
Abstract
The contribution of leukocyte infiltration to brain damage after permanent focal cerebral ischemia and the underlying molecular mechanisms are still unclear. Therefore, the aim of this study was to establish a mouse model for the visualization of leukocytes in the cerebral microcirculation in vivo and to investigate leukocyte-endothelial interaction (LEI) after permanent middle cerebral artery occlusion (MCAO). Sham-operated 129/Sv mice showed physiologic LEI in pial venules as observed by intravital fluorescent microscopy. Permanent focal cerebral ischemia induced a significant increase of LEI predominantly in pial venules. The number of rolling and adherent leukocytes reached 36.5 ± 13.2/100 μm × min and 22.5 ± 7.9/100 μm × min, respectively at 120 minutes after MCAO (P = 0.016 vs. control). Of note, rolling and adherent leukocytes were also observed in arterioles of ischemic animals (7.3 ± 3.0/100 μm × min rolling and 3.0 ± 3.6/100 μm × min adherent). Capillary density was not different between groups. These results demonstrate that leukocytes accumulate in the brain not only after transient but also after permanent focal cerebral ischemia and may therefore contribute to brain damage after stroke without reperfusion.
The activation of leukocytes during ischemia is a pathologic phenomenon that occurs in various peripheral organs (Engler et al., 1983; Menger et al., 1992; Vollmar et al., 1994), as well as in the brain (del Zoppo et al., 1991; Kochanek and Hallenbeck, 1992). Leukocyte infiltration has been well documented after cerebral ischemia in experimental animals (Barone et al., 1992) and in humans (Akopov et al., 1996). Neutrophil accumulation in stroke patients has been associated with poor clinical outcome (Pozzilli et al., 1985).
Leukocyte adhesion and extravasation are mediated by adhesion molecules expressed on leukocytes and endothelial cells. Intercellular cell adhesion molecule-1 (ICAM-1) is a cell surface glycoprotein expressed on vascular endothelium that plays an important role for the firm adherence of leukocytes (Rothlein et al., 1986; Smith et al., 1988). Macrophage-1 antigen (Mac-1; CD11b/CD18), a β2 integrin, is the couterligand of ICAM-1 expressed on the surface of leukocytes (Diamond and Springer, 1993). Administration of neutralizing antibodies to these molecules decreased infarct volume in models of transient focal ischemia (Chopp et al., 1994; Zhang et al., 1995), and both ICAM-1 (Connolly et al., 1996) and Mac-1 deficient mice (Soriano et al., 1999) showed less susceptibility to tissue injury after focal cerebral ischemia and reperfusion. These data suggest that neutrophil accumulation mediates brain damage after cerebral ischemia. Although this hypothesis is supported by histopathologic studies and investigations on antileukocyte treatment, only very few studies directly analyzing the kinetic of leukocyte-endothelial interaction (LEI) in vivo have been published to date (Dirnagl et al., 1994; Ishikawa et al., 1999, 2003; Ritter et al., 2000; Santizo and Pelligrino, 1999; Uhl et al., 2000). Most of these studies have been conducted in rats (Dirnagl et al., 1994; Ishikawa et al., 1999; Ritter et al., 2000; Santizo and Pelligrino, 1999) or gerbils (Uhl et al., 2000). However, for molecularly based investigations, a mouse model suitable for the use of transgenic and knockout mice would be more desirable. Moreover, all previous studies on LEI focused upon the reperfusion period after focal (Ishikawa et al., 1999; Ritter et al., 2000; Santizo and Pelligrino, 1999) or global ischemia (Dirnagl et al., 1994; Ishikawa et al., 2003; Uhl et al., 2000). With regard to permanent focal cerebral ischemia, the significance of leukocyte activation and adhesion remains to be discovered.
Therefore, the purpose of this study was (1) to establish an in vivo model for the direct observation of the cerebral microcirculation after focal cerebral ischemia in mice, and (2) to investigate LEI after permanent focal cerebral ischemia.
MATERIALS AND METHODS
Animals
Male 129/SvPasIcoCrlBR mice (25–30 g body weight) were used in this study. The animals had free access to tap water and pellet food. Animal experiments were conducted in accordance with institutional guidelines approved by the government of Upper Bavaria.
Model of focal cerebral ischemia
Animals were anesthetized with an intraperitoneal injection of Medetomidin (0.5 mg/kg b.w., Domitor®, Dr. E. Graeub AG, Basel, Switzerland), Fentanyl (0.05 mg/kg b.w., Janseen-Cilag, Neuss, Germany), and Midazolam (5 mg/kg b.w., Dormicum®, Roche, Basel, Switzerland), intubated, and mechanically ventilated during the whole experiment. Anesthesia was maintained for up to 4 hours by hourly injections of one third of the dose necessary for initial anesthesia induction. Body temperature was maintained at 37.0 ± 0.1°C with a temperature control unit (FHC, Bowdoinham, Maine, U.S.A.). The left femoral artery was cannulated for blood pressure monitoring and blood gas analysis, and the left femoral vein was cannulated for injection of fluorescent dyes. Middle cerebral artery occlusion (MCAO) was induced by the intraluminal filament method as described previously (Plesnila et al., 2001). Briefly, the left common carotid artery (CCA) was exposed through a midline neck incision. A silicone-coated 8-0 monofilament was inserted into the internal carotid artery (ICA) and advanced towards the origin of the middle cerebral artery (MCA) until occlusion occurred. MCA occlusion was monitored by Laser-Doppler fluxmetry (Perimed, Stockholm, Sweden). Sham-operated control animals underwent the same surgical procedure, except that the filament was not advanced into the ICA.
Intravital microscopy
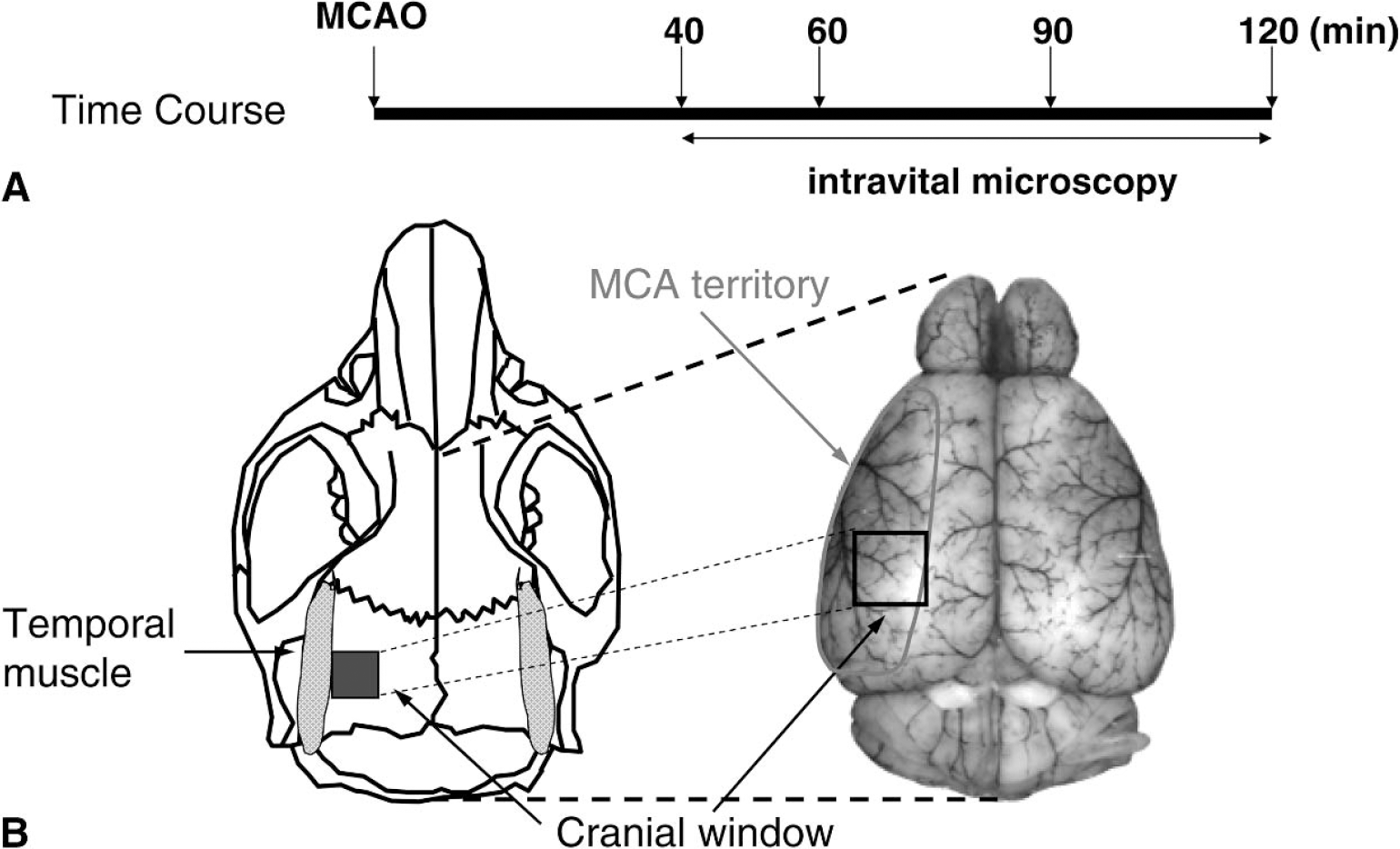
For intravital microscopy, the animals were placed on a computer-controlled microscope stage for repeated analysis of identical vessel segments. A rectangular 2 × 2-mm cranial window was made over the left parietal cortex, leaving the dura mater intact so that the window contained some terminal branches of the middle cerebral artery (Fig. 1B). In the coronal direction, the cranial window extended from the attachment of the temporal muscle to the midpoint between the sagittal suture and the attachment of the temporal muscle. In the sagittal direction, the window was aligned to the middle of the sagittal suture. The exposed dura matter was rinsed continuously with isotonic 0.9% saline at 37.0°C. The intravital microscope (Leitz, Wetzlar, Germany) was equipped with a 75-W xenon lamp and a Ploemo-Pack filter block for epi-illumination. Visualization of microvessels was achieved by an intravenous injection of fluorescein isothiocyanate-labeled dextran (FITC-dextran; molecular weight 150,000) (Sigma Chemical, St. Louis, Missouri, U.S.A.) before the first measurement (0.1 mL of a 0.5% solution). Leukocytes were stained in vivo before each measurement by intravenous injection of 0.05 mL of a 0.01% solution of the fluorescent dye Rhodamine 6G (Merck, Darmstadt, Germany). Leukocyte-endothelium interactions and vessel diameters were observed with a salt water immersion objective (W25 × /0.6 Leitz, Leica Microsystems, Wetzlar, Germany) using the Leitz/Leica + N3 (excitation: BP 546 ± 12 nm; emission: BP 600 ± 40 nm) and the Leitz/Leica I3 filter block (excitation: BP 470 ± 20 nm; emission: LP > 515 nm), respectively. Intravital microscopic images were recorded by a silicone intensified tube (SIT) video camera (C2400, Hamamatsu Photonics, Herrsching, Germany) and recorded on videotapes (MQSE-120, Sony, Tokyo, Japan).
Analysis of microcirculatory parameters
Quantification of intravital microscopic images was performed off-line by frame-to-frame analysis at a total magnification of 625× using a computer-assisted microcirculation analysis system (Capimage; Ingenieurbüro Dr. Zeintl, Heidelberg, Germany). Arteriolar and venular diameters, the number of rolling and adherent leukocytes in venules and arterioles, and the capillary density were analyzed. Rolling leukocytes were defined by their multiple intermittent contacts with the vascular endothelium, thereby moving significantly slower than the freely moving leukocytes in the center flow of the vessel. Adherent leukocytes were defined by their attachment to the vascular wall for more than 30 seconds. One vessel segment of 100 μm in length for each region of interest was studied for 30 seconds. The capillary density, defined as capillaries perfused with plasma tracer and red blood cells (capillary length/area), was also quantified by the Capimage system.
Experimental design
Five animals per group were randomly assigned to the sham-operated or the ischemic group. Immediately after MCA occlusion, the animals were immobilized in a stereotactic frame (Stoelting, Wood Dale, Illinois, U.S.A.), and the cranial window was prepared. The animals were transferred to the intra-vital microscope, and six regions of interest (ROI) were selected: two containing at least one pial arteriole each, two containing at least one pial venule each, and two containing capillaries. Accordingly, a total of 10 pial venules and 10 pial arterioles were examined in each group. After MCA occlusion (40, 60, 90, and 120 minutes), each ROI was observed for 40 seconds by intravital microscopy (Fig. 1A): 30 seconds using the +N3 filter block for visualization of Rhodamine 6G labeled leukocytes and 10 seconds using the I3 filter block for visualization of the FITC-Dextran labeled vessel lumen.
Statistical analysis
The Mann-Whitney U-test was used for the analysis of differences between groups. Friedman one-way analysis of variance on ranks followed by the Student-Newman-Keuls test was used for analyzing differences over time. Data are presented as means ± standard deviation (SD). A statistically significant difference was assumed at P < 0.05.
RESULTS
Physiologic parameters
In the present study, we investigated the interaction of leukocytes with the vascular endothelium during permanent focal cerebral ischemia in mice by direct observation of the cerebral microcirculation by intravital microscopy. As indicated in Table 1, key physiologic parameters of animals in the ischemic group were maintained within the physiologic range and did not show any differences from those in sham-operated animals during the whole experiment.
Phisological parameters
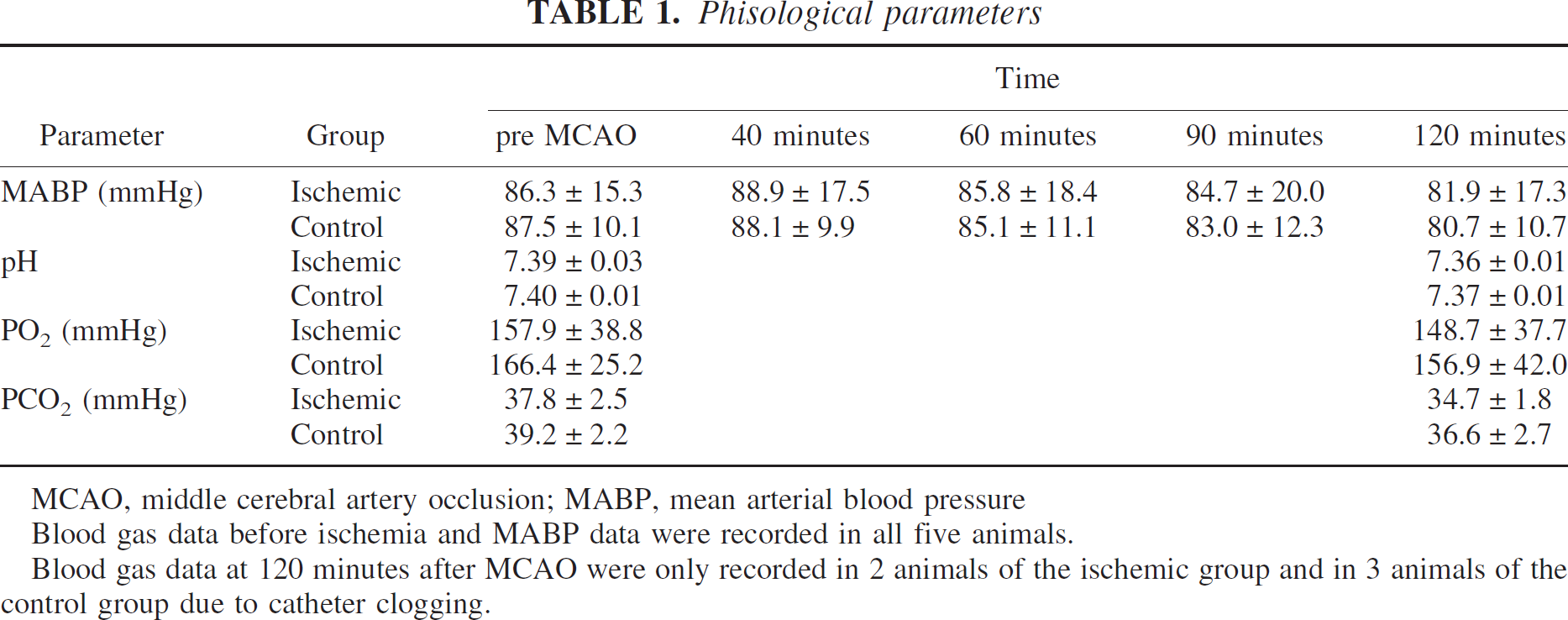
MCAO, middle cerebral artery occlusion; MABP, mean arterial blood pressure
Blood gas data before ischemia and MABP data were recorded in all five animals.
Blood gas data at 120 minutes after MCAO were only recorded in 2 animals of the ischemic group and in 3 animals of the control group due to catheter clogging.
Regional cerebral blood flow
To document permanent occlusion of the MCA during the whole experiment, regional cerebral blood flow (rCBF) was recorded over the MCA territory by laser Doppler fluxmetry (Fig. 2). In the ischemic group, rCBF decreased immediately after MCA occlusion to 11.5 ± 4.5% of baseline and remained at this level until the end of the observation period. The decrease of rCBF was confirmed by intravital microscopy. Direct visualization of pial arterioles demonstrated low flow conditions or even retrograde flow in all investigated vessel segments. It is interesting to note that no microvascular stasis was observed in any vessel during the entire duration of the experiment.
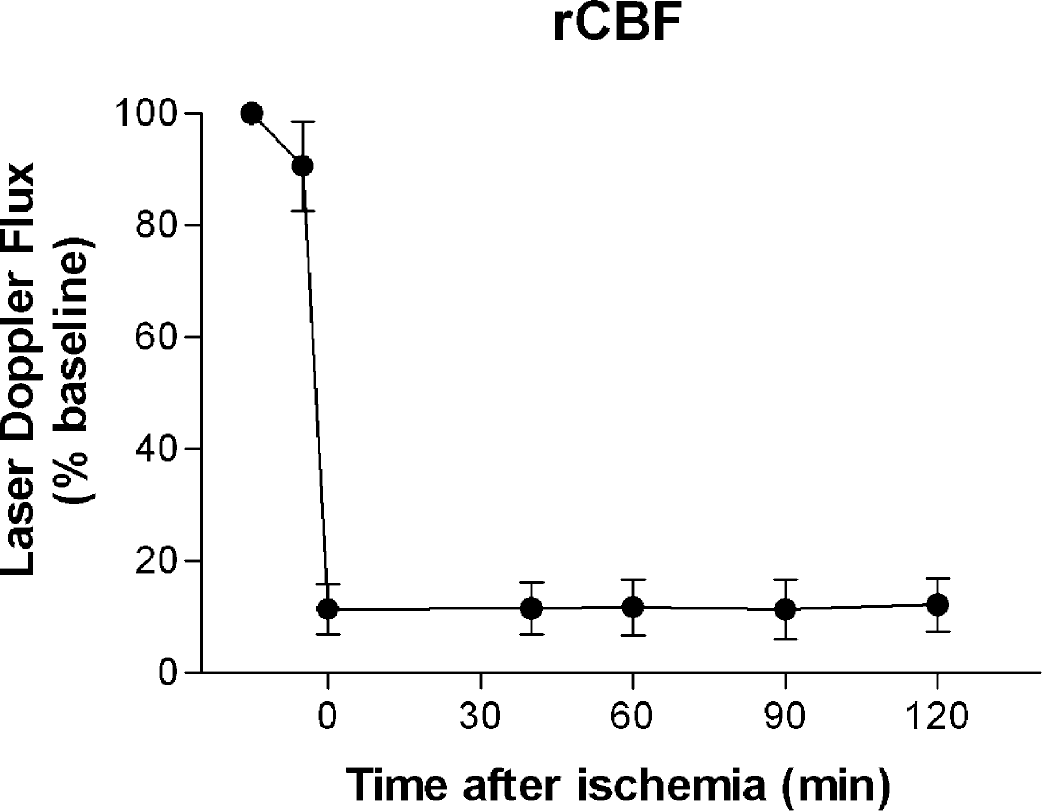
rCBF in percent of baseline assessed by laser Doppler flowmetry before and after MCAO. Data are presented as mean ± SD. n = 5. Immediately after MCAO, the relative value of rCBF decreased to 11 ± 5% of baseline. rCBF did not change significantly during ischemia. rCBF, regional cerebral blood flow; MCAO, middle cerebral artery occlusion.
Vessel diameters
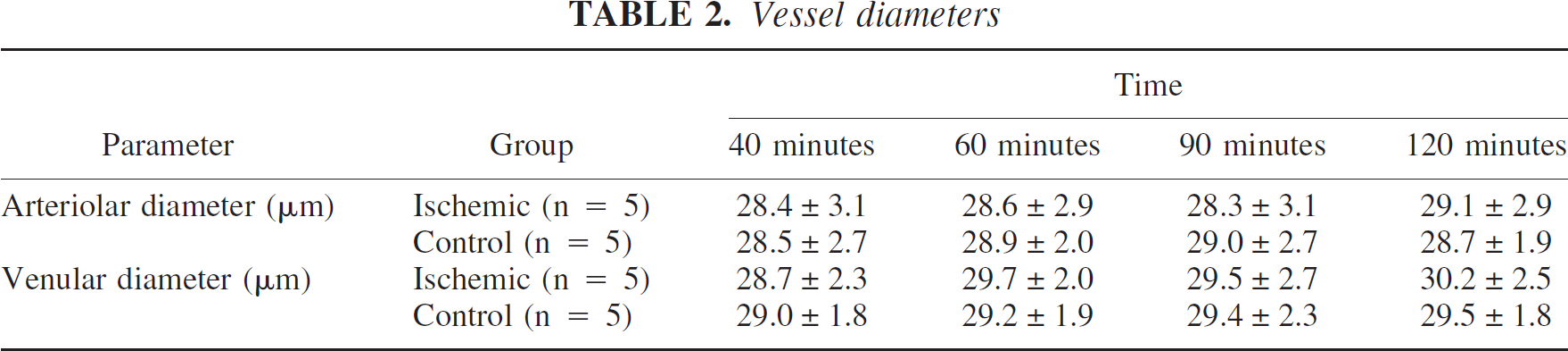
Although the venular and arteriolar diameters slightly increased during the observation period in the ischemic group, there is no statistically significant difference over time (Table 2). In the sham-operated group, the venular and arteriolar diameters remained unchanged during the whole observation period. There was no significant difference between the ischemic and the sham operated group.
Vessel diameters
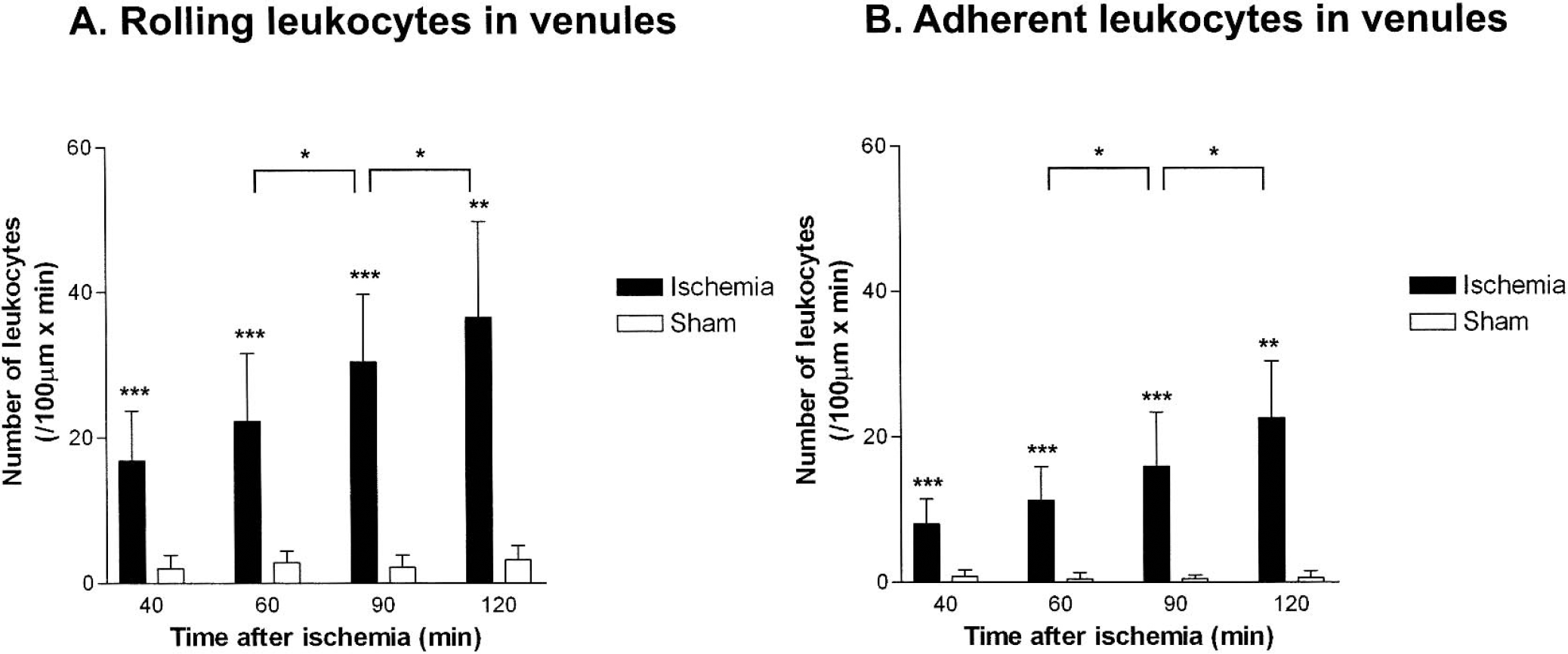
Leukocytes-endothelium interaction in venules
In ischemic animals, the number of rolling and adherent leukocytes increased continuously during MCAO (Figs. 3 and 4). Forty minutes after induction of MCAO, the number of rolling and adherent leukocytes (expressed as number of cells per 100 μm × minute) increased eightfold as compared to sham-operated control animals (rolling leukocytes: 16.8 ± 6.8 vs. 2.0 ± 1.9, P = 0.008; adherent leukocytes: 8.0 ± 3.4 vs. 0.8 ± 0.8, P = 0.008). Both parameters increased significantly over time in ischemic animals, whereas no significant increase of LEI was detected in sham-operated animals. In ischemic animals, both parameters more than doubled between 40 and 120 minutes after ischemia and reached their maximum at the end of the observation period, 120 minutes after the beginning of MCAO (rolling leukocytes: 36.5 ± 13.2 vs. 3.2 ± 1.9, P = 0.016; adherent leukocytes: 22.5 ± 7.9 vs. 0.6 ± 0.9, P = 0.016).
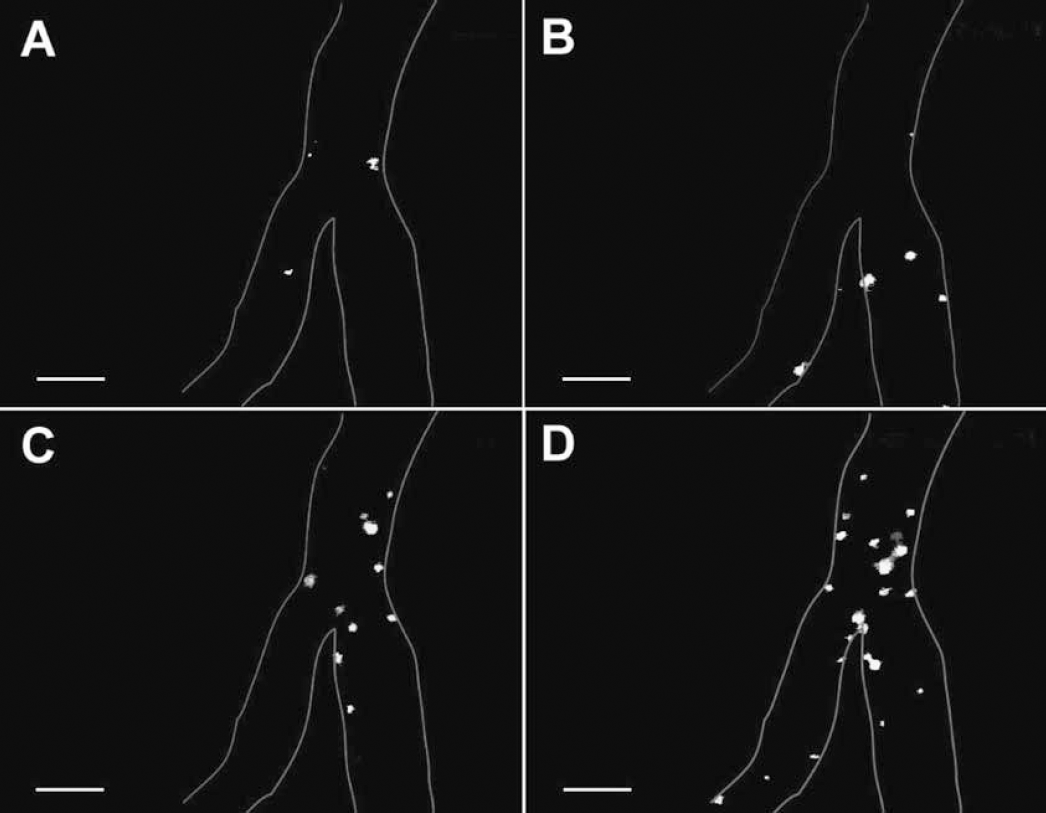
Representative images of rhodamine-labeled leukocytes in pial venules as observed by intravital microscopy 40
Leukocytes-endothelium interaction in arterioles
Although LEI was mainly observed in venules, some rolling and adherent leukocytes were also detected in arterioles of ischemic animals (Fig. 5). Forty minutes after the beginning of MCAO, the number of rolling leukocytes in arterioles increased 25-fold as compared to sham-operated control animals (5.0 ± 2.1 vs. 0.2 ± 0.5, P = 0.008). However, in contrast to venules, in arterioles the number of rolling leukocytes did not increase over time.
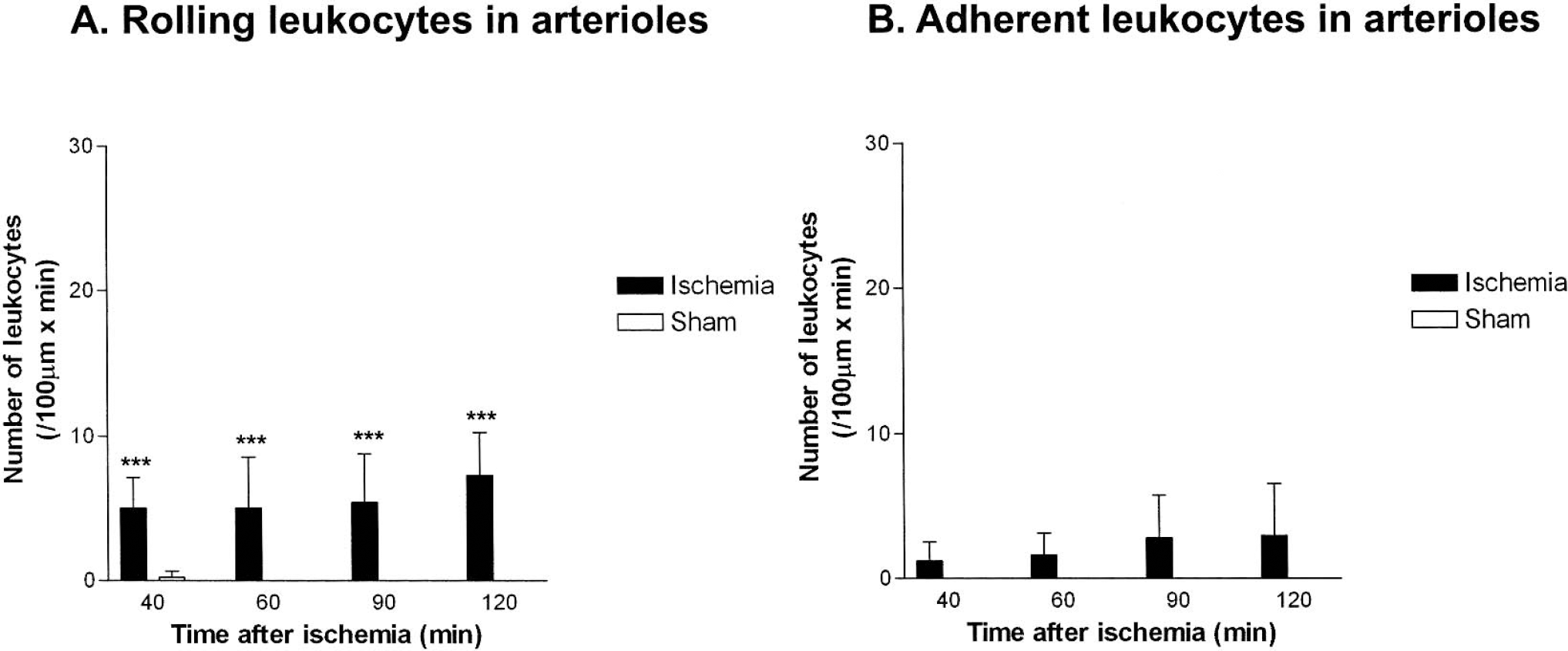
Leukocytes adherent to the cerebral endothelium were observed only in ischemic animals (Fig. 5). In contrast to venules and in parallel to the number of rolling leukocytes in arterioles, the number of adherent leukocytes also remained constant during the entire observation period. No adherent leukocytes were detected in sham-operated control animals.
Capillary density
Nine regions of interest were examined in each group. Capillary density remained unchanged throughout the observation period in every single investigated region of interest and in both groups as a total (Fig. 6). Capillary density ranged between 127 ± 23 and 131 ± 22 cm/cm2 in the sham-operated group and between 130 ± 19 and 133 ± 19 cm/cm2 in the ischemic group (Fig. 6C), which are values found in normally perfused mouse brain (data not shown). Although severe ischemia was present in the investigated cerebral cortex during the whole time of intravital microscopy, all capillaries remained perfused with plasma and with red blood cells. The previously reported phenomenon of “capillary plugging” (del Zoppo et al., 1991; Grogaard et al., 1989; Hallenbeck et al., 1986) was not observed.
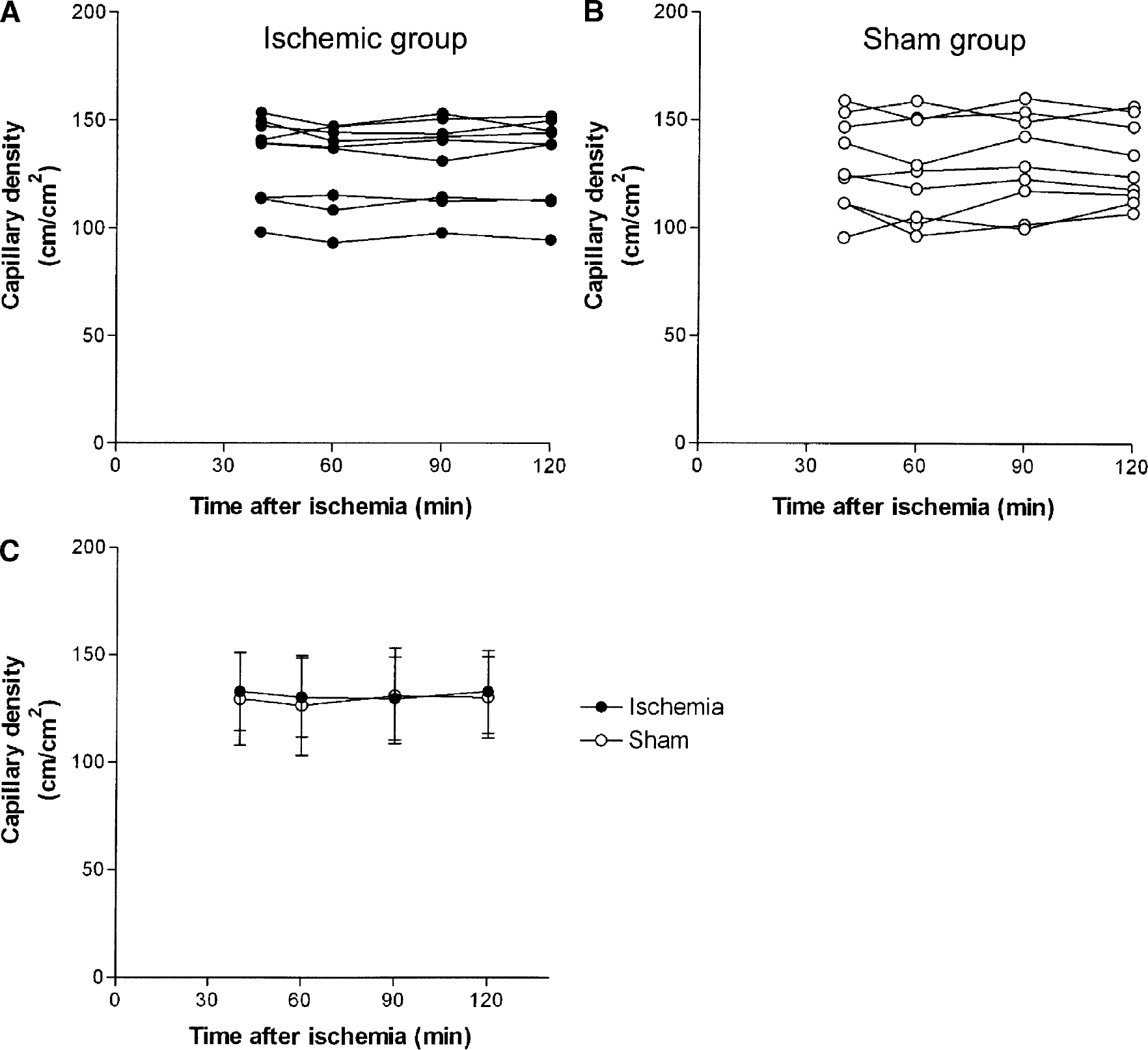
Capillary density after permanent MCAO
DISCUSSION
Experimental model
In this study, we describe for the first time to our knowledge a model for the intravital microscopic analysis of leukocyte-endothelial interactions (LEI) after focal cerebral ischemia in mice. Only a few intravital microscopic studies on LEIs after cerebral ischemia have been reported to date, most of them using a closed cranial window preparation, which is technically demanding (Dirnagl et al., 1994). This technique requires opening of the dura and irrigation of the brain surface with artificial cerebrospinal fluid. Compared with this method, the transdural window technique, previously used in our laboratory after global cerebral ischemia in gerbils (Uhl et al., 2000), is simple and unlikely to disturb the environment around microvessels and, thus, the microcirculation. Recently, Ishikawa and colleagues (2003) reported a model for the visualization of LEIs by intravital microscopy after global cerebral ischemia in mice, thereby demonstrating that the transdural window technique is also feasible in this species. We were able to confirm these findings using the transdural window technique for intravital microscopy after focal cerebral ischemia. In our model, microcirculatory parameters and animal physiology remained stable (Tables 1 and 2). In mice, the dura mater is translucent so that the cerebral microcirculation can be observed with excellent optical quality down to the capillary level. Intravital microscopy in sham-operated animals demonstrates that removal of the skull bone, irrigation of the dura mater with normal saline, and repeated illumination of the brain does not damage the brain surface (data not shown) and does not induce pathologic LEI in venules or arterioles (Figs. 4 and 5). As reported previously (Uhl et al., 1999), only physiologic leukocyte rolling was observed in pial venules of these animals (Fig. 4A). Because a variety of molecular biological tools for the study of LEI became available recently, such as transgenic mice (Bullard et al., 1995; Mayadas et al., 1993), the currently established intravital microscopy model offers multiple options for the identification of molecular mechanisms associated with LEIs after cerebral ischemia.
A general and inherent disadvantage of intravital microscopy of the brain is that the observation of vessels is limited to the pial surface. Therefore, changes observed in pial vessels may not necessarily be representative for the whole brain. This is particularly true for the situation after middle cerebral artery occlusion where deep brain areas (e.g., the striatum), which represent the core of the infarct, have completely different blood flow characteristics as compared with the pial territory that is superficial to the ischemic core and has the advantage of a robust collateralization. Therefore, it should be taken into consideration that there may be differences in responses of inflammatory cells to ischemia between the pial vasculature and vessels in deep brain structures. However, this may be of minor importance for the current study, which has the main goal to investigate penumbral tissue, which is also superficial and therefore accessible by intravital microscopy.
Leukocyte-endothelium interaction after permanent focal cerebral ischemia in venules
The current study is the first report to our knowledge on in vivo observation of LEIs after permanent focal cerebral ischemia. Leukocytes rolling on or adherent to the vascular endothelium after MCAO was typically observed in venules of the ischemic brain receiving collateral blood flow. Activation of leukocytes was detected already after 40 minutes of MCAO and the number of leukocytes rolling or adhering increased over time. The time course of leukocyte activation was more pronounced than that described during reperfusion following global cerebral ischemia (Ishikawa et al., 2003; Uhl et al., 2000). We assume that the early occurrence of LEIs was caused by a combination of low blood flow velocity and of constitutively expressed cell adhesion molecules, such as ICAM-1 or ICAM-2 (Brayton et al., 1998). In ischemic animals, retrograde flow or antero-grade low flow was observed in terminal branches of MCA. This mechanical factor is considered one main factor to provoke LEIs (Schmid-Schoenbein et al., 1975). The subsequent increase in the number of activated leukocytes is most likely caused by upregulation of cell adhesion molecule expression on the endothelium of cerebral microvessels after ischemia, such as ICAM-1, P-selectin, and E-selectin. It has been shown that P-selectin (Okada et al., 1994), E-selectin (Zhang et al., 1996), and ICAM-1 (Okada et al., 1994; Wang et al., 1994) are expressed in the brain after ischemia. P-selectin expression begins to increase after 1 hour of MCAO (Okada et al., 1994), and ICAM-1 expression increases significantly 3 hours after permanent MCAO (Wang et al., 1994). Therefore, it is possible that upregulation of these molecules contributes to the increase of the number of rolling or adherent leukocytes over time. Future studies using the present model together with transgenic mice may reveal the underlying mechanisms in more detail.
Leukocyte accumulation in microvessels may cause disturbances of cerebral blood flow (del Zoppo et al., 1991). In addition, activated leukocytes could release inflammatory cytokines (Ferrante et al., 1992) and produce oxygen free radicals (Clark et al., 2001), resulting in the further damage of endothelium and neuronal tissue. There is a strong possibility that leukocyte activation after cerebral ischemia contributes to secondary brain damage (Hallenbeck, 1996). However, so far, the pathophysiologic significance of leukocyte accumulation in brain tissue has only been emphasized for ischemia followed by reperfusion. In their review, Härtl and colleagues (1996) concluded that “WBCs clearly contribute to ischemic cell damage in the scenario of experimental focal cerebral ischemia followed by reperfusion, if the ischemic insult is not too severe. However, their role in global ischemia, permanent focal ischemia and severe transient focal ischemia has not yet been clarified.” (Hartl et al, 1996, p. 1114) In models of transient MCAO, several antileukocytic interventions reduced cerebral leukocyte infiltration and infarct size (Chopp et al., 1994). ICAM-1 deficient transgenic mice and Mac-1 deficient transgenic mice showed a reduction of infarct volume accompanied by a reduction of the number of leukocyte infiltration after MCAO and reperfusion (Connolly et al., 1996; Soriano et al., 1999). On the other hand, antileukocytic intervention did not reduce stroke volume after permanent MCAO (Garcia et al., 1996; Zhang et al., 1995), and CD18 deficiency contributed to cerebral protection only in transient ischemia models but not in models of permanent MCAO (Prestigiacomo et al., 1999). However, our current data show leukocyte accumulation in low flow areas adjacent to the ischemic focus after permanent MCA occlusion. Because the evolution of post-ischemic brain damage takes place mainly in low flow areas surrounding the core of the infarct (Obrenovitch, 1995), leukocytes may well exacerbate tissue damage also after permanent ischemia. Clear-cut evidence as to the mechanism of leukocyte-mediated injury to the ischemic brain has not yet been reported, except for one report about a positive correlation between leukocyte accumulation and brain edema formation after focal cerebral ischemia (Matsuo et al., 1994). The currently established model of intravital microscopy after MCAO in mice may thus be highly useful to further clarify this point.
Leukocyte-endothelium interaction after permanent focal cerebral ischemia in arterioles
Although, in most organs, leukocyte rolling and adherence occur mainly in venules, some studies also report LEIs in arterioles, although at a significantly lower level than in venules (Ley and Gaehtgens, 1991; Nazziola and House, 1992; Perry and Granger, 1991). Similar observations were reported in the brain for models of cerebral ischemia followed by reperfusion (Ishikawa et al., 1999; Ritter et al., 2000). In the present study, we also observed a small number of rolling and adherent leukocytes in cerebral arterioles. One possible explanation for this observation is the difference in the expression level of cell adhesion molecules between venules and arterioles, for example, ICAM-1 expression in arterioles is one tenth of that in venules (Iigo et al., 1997). Another possible explanation is the drastic reduction of shear stress in arterioles after MCAO. In the present study, retrograde flow and dramatic reductions of flow velocity were observed in the terminal branches of the MCA. Under similar conditions, LEIs were preferentially observed in arterioles of the rat mesentery (Nazziola and House, 1992). In vitro studies on the expression of ICAM-1 suggest that expression of adhesion molecules in arterioles may be upregulated by reduced shear stress (Nagel et al., 1994). Whether this or other mechanisms, such as E-selectin, may also be responsible for LEI in brain arterioles after cerebral ischemia needs to be addressed in future studies, such as those using ICAM-1 or E-selectin knockout mice.
Capillary perfusion after permanent focal cerebral ischemia
The phenomenon of “capillary plugging” was initially postulated to explain hypoperfusion during early reperfusion after cerebral ischemia (Grogaard et al., 1989; Hallenbeck et al., 1986). Leukocytes accumulated in the low flow regions of the brain after cerebral ischemia induced by air embolism in a canine model (Hallenbeck et al., 1986) and occluded capillaries following MCAO and reperfusion in baboons (del Zoppo et al., 1991). However, this phenomenon was not observed during the reperfusion period after global cerebral ischemia in gerbils (Uhl et al., 2000) and after focal cerebral ischemia in rats (Ritter et al., 2000). In the present study, capillary density in permanently ischemic mouse brain was not different from that in sham-operated animals. Because we used a fluorescent plasma marker, we cannot exclude the presence of plugging leukocytes in the microvasculature. Our data only demonstrate that plasma flow is not disturbed in pial capillaries after permanent cerebral ischemia, at least during the first 2 hours after onset of ischemia. The difference between the findings described previously may derive from the different vascular anatomy among the species used for the respective studies and the methods used for ischemia induction. Another explanation may be that “capillary plugging” may occur later than 120 minutes after onset of ischemia, that is, after the end of the observation time of the current experiments, or that “capillary plugging” occurs only in the infarct core, such as in deep cortical layers or in the striatum, which are not accessible by intravital microscopy. Because infarct expansion, however, takes place in the penumbra, which is directly visualized in the current study, our findings raise the question of whether “capillary plugging” plays a significant role for post-ischemic secondary brain damage in the penumbra.
CONCLUSION
Taken together, we established an in vivo model for the observation of leukocyte-endothelial interactions in the cerebral microcirculation of mice and used this approach for the investigation of leukocyte accumulation in the brain during permanent focal ischemia. Increasing numbers of rolling and adherent leukocytes were observed in the pial microcirculation after permanent MCAO. Activated leukocytes were mainly observed in venules, but small numbers of leukocytes were also found in arterioles. Capillary density was not different between ischemic and sham-operated animals. These data demonstrate that leukocyte-endothelial interactions do not occur only after transient but also after permanent cerebral ischemia. The currently established animal model of intravital microscopy in mice may help to enhance our understanding of the molecular mechanisms involved in microcirculatory changes and leukocyte accumulation observed after cerebral ischemia.