sections with 3 mL MeOH: CHCl3 (2:1) and 0.7 mL alkaline water (0.8-mmol/L NH4OH, pH 8–10). Samples were centrifuged (2,000 g, 10 minutes); organic phase was collected and dried under nitrogen. The lipids were dissolved in 1 mL of CHCl3 and stored at −70°C until use. Ceramide levels were measured by reversed-phase high-pressure liquid chromatography as described previously (Zimmermann et al., 2001). Ceramide values were normalized per nanomole lipid phosphate (LP).
GCS assay
Brain sections were minced and homogenized in ice-cold 0.25-mol/L sucrose solution (50-mmol/L Tris-HCl, pH 7.4; 25-mmol/L KCl) with protease inhibitors (Complete, 1T/10 mL; Roche, Indianapolis, IN, U.S.A.) and 1-mmol/L dithiothreitol. After sonication (3 × 10-second pulse, 50% efficiency), a detergent solution (0.1% Triton X-100, 150-mmol/L NaCl, 0.5-mmol/L edetic acid) was added to the homogenate. One hour later, the homogenate was centrifuged for 30 minutes at 14,000 g, and the supernatant was collected and stored at −70°C until use. Protein levels of the lysates were measured by Bradford method. The enzyme assay was performed by a modified method of Marks et al. (2000). GCS activity was determined as a conversion ratio (% conversion) from C6-NBD-ceramide to C6-NBD-glucosylceramide.
Statistical analysis
Data are expressed as mean ± SD. Statistical analysis was by analysis of variance followed by Fisher PLSD test.
RESULTS
Time course of ceramide accumulation after pMCAO
Brain ceramide levels increased after pMCAO in the ipsilateral hemisphere (core: 4.3 ± 0.3 [baseline], 10.1 ± 4.1 [4 hours], 11.4 ± 3.0 [6 hours], 25.6 ± 9.3 [24 hours]; perifocal/penumbral: 6.8 ± 2.4 [4 hours], 7.8 ± 1.5 [6 hours], 19.8 ± 5.3 [24 hours]; pmol/nmol LP, n = 3–5). There were no significant changes in contralateral hemisphere ceramide levels.
Effects of ischemic preconditioning on ceramide accumulation after pMCAO
In the ipsilateral hemisphere, ischemic preconditioning for 10 minutes performed 24 hours before pMCAO decreased brain ceramide levels from those of non-preconditioned animals to 10.6 ± 2.7 and 11.6 ± 5.7 pmol/nmol LP (P < 0.05) in ischemic core and perifocal areas, respectively (Fig. 1). Brain ceramide levels in the contralateral hemisphere were unaffected by the preconditioning (Fig. 1).
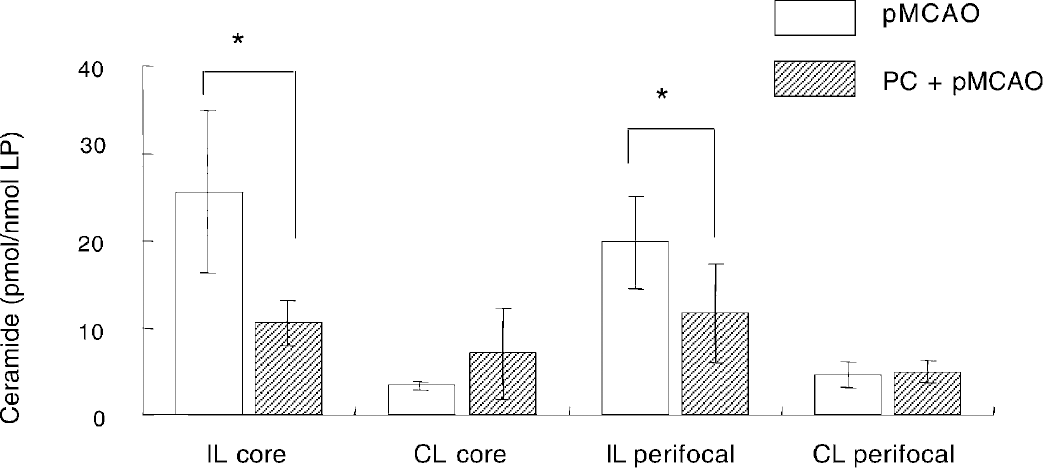
Brain ceramide level in ipsilateral (IL) core, contralateral (CL) core, IL perifocal/penumbral, and CL perifocal/penumbral areas at 24 hours after pMCAO. The ischemic preconditioning for 10 minutes was performed 24 hours before pMCAO. Results are means ± SD of five animals in each group. *P < 0.05 compared with pMCAO group (Fisher PLSD test).
Effects of p MCAO and ischemic preconditioning on GCS activity
In nine animals that were not exposed to ischemia, baseline control activity expressed as conversion ratio (% conversion) from C6-NBD-ceramide to C6-NBD-glucosylceramide was 4.8% ± 0.5%. In the ipsilateral hemisphere, brain GCS activities in ischemic core and perifocal areas were decreased to 32% (from 4.7% ± 1.5% to 1.5% ± 1.2% conversion; P < 0.05) and 48% (from 4.2% ± 0.9% to 2.0% ± 1.3% conversion; P < 0.05), respectively, of the corresponding values in the contralateral hemisphere 24 hours after pMCAO (Fig. 2). In the ipsilateral hemisphere, ischemic preconditioning for 10 minutes performed 24 hours before pMCAO elicited a trend toward increased brain GCS activities from 1.5% ± 1.2% to 2.7% ± 1.3% conversion; (P = 0.14) in the ischemic core, and induced a significant increase of GCS activity from 2.0% ± 1.3% to 4.0% ± 0.9% conversion; (P < 0.05) in perifocal areas (Fig. 2). Brain GCS activities in the nonischemic contralateral hemisphere of preconditioned animals were not significantly different than corresponding activities in nonpreconditioned animals.
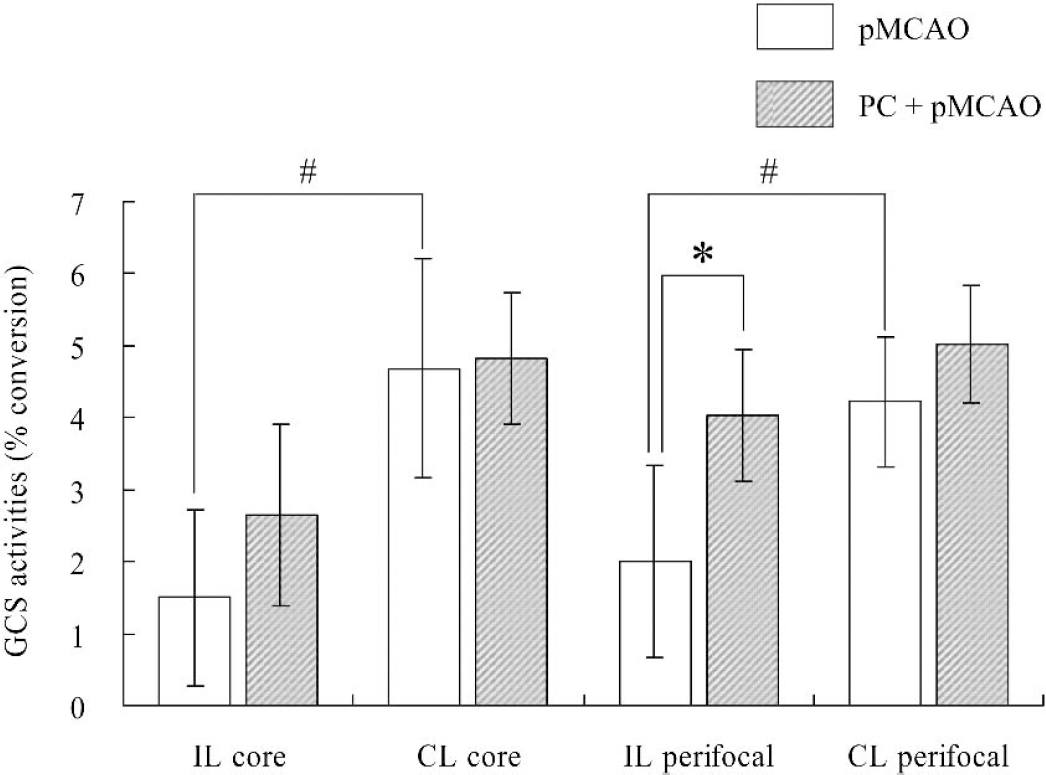
Brain GCS activity in ipsilateral (IL) core, contralateral (CL) core, IL perifocal/penumbral, and CL perifocal/penumbral areas at 24 hours after pMCAO. The preconditioning for 10 minutes was performed 24 hours before pMCAO. Results are means ± SD of four or five animals in each group. #P < 0.05 compared with each corresponding value in CL hemisphere (Fisher PLSD test); *P < 0.05 compared with pMCAO group (Fisher PLSD test).
Effects of ischemic preconditioning alone on ceramide level and GCS activity
Measurements were performed 6, 18, and 24 hours after the 10-minute ischemic preconditioning. As shown in Table 1, there were no changes in mean brain ceramide levels in any area 6 or 18 hours after ischemic preconditioning, but at 24 hours, a trend toward mild increase in brain ceramide level (P = 0.06) was observed in the ipsilateral ischemic areas (6.4 ± 5.6 pmol/nmol LP and 5.5 ± 3.7 pmol/nmol LP, core and perifocal region, respectively; average overall 5.9 ± 4.5 pmol/nmol LP) compared with those in the corresponding regions contralaterally (2.7 ± 1.2 pmol/nmol LP and 3.9 ± 1.8 pmol/nmol LP, core and perifocal, respectively; average overall 3.3 ± 1.6 pmol/nmol LP). This trend was due to two outliers in the 24-hour sample with ceramide levels that averaged 13.8 pmol/nmol LP. There were no changes in GCS activity of any brain region at any time point after ischemic preconditioning (Table 1).
Effects of preconditioning alone on brain ceramide level and GCS activity
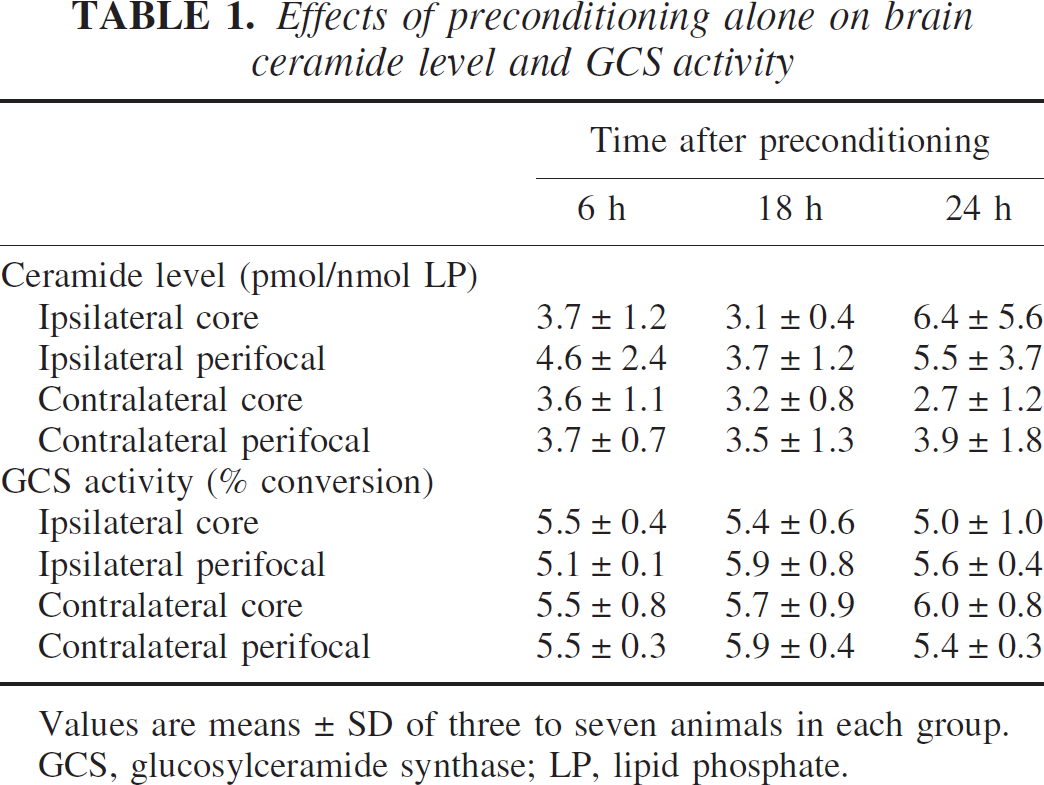
Values are means ± SD of three to seven animals in each group.
GCS, glucosylceramide synthase; LP, lipid phosphate.
DISCUSSION
The cellular level of ceramide strongly influences its neuroprotective or neurodegenerative effects. In general, low concentrations of ceramide promote cell survival in hippocampal (Goodman and Mattson, 1996; Mitoma et al., 1998), cerebellar Purkinje (Furuya et al., 1998), and sympathetic neurons (Ito and Horigome, 1995), whereas higher concentrations of ceramide induce cell death in hippocampal (Mitoma et al., 1998), cortical (Willaime et al., 2001) and mesencephalic neurons (Brugg et al., 1996).
In our model of focal ischemia and focal preconditioning with well-defined areas of infarction core and perifocal/penumbral regions in brains of both intact and preconditioned animals (Barone et al., 1992, 1998), we have measured ceramide levels and found a time-dependent accumulation of ceramide. By 24 hours, ceramide became 6 times higher than preischemic levels in the core of infarction and 4.6 times higher in the surrounding tissue. This agrees with a previous report demonstrating a 4.2-fold increase in ceramide in rat cerebral cortex during cell-damaging focal ischemia (Kubota et al., 1996) and studies in culture systems linking the same order of ceramide elevation to apoptosis in neurons and other brain cells (Goswami and Dawson, 2000; Toman et al., 2002). Ischemic preconditioning resulted in inhibition of ceramide levels by 58% and 41% in ischemic core and perifocal areas, respectively. Ceramide rose in two of seven animals 24 hours after preconditioning. Our failure to show ceramide production in other animals could be explained by its transient elevation as observed in our in vitro studies (Liu et al., 2000).
Ceramide glucosylation by GCS protects cells from ceramide-induced death (Bleicher and Cabot, 2002). Until now, the relationship between GCS activity and brain ischemia has not been directly examined, although an activator of GCS has been used 1 to 7 days after MCAO in rats (Inokuchi et al., 1998; Kubota et al., 2000) with beneficial effects. Although preconditioning-induced preservation of GCS activity accords with the observed modulation of postischemic ceramide increase, participation of other mechanisms in ceramide control by preconditioning animals has not been ruled out.
We infer that regulation of ceramide levels to permit only a modest rise associated with relative preservation of GCS activity constitutes one aspect of ischemic tolerance in brain.
Footnotes
Acknowledgements
The authors thank Dr. David Marks (Mayo Clinic, Rochester, MN, U.S.A.) for his helpful advice related to these studies.