
Abstract
Cerebral ischemia triggers an inflammatory process involving the infiltration of leukocytes to the parenchyma. Circulating leukocytes adhere to the vascular wall through adhesion molecules. Here we quantified the in vivo expression of vascular cell adhesion molecule-1 (VCAM-1) in the brain, heart and lungs from 6 to 48 h after transient middle cerebral artery (MCA) occlusion in rats, by intravenous injection of a tracer radiolabeled anti-VCAM-1 antibody. The vascular localization of VCAM-1 was verified by immunohistochemistry after in vivo injection of the antibody. Vascular cell adhesion molecule-1 was strongly induced (4-fold at 24 h) in the microvasculature of the ischemic area, and, to a lesser extent, in the contralateral hemisphere and in a remote organ, the heart, but not in the lungs, indicating that the inflammatory process propagates beyond the injured brain. We injected intravenously either blocking doses of anti-VCAM-1 antibodies or control antibodies after MCA occlusion in rats and mice. We evaluated the neurological score in rats, and infarct volume at 2 days in rats and at 4 days in mice. Anti-VCAM-1 did not protect against ischemic damage either in rats or in mice. Vascular cell adhesion molecule-1 blockade significantly decreased the number of ED1 (labeling monocytes/macrophages/reactive microglia)-positive cells in the ischemic rat brain. However, it did not reduce the numbers of infiltrating neutrophils and lymphocytes, and total leukocytes (CD45 positive), which showed a trend to increase. The results show vascular upregulation of VCAM-1 after transient focal ischemia, but no benefits of blocking VCAM-1, suggesting that this is not a therapeutical strategy for stroke treatment.
Keywords
Introduction
The view is now growing that inflammatory molecules are involved in the pathogenesis of cerebral ischemia and contribute to secondary neuronal injury (Frijns and Kappelle, 2002; Fassbender et al, 1999). Cell adhesion molecules play an important role in rolling, adhesion and recruitment of leukocytes in response to inflammation (Panés and Granger, 1998). Antigen-4 (VLA-4)/vascular cell adhesion molecule-1 (VCAM-1) interactions are essential for leukocyte transmigration across the endothelium (Panés and Granger, 1998). Vascular cell adhesion molecule-1 supports the adhesion of the primary leukocyte populations present in atherosclerotic plaques and may contribute to the quantitative predominance of monocytes over lymphocytes (Gerszten et al, 1998). Vascular cell adhesion molecule-1 facilitates transendothelial entry of circulating monocytes through human brain endothelial cells (MacIntyre et al, 2003), and it plays an important role in leukocyte recruitment in brain diseases, as shown in experimental models of systemic lupus erythematosus (James et al, 2003), and in chronic graft-versus-host disease (Sostak et al, 2004). At the bedside, soluble VCAM-1 concentration in plasma is raised in patients with in acute stroke (Blann et al, 1999), and a two-fold increase of this parameter was found independently associated to poor outcome after stroke (Chamorro et al, 2002). Postmortem examination of brain tissue from stroke patients showed intense expression of VCAM-1 by astrocytes and endothelial cells within the infarcted tissue only (Blann et al, 1999). Increased VCAM-1 staining in the brain endothelium after stroke was associated with enhanced angiogenesis (Krupinski et al, 1994). However, selective upregulation of VCAM-1 mRNA in the ischemic brain was not found in spontaneously hypertensive rats (Vemuganti et al, 2004) and in Sprague-Dawley rats (Berti et al, 2002), although the latter study showed an increase in the cerebral expression of VCAM-1 mRNA 6 h after the onset of ischemia, with similar increases in the ipsilateral and contralateral hemispheres. Yet, quantification and time course of the vascular expression of VCAM-1 protein at the early hours after ischemia have not been reported.
Here we study the time course of VCAM-1 expression after focal ischemia/reperfusion using a dual radiolabeled antibody technique that allows in vivo quantification of adhesion molecule expression in the vascular endothelium (Panés et al, 1995; Sans et al, 1999), and we administered intravenous a saturating dose of anti-VCAM-1 monoclonal antibody (mAb) to block VCAM-1. The results showed increased vascular VCAM-1 expression peaking at 24 h of reperfusion, but no benefits of the anti-VCAM-1 treatment.
Materials and methods
Experiments with Animals
Animals were housed under a 12-h day/night light cycle, and they had free access to food and water. Animal experiments were conducted with the approval of the ethical committee of our Institutions, and in compliance with the regulation of our local authority and the Spanish and European legislations.
Transient Focal Cerebral Ischemia in Rats
Adult male Sprague-Dawley rats (Iffa-Credo, Lyon, France) (n = 67) (280 to 320g of body weight) were used. Focal brain ischemia was produced by 1-h intraluminal occlusion of the right middle cerebral artery (MCA) with reperfusion, as reported (Justicia et al, 2001). Briefly, rats were anesthetized with halothane and intubated through the trachea for controlled ventilation. Mean arterial blood pressure was monitored and body temperature was maintained between 36.5°C and 37.5°C during surgery. A filament (nylon monofilament 4/0, Suturas Aragó, Spain) was introduced (24 mm) through the external carotid artery to the level where the MCA branches out. In addition, the ipsilateral and contralateral common carotid arteries (CCA) were clamped out. The microclip on the contralateral CCA was removed 50 mins later to minimize the risk of hemorrhage at reperfusion. After l0 mins, that is, 1 h after MCA occlusion, the filament was cautiously removed and the clip of the ipsilateral CCA was taken off to allow reperfusion. Rats were allowed to recover and were then killed at different time points ranging from 6 h to 3 days.
Transient Focal Cerebral Ischemia in Mice
Adult male Swiss CD-I mice (Charles River) (n = 3l) (mean ± s.d. body weight = 24.68 ±1.99 g) were used. Focal brain ischemia was produced by 90-min occlusion of the right MCA, followed by reperfusion. Mice were anesthetized with halothane by the aid of a facial mask. A filament (nylon monofilament 6/0, Suturas Aragó, Spain) was introduced (11 mm) through the external carotid artery to the level where the MCA branches out. In addition, the ipsilateral CCA was clamped out. Mice were allowed to recover from the anesthesia, and 90 mins later mice were anesthetized, the filament was cautiously removed, and the suture of the ipsilateral CCA was taken off to allow reperfusion. Animals were allowed to recover and were killed at 4 days.
Western Blot
Control rats (_n = 3) and rats subjected to 1-h MCA occlusion and 24 h reperfusion (n = 4) were anesthetized and subjected to euthanasia, the brain was removed, and the ipsilateral and contralateral cortex and striatum were dissected out and rapidly frozen and kept at −80°C. Tissue samples were homogenized in radioimmunoprecipitation assay buffer (RIPA), containing 0.01mol/L phosphate-buffered saline (PBS), sodium dodecyl sulfate, sodium deoxycholate, the nonionic detergent Igepal and a cocktail of protease inhibitors (Complete, Boehringer Mannheim, Germany). All products and reagents, unless otherwise stated, were from Sigma. Samples were kept on ice for 30 mins and then centrifuged at 12,000g at 4°C for 15 mins, and the supernatants were used as the total protein fraction. The protein concentration was determined with the Bradford assay (Bio-Rad, Hercules, CA, USA). In all, 50μ of the protein extracts was denatured at 100°C for 5 mins and then loaded in 10% Polyacrylamide gels. Proteins were then transferred to a polyvinylidene difluoride membrane (Immobilon-P, Millipore, Bedford, MA, USA), which was incubated overnight at 4°C with a purified mouse anti-rat mAb directed against VCAM-1 (#559165, BD Pharmingen) diluted 1:500. On the following day, membranes were incubated for 1 h with an anti-mouse Ig peroxidase-linked secondary antibody (1:2000) (Amersham). The reaction was visualized using a chemiluminescence detection system based on the luminol reaction. Membranes were then reacted with a rabbit polyclonal antibody against Actin (Sigma) to check for equal protein loading in each lane. The intensity of the bands was measured by densitometric analysis (Kodak, DC-120 camera and KdslD, Digital Science System software). The ratio between VCAM-1 band intensity to the corresponding actin band intensity was calculated to correct for any variation in protein gel loading.
Quantification of Vascular Cell Adhesion Molecule-1 Expression
Vascular cell adhesion molecule-1 was studied by a double-labeling isotopic technique described by Sans et al (1999), that was adapted to our model of focal cerebral ischemia-reperfusion in rats. A murine mAb Ig G2a against rat VCAM-1 (5F10, Biogen Inc., Cambridge, MA, USA) was labelled with 125I (Amersham Iberica, Madrid, Spain), and an isotype-matched mAb (UPC-10, Sigma), which does not bind to rat VCAM-1, was labelled with 131I (Amersham Iberica) by using the iodogen method (Sans et al, 1999). Controls (n = 6) and rats subjected to MCA occlusion (n = 20) were anesthetized (ketamine/xylazine) at 6 (n = 4), 16 (n = 5), 24 (n = 7) and 48 (n = 4) h after MCA occlusion, and then had the femoral artery and vein cannulated. A mixture containing 20μ of 125I-5F10 and 5 μ of 131I-UPC-10 was injected in vivo through the vein. This dose of anti-VCAM-1 mAb has been shown to be saturating in the previous assays (Sans et al, 1999). After 5 mins, the rat was exsanguinated, the brain was removed, the ipsilateral and contralateral MCA territories (including cortex and striatum) were dissected out, and radioactivity was measured in a gamma-counter. The heart and lungs were also dissected out to measure the radioactivity content. Specific binding of mAb (ng/g of tissue) was calculated by correcting for nonspecific binding, as reported (Sans et al, 1999).
Immunohistochemical Location of the Systemically Injected Monoclonal Antibody Against Vascular Cell Adhesion Molecule-1
At 24 h after MCA occlusion/reperfusion (n = 3) and control (n = 3), rats were anesthetized (ketamine/xylazine), 20 μg of nonradioactive 5F10 mAb against VCAM-1 was injected through the femoral vein, and 5 mins later the rat was exsanguinated as above. The brain was removed from the skull, cryoprotected by immersion in sucrose (30%) for 1 day, frozen on dry ice, and 20-μ-thick coronal sections were obtained in a cryostat. Immunohistochemistry to detect the presence of the intravenously injected mAb against VCAM-1 was performed using a biotinylated anti-mouse secondary antibody, and an enhancing detection system (Envision + Dual Link System-peroxidase (DAB +), DakoCytomation, Carpinteria, CA, USA) based on the streptavidin-biotin reaction followed by diaminobenzidine was used. Sections were then counterstained with hematoxylin, dehydrated, mounted and observed under the light microscope. Immunohistochemistry controls included ischemic rats that did not receive the intravenous injection of the mAb against VCAM-1 (n = 3), ischemic rats injected with the nonspecific control mAb (n = 2) and nonischemic rats that received the anti-VCAM-1 mAb (n = 2).
Assessment of Leukocyte Infiltration in Brain and Heart
At 3 days after MCA occlusion/reperfusion (n = 2] and control (n = 2), rats were anesthetized (halothane) and perfused through the heart with 4% paraformaldehyde. The brain, heart and lungs were removed and postfixed overnight with the same fixative. Tissues were embedded in paraffin and sectioned (5-μ thick) in a microtome. Sections were deparaffined and subjected to antigen retrieval by immersing in citrate buffer, pH 6 (BioSystems, Barcelona, Spain) and heating to 121°C in an autoclave for 10 mins. All products and reagents, unless otherwise stated, were from Sigma. Endogenous peroxidases were blocked with methanol-H2O2, and unspecific binding sites were blocked with 3% normal horse serum for 2 h and the dual endogenous enzyme block (Envision + Dual Link System, DakoCytomation) for 30 mins. Sections were then incubated overnight in a humidified chamber at 4°C with a purified mouse anti-rat mAb against the common leukocyte antigen CD45 (#550566, BD Pharmingen) diluted 1:10. This was followed by a biotinylated secondary antibody and the streptavidin-biotin method using the Envision + kit (DakoCytomation), as above.
In vivo Blockade of Vascular Cell Adhesion Molecule-1 in Rats and Mice
Rats (n = 8) received an intravenous bolus injection of purified mouse anti-rat CD 106 (VCAM-1)-blocking mAb (# 559165; Pharmingen, BD Biosciences, Madrid, Spain) (0.8mg/kg of body weight), 30 mins after reperfusion after 1-h MCA occlusion. This dose of the anti-VCAM-1 antibody has been shown to be saturating in rats, and prevents leukocyte adhesion in colonic venules of rats with severe colitis (Sans et al, 1999). For treatment control, rats received the same dose of a nonrelated isotype-matched mAb (Mouse IgG2a, κ, UPC-10, #M 9144; Sigma, Madrid, Spain), which was previously shown to be adequate as a control for anti-VCAM-1 treatment (Soriano et al, 2000). Before injection into rats, the commercial antibodies were dialyzed overnight in PBS using Slide-A-Lyzer® Cassettes (#66425; Pierce, Rockford, IL, USA) to remove azide. The half-life of anti-VCAM-1 mAb in rats is 24 to 30 h, as previously assessed (Sans et al, 1999). Rats were killed at 48 h.
In mice, we blocked VCAM-1 by daily intravenous injections (1 mg/kg) of a different mAb against mouse VCAM-1 (purified anti-mouse CD106 (VCAM-1) NA/LE™, clone: 429 (MVCAM.A), isotype: rat IgGa; #553329; BD Pharmingen) (n = 15). This antibody blocks mouse VCAM-1 (see, for instance, Belcher et al, 2005). For treatment control, mice (n = 16) received the same dose of an isotype-matched control antibody (purified rat IgG2a, k isotype control, NA/LE™, R35–95; #554687; BD Pharmingen) (Belcher et al, 2005). Treatment was started at 90 mins of reperfusion after MCA occlusion, and was administered three times more every 24 h. Mice were killed at 4 days.
Assessment of Brain Damage after Antibody Treatments
At 48 h, a simple neurological test in a 9-point scale (0 = normal to 9 = highest handicap) was performed in the rats, as a modification of previously reported tests (Bederson et al, 1986; Garcia et al, 1995; Menzies et al, 1992). We performed four tests to assess: (a) alteration in spontaneous ability for movement (normal movement and exploration activity = 0; movement without exploration = 1; no displacement = 2); (b) laterality in movement (symmetrical = 0; left drifting when elevated by the tail and pushed or pulled = 1; spontaneous left drifting = 2; circling without displacement or spinning to the left = 3); (c) asymmetry in the use of front legs by the ‘parachute’ reflex (movement of front legs while maintaining the rat in the air by subjecting its tail; symmetrical movement = 0; asymmetrical movement = 1; retraction of the left forelimb towards the body = 2); (d) ability to use the left forepaw (we assessed whether the rat did offer resistance while holding the left front leg: rat did not permit stretching = 0; rat allowed stretching after several attempts = 1; rat did not oppose any resistance = 2). Scores obtained in each test were added to obtain the final neurological score.
For measurement of infarct volume, rats and mice were anesthetized with halothane and killed by decapitation. The brain was removed and sliced in 2-mm-thick coronal sections that were stained with a 1% solution of 2,3,5-triphenyltetrazolium chloride (TTC) for lOmins at 37°C. Sections were then immersed overnight in a 4% paraformaldehyde solution in phosphate buffer and washed in phosphate buffer. Ischemic tissue gave no reaction with the mitochondrial TTC staining, which turns red in healthy tissue. Eight sections of the rat brain, between + 6 and −8 mm from Bregma, and four sections of the mouse brain, between + 2 and −4 mm from Bregma, were recorded with a video camera and analyzed with an image analysis system (AIS, Imaging Research Inc., Canada). White or pale areas in each section were measured and integrated to calculate infarct volume. In addition, contralateral and ipsilateral hemisphere areas were measured, and the difference between ipsilateral and contralateral areas in each section was used to calculate the volume of brain swelling. Measures were performed by one of the investigators who was masked to the treatment of each particular rat.
Assessment of Leukocyte Infiltration in the Brain of Rats Treated with the Antibodies
One of the 2-mm brain sections (at the level of Bregma) was embedded in paraffin and cut into 5-μ sections with a microtome. A consecutive 2-mm brain section was cut in 50-μ-thick sections in a vibratome. Paraffin sections were stained with hematoxylin and eosin, or subjected to immunohistochemistry. The following monoclonal antibodies were used: anti-myeloperoxidase (BD Pharmingen) diluted 1:800 for neutrophil detection; anti-EDl (BD Pharmingen) diluted 1:50, which mainly labels monocytes/macrophages/reactive microglia, and, to a lesser extent, neutrophils; the anti-CD45 (BD Pharmingen) 1:50 for total leukocyte labelling (see above). The latter antibody was used free-floating with the vibratome sections, using standard procedures (Planas et al, 2001).
Cell Counts
The infiltration of CD45-, EDl- and MPO-immunopositive cells within the ischemic cortex and striatum was assessed by counting the number of stained cells at 48 h after ischemia. For EDl and MPO, cells were counted under the optical microscope in five different fields (0.28 mm2) per region using the × 40 magnification objective. Cells were identified and counted by a researcher who was masked to treatment, and counts were performed in areas showing the highest staining density. For CD45-positive cells the × 20 magnification objective was used, as this lower magnification allowed a better focus in the immunohistochemistry with the thicker vibratome sections. Two fields per area (0.14 mm2) were captured with a camera and cells were counted in the corresponding images by touch count using the AnalySIS software (Soft Imaging System, Münster, Germany). The presence of lymphocytes in the ischemic tissue was assessed by counting the cells in the hematoxylin-and-eosin-stained sections in 5 fields per area (0.11 mm2) under the optical microscope, using the × 63 magnification immersion objective. Lymphocytes were identified by their particular morphology and staining features, as they possess a circular very dark-staining nucleus that can occupy most of the cell surface. Results are expressed as the average number of cells per area.
Statistics
Change in VCAM-1 expression at the different time points after MCA occlusion in relation to controls was analyzed with the nonparametric Kruskal-Wallis test, followed by post hoc analysis with Dunn's multiple comparison test. Comparisons between two groups were performed with the Student's t-test. Correlations were studied by linear regression analysis.
Results
Physiological Parameters
Physiological parameters were monitored during the 1-h MCA occlusion period. Mean ± s.d. values in the nontreated group of rats (n = 25) were as follows: body temperature = 36.57°C ± 0.51°C; mean arterial blood pressure = 100.0 ± 7.8 mmHg; blood pH = 7.427 ± 0.038; pCO2 = 138.34 ± 26.94; pC2 = 39.45 ± 4.65. Physiological parameters were not different in the rats treated with anti-VCAM-1 antibody or with control antibody. In the anti-VCAM-1 group (n = 8), mean ± s.d. values were: body temperature = 36.98°C ± 0.61°C; mean arterial blood pressure = 102.2 ± 11.4 mm Hg; blood pH = 7.421 ± 0.077; pO2 = 103.4 ± 16.85; pCO2 = 39.49 ± 7.69. In the control antibody group (n = 7), mean ± s.d. values were: body temperature = 36.48°C ± 0.53°C; mean arterial blood pressure = 98.95 7.7 mm Hg; blood pH = ±7.407 ± 0.053; pO2 = 104.7 ± 16.64; pCQ2 = 37.10 ± 3.55.
Induction of Brain Vascular Cell Adhesion Molecule-1 after Ischemia/Reperfusion
The cerebral expression of VCAM-1 increased at 24 h after ischemia, as assessed by Western blot in tissue extracts of the ischemic rat brain (Figure 1A). The ratio of VCAM-1 band intensity in the Western blot membranes to the corresponding band intensity of actin was calculated to correct for any differences in the amount of protein loaded to each lane. A significant (P < 0.05) increase of VCAM-1 expression was found in the ischemic rat brain in relation to the control rat brain (Figure IB).
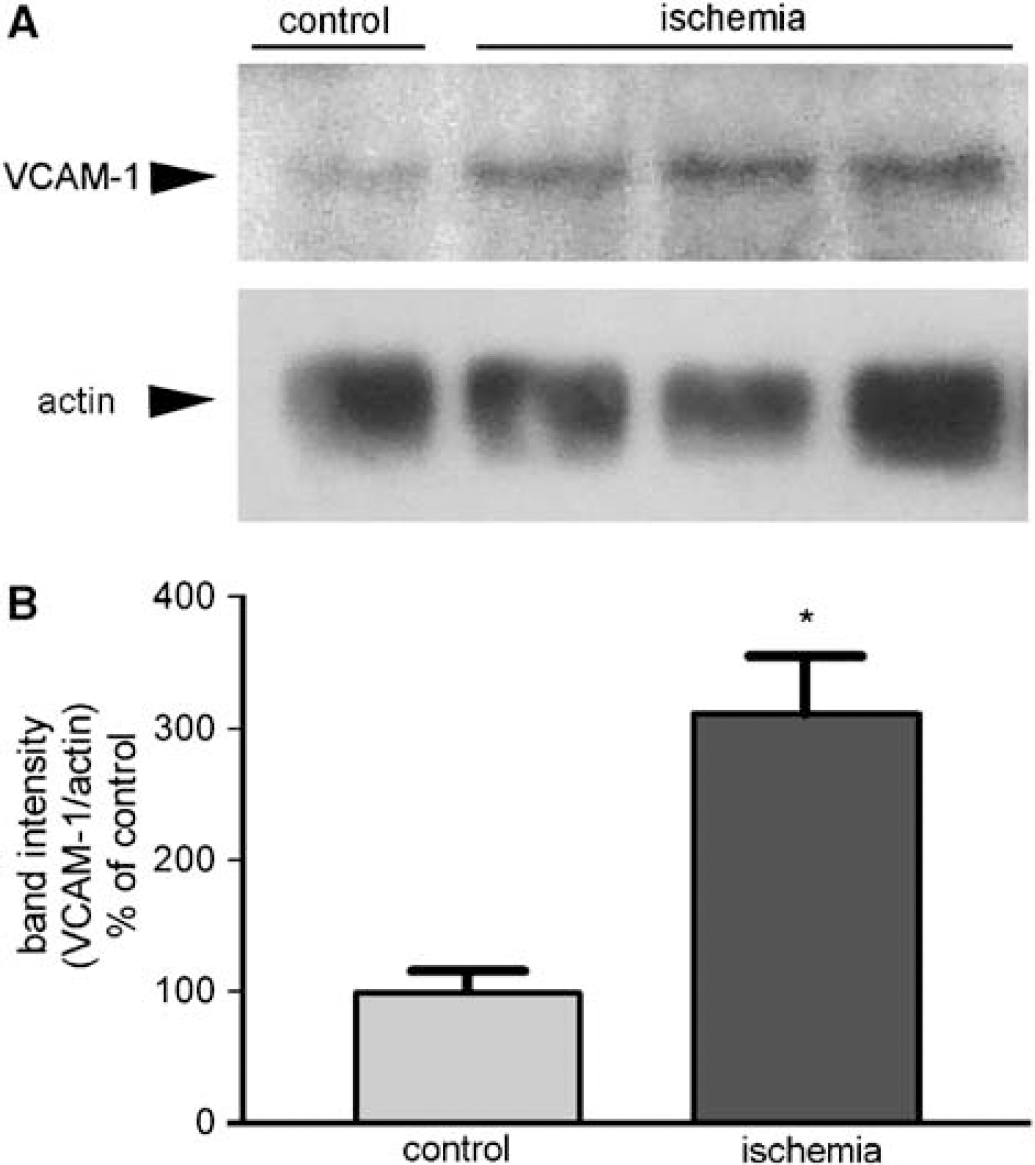
Expression of VCAM-1 in brain homogenates. (
Vascular Cell Adhesion Molecule-1 Expression is Increased in the Vasculature of the Ischemic Brain Tissue at Different Time Points after Middle Cerebral Artery Occlusion, as Measured with the Dual Radiolabelled Antibody Technique
Significant increases of VCAM-1 in relation to the controls were observed in the ischemic ipsilateral hemisphere at 16 h (P < 0.05) and 24 h (P < 0.001) after MCA occlusion in rats (Figure 2A). Values, which were expressed as ng of VCAM-1 mAb/g of brain tissue, increased in the ipsilateral hemisphere 2.5- and 3.7-fold above controls (mean ± s.e.m. control value = 4.53 + 0.32 ng mAb/g of brain tissue) at 16 and 24 h after ischemia, respectively. In addition, moderate increases of VCAM-1 expression were also observed at 16 and 24 h in the contralateral hemisphere (Figure 2A) corresponding to 1.8-fold (P < 0.01) and 1.7-fold (P < 0.05) increase, respectively.
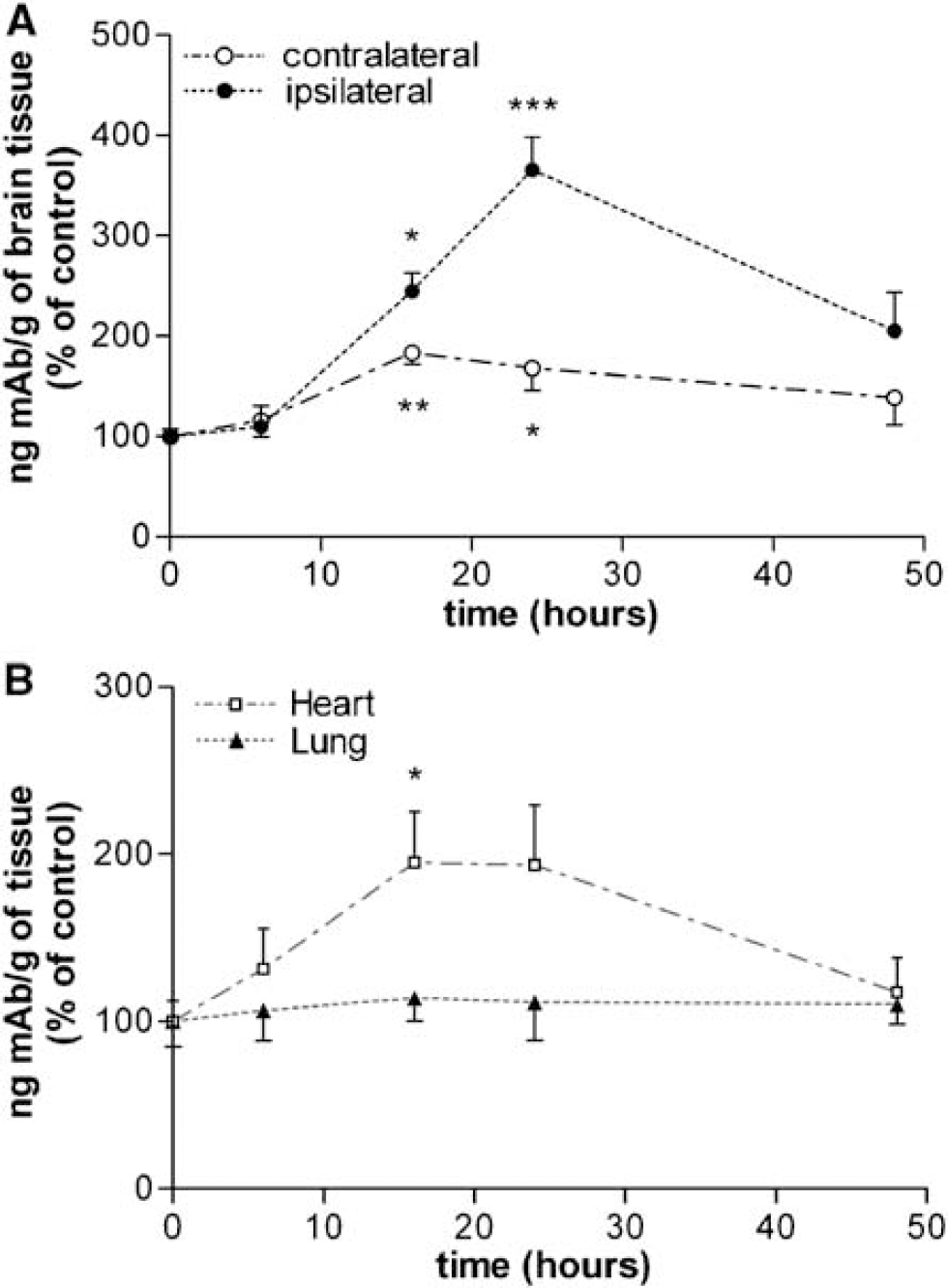
Time course of VCAM-1 induction after MCA occlusion using the quantitative double radiotracer antibody technique. (
Localization of Vascular Cell Adhesion Molecule-1 Induction in the Cerebral Microvessels
Immunohistochemistry in rat brain tissue sections after in vivo intravenous injection of the m Ab against VCAM-1 showed staining in the endothelium of brain microvessels in the ischemic cortex and striatum (Figures 3B-3D) at 24 h. A few microvessels showed some lower intensity of staining in the contralateral nonischemic hemisphere (Figures 3E-3F). Staining was not detected in animals that did not receive intravenous injection of the antibody or received the control mAb (not shown), nor in control rats that received the anti-VCAM-1 mAb injection but that were not subjected to ischemia (Figure 3A). This shows that the increase in cerebral VCAM-1 expression that we detected with the quantitative dual radiolabeled antibody technique (Figure 2A) was located in brain microvasculature.
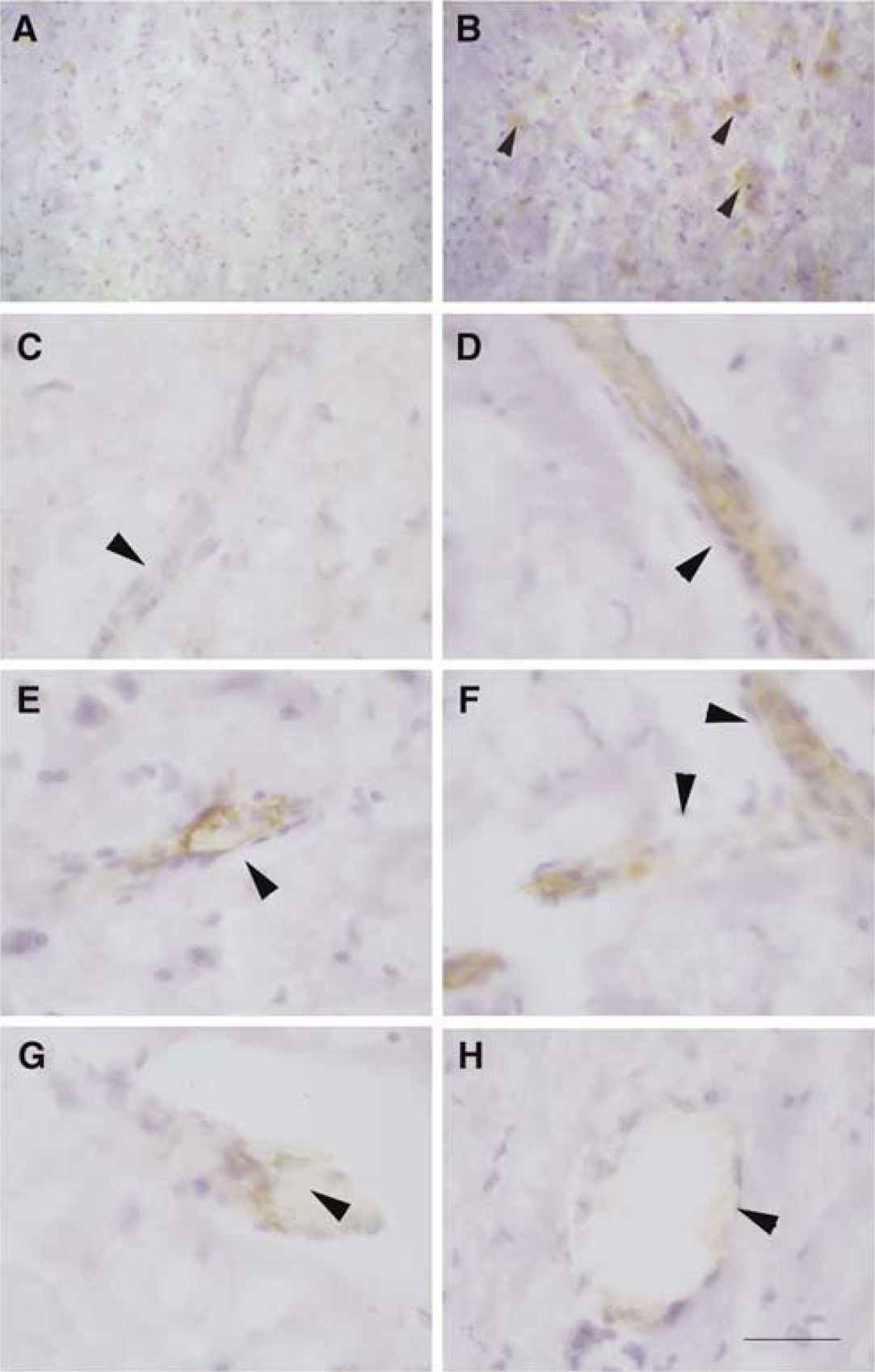
The measured induction of VCAM-1 after ischemia is located in brain microvessels. Rats (controls or at 24 h after MCA occlusion) received an intravenous injection of mAb against VCAM-1 (see Materials and methods). The mAb was detected in brain sections by immunohistochemistry. Arrows indicate brain microvessels. Staining is not detected in the control brain (
Mild and Transient Increase of Vascular Cell Adhesion Molecule-1 Expression in Heart, but not Lungs, after Brain Ischemia
Besides the brain, we also quantified the expression of VCAM-1 in other organs in the rats subjected to MCA occlusion. Under physiological conditions, the lungs expressed high levels of VCAM-1 per tissue weight (around 10- and 30-fold the levels expressed by the control heart and brain, respectively), likely attributable to the very large endothelial surface of the lungs (Panés et al, 1995). A significant 2-fold increase of VCAM-1 expression was observed in the heart 16 h (P < 0.05) after ischemia in relation to control values, whereas no alterations were observed in the lungs (Figure 2B). This implies that some stimuli derived from the ischemic brain triggers induction of VCAM-1 in certain peripheral organs, but not in others.
Leukocytes Infiltrate the Ischemic Tissue, but not the Contralateral Hemisphere or the Heart
We examined leukocyte infiltration 3 days after MCA occlusion in paraffin rat tissue sections by means of immunohistochemistry with a CD45 mAb, which recognizes a leukocyte common antigen. Leukocyte infiltration was restricted to the ischemic cortex and striatum (Figure 4B), but was not observed in the control (Figure 4A), the contralateral brain hemisphere or in the heart (not shown), in spite of the increased VCAM-1 expression reported above (Figure 2B).
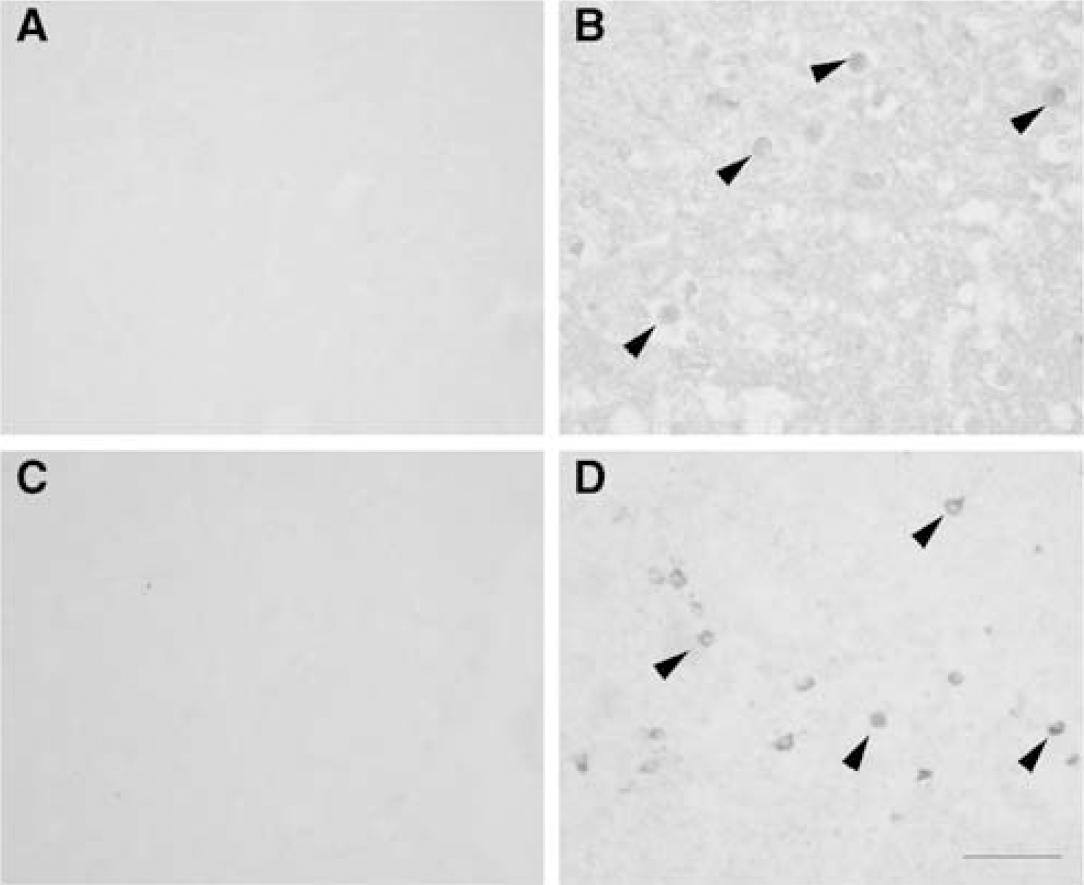
Postischemic leukocyte infiltration. CD45-positive cells (arrows) are not detected in the control brain (
Systemic Blockade of Vascular Cell Adhesion Molecule-1 at Reperfusion was not Protective in Rats
Rats received an intravenous saturating dose of the blocking mAb against VCAM-1 (n = 10) or a nonrelated control mAb (n = 9) at 30 mins of reperfusion, and infarct volume was evaluated at 48 h. Three rats died before 48 h (1 of the control group and 2 of the anti-VCAM-1 group). One rat belonging to the anti-VCAM-1 group showed no signs of infarction. In the remaining rats (n = 15), anti-VCAM-1 treatment caused no reduction of infarct volume (Figure 5A). On the contrary, rats treated with the anti-VCAM-1 mAb showed a nonsignificant tendency to a higher infarct volume compared with rats receiving the control mAb. This effect was apparent in the cortex rather than in the striatum, as the volume of striatal infarction was unchanged after treatment (Figure 5A). Brain swelling was significantly higher (P < 0.05) in the anti-VCAM-1-treated group than in the group receiving the control mAb. Treatment affected most brain levels, as revealed by comparison of infarcted areas in the different brain sections (Figure 5B). The neurological score was positively correlated with infarct volume by linear regression analysis (P < 0.005) (Figure 5C), and this functional parameter was not ameliorated either in the rats treated with anti-VCAM-1 m Ab (Figure 5D).
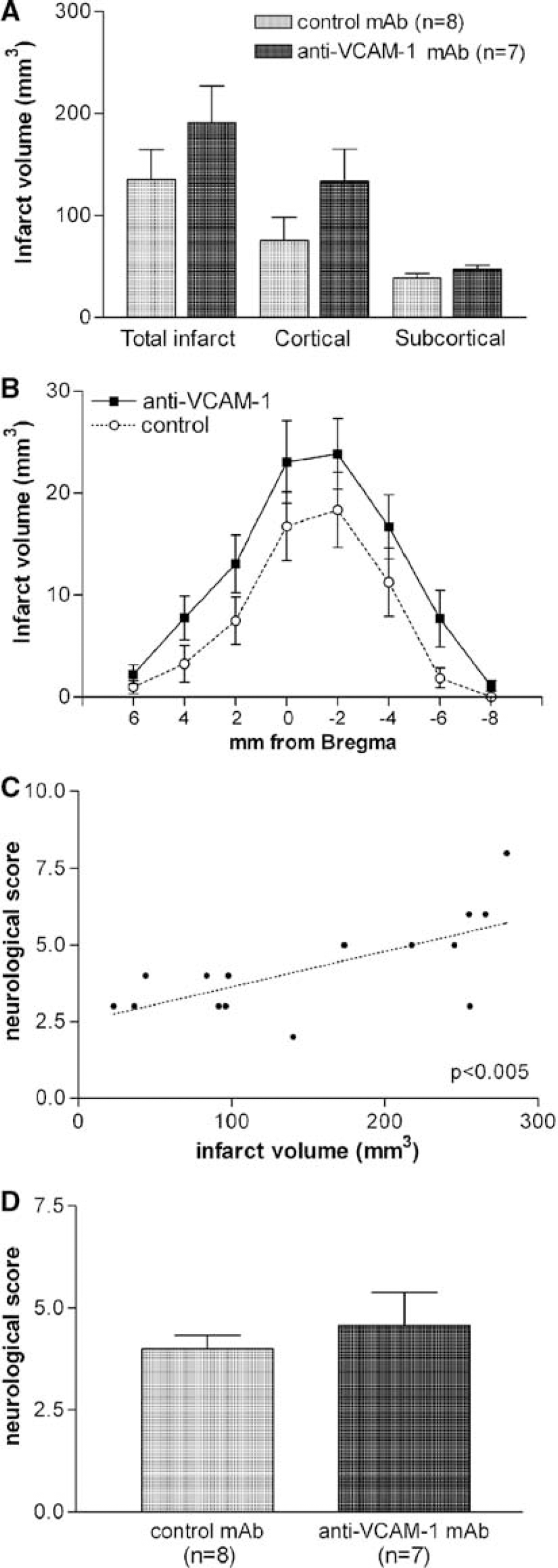
Systemic injection of anti-VCAM-1 mAb to ischemic rats is not protective. (
Changes in Leukocyte Infiltration in the Rat Brain after anti-Vascular Cell Adhesion Molecule-1 Treatment
The anti-VCAM-1 treatment did not prevent leukocyte infiltration in the ischemic tissue at 48 h. Furthermore, the counts of cells positive to the common leukocyte antigen CD45 (Figure 6A) showed a nonsignificant trend to increase after this treatment in both cortex and striatum. The mean number of infiltrating leukocytes per area was positively correlated (P < 0.05) to infarct volume (Figure 6B). However, the number of EDI-positive cells in the ischemic tissue was reduced after anti-VCAM-1 antibody administration (P < 0.05 in the striatum) (Figure 6C), while this treatment did not prevent neutrophil infiltration, as revealed by the number of MPO-positive cells (neutrophils) (Figure 6D). The number of lymphocytes (Figure 6E) per area was significantly increased in the striatum in the anti-VCAM-1-treated group (Figure 6F). These results are compatible with reduced monocytes, but not global leukocyte, infiltration after VCAM-1 blockade in the ischemic brain.
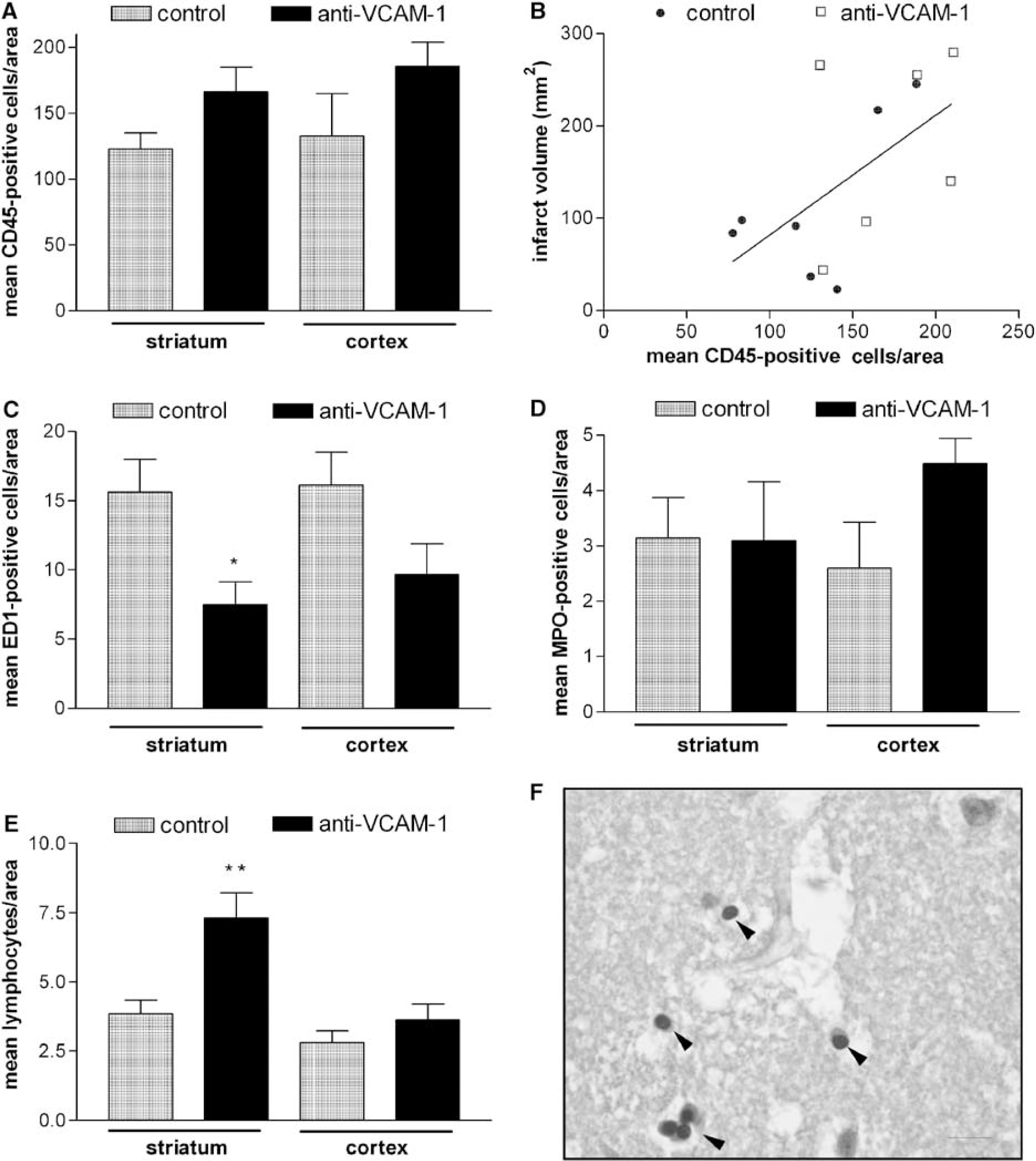
Alteration in leukocyte infiltration at 48 h after anti-VCAM-1 mAb treatment. (
Systemic Blockade of Vascular Cell Adhesion Molecule-1 at Reperfusion was not Protective in Mice
To further confirm our finding of lack of protection of VCAM-1 blockade in the ischemic rat brain, we blocked VCAM-1 in mice subjected to transient MCA occlusion with a different blocking antibody (see Materials and methods), and evaluated the infarct volume at 4 days. Mice subjected to 90 mins MCA occlusion received an intravenous injection of the anti-VCAM-1 antibody at 90 mins of reperfusion, and three times more every 24 h (n = 15). One mouse died, 3 mice showed no signs of infarction and 11 mice showed brain infarction as assessed with TTC staining. Treatment controls (n = 16) received a similar treatment protocol with an isotype-matched control antibody. Three control mice died, 5 mice showed no infarction, and 8 mice showed brain infarction. Infarct volume showed a nonsignificant tendency to increase after anti-VCAM-1 treatment compared with treatment with the corresponding control antibody (Figure 7A), as occurred in the rat brain (Figure 5?). Evaluation of the infarcted areas in the different brain sections showed a similar effect of treatment in most sections (Figure 7B). The mean volume of swelling, as assessed by the volume difference between the contralateral and ipsilateral hemispheres, was around five times higher in the anti-VCAM-1-treated group than in the control antibody group, but this difference reached no statistical significance.
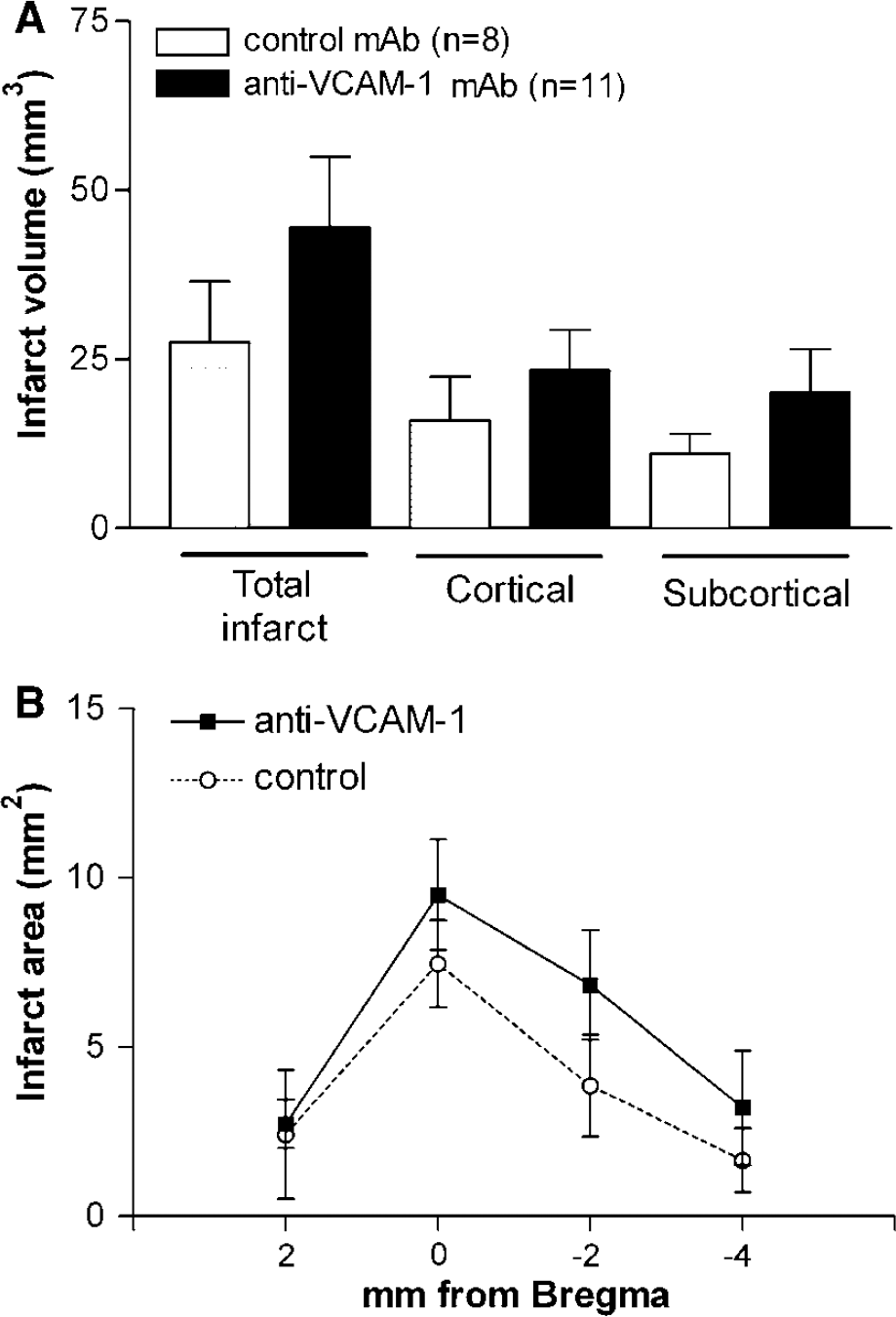
Systemic injection of anti-VCAM-1 mAb to ischemic mice is not protective. (
Discussion
Here we quantified the induction of VCAM-1 in the brain after cerebral ischemia by a dual radiolabeled antibody technique (Sans et al, 1999) and performed immunohistochemical detection after systemic injection of the mAb to identify the cellular target measured in our quantitative study. We detected a significant increase of VCAM-1 expression at 24 h of reperfusion, and then aimed to investigate whether this contributed to the neuropathological outcome of ischemia by administering a blocking dose of the anti-VCAM-1 mAb. Sustained preclinical evidence gives support to the possibility that blocking endothelial adhesion molecules might have a therapeutical value by reducing leukocyte infiltration. Blocking VCAM-1 reduces the histological signs of inflammatory damage in a rat model of experimental colitis (Sans et al, 1999). However, this treatment afforded no benefit in cerebral ischemia at 48 h after MCA occlusion in rats, as infarct volume was not reduced and the neurological score was not ameliorated either, in comparison with a group of rats receiving a control mAb. Likewise, we found no protection at 4 days after transient ischemia by injecting anti-VCAM-1 mAb to mice. In contrast to the present findings with anti-VCAM-1 mAb, treatment with anti-ICAM-1 mAb gave favorable results in preclinical studies of ischemia/reperfusion, as it reduced infarct volume (Zhang et al, 1994, 1995) and apoptosis (Chopp et al, 1996) in rats, and neurological damage in rabbits (Bowes et al, 1993). Also, microcirculatory disturbance and infarct volume were attenuated in ICAM-1-knockout mice subjected to transient focal cerebral ischemia (Connolly et al, 1996), and treatment with antisense oligonucleotides reduced the infarct volume and improved the neurological score in spontaneously hypertensive rats after ischemia/reperfusion (Vemuganti et al, 2004). Yet, ICAM-1 antibody treatment was not beneficial in rats subjected to permanent ischemia (Zhang et al, 1995). Also, no clear benefit of this therapy was detected in the Enlimomab Acute Stroke Trial (2001), and several potential mechanisms for the negative outcome of this trial have been proposed (Furuya et al, 2001). To our knowledge, therapies directed against VCAM-1 were not explored in cerebral ischemia before the present study, and our results do not support this as a beneficial strategy for the treatment of stroke. Our findings are compatible with reduced monocyte infiltration after treatment with the mAb against VCAM-1, showing that this treatment was effective in blocking VCAM-1. However, this effect on monocytes was not accompanied by a decrease in global leukocyte infiltration to the ischemic tissue. On the contrary, a nonsignificant tendency to enhanced leukocyte infiltration was observed after this treatment, with significant increases in the numbers of neutrophils and lymphocytes per area, while EDl-positive cells were reduced. Both blood-born macrophages and reactive microglia are EDl-positive. Vascular cell adhesion molecule-1 was reported to be more effective at mediating the initial attachment of monocytes than of lymphocytes, and to be sufficient to initiate events culminating in monocyte, but not lymphocyte, transmigration (Gerszten et al, 1998). Also, VCAM-1 supports monocyte adhesion to endothelial cells better than ICAM-1 and selectins (Zhang et al, 2004). Therefore, attenuation of blood monocyte infiltration may account for the reduced number of EDl-positive cells after VCAM-1 blockade. Taken altogether, these results suggest that unbalanced adhesion molecule availability at the endothelium by reduced VCAM-1 alters the composition of the infiltrating leukocytes (by reducing monocyte infiltration and increasing neutrophils and lymphocytes), which might not necessarily be beneficial for the outcome of stroke. Yet, we cannot discard that the tendency of increased leukocyte infiltration may just reflect the tendency to increased infarct volume that was observed in the group treated with anti-VCAM-1 mAb, as these two parameters were correlated.
A 1-h focal ischemia triggered a transient induction of VCAM-1 in the ischemic tissue, as quantified with a double radiolabeled antibody technique. Using this technique, Sans et al (1999) showed in colonic tissue that the accumulation of mAbs after intravenous injection (with a circulation time limited to 5 mins, as we used here) occurs at the luminal side of the vessels. Here we performed immunohistochemistry to identify the cellular target of the injected mAb. The results showed that the intravenous injected anti-VCAM-1 mAb was bound to the endothelial surface of brain microvessels. Thus, the induction of VCAM-1 expression measured here was located in the microvessels. This finding agrees with the increased endothelial expression of VCAM-1 in the postmortem tissue of stroke patients (Krupinski et al, 1994; Blann et al, 1999). Previous studies examining the expression of VCAM-1 mRNA in rat brain tissue failed to detect a differential increase in the ischemic area compared with the contralateral (Berti et al, 2002; Vemuganti et al, 2004). Several reasons may account for this apparent discrepancy. First, besides the increase in VCAM-1 in the ischemic area, we also found a low but significant induction of VCAM-1 expression in vessels of the contralateral hemisphere at 16 and 24 h after MCA occlusion. Although this latter raise of VCAM-1 protein was significantly lower than that in the ischemic area, it might be high enough to confound the results at the level of mRNA. Second, neural cells other than endothelial cells express VCAM-1. Indeed, astrocytes and microglia express VCAM-1 on activation (Lee and Benveniste, 1999). Therefore, in studies with brain tissue homogenates, any increases in glial VCAM-1 after ischemia may mask the induction of VCAM-1 in the vessels, which is the one involved in leukocyte adhesion to the endothelium for further transmigration. Third, the intravenously injected antibody against VCAM-1 was allowed to circulate only during 5 mins, therefore interacting with the VCAM-1 molecules expressed in the luminal side of the vessels (Sans et al, 1999). Thus, the possibility that preexisting VCAM-1 protein in the endothelial cells was recruited to the cell membrane after ischemia cannot be discarded. In this latter case, induction of VCAM-1 mRNA would not be necessary to explain upregulation of VCAM-1 in the vessel wall.
Interestingly, cerebral induction of VCAM-1 was accompanied with a significant increase in the expression of this adhesion molecule in the heart at 16 h after ischemia. However, no changes in VCAM-1 expression were observed after cerebral ischemia in the lungs. Upregulation of VCAM-1 in the heart in the absence of changes in lung is in keeping with previous evidence showing a higher sensitivity of heart endothelium to proinflammatory stimuli that induce adhesion molecule expression (Panés et al, 1995). It is recognized that the severity of the inflammatory reactions generated in a tissue by ischemia/reperfusion can be so intense that the inflammatory response is even apparent in distant organs (Carden and Granger, 2000). For instance, remote effects of ischemia/reperfusion are frequently observed in the lung and cardiovascular system, resulting in the development of systemic inflammatory response syndrome and multiple organ dysfunction syndrome (Carden and Granger, 2000). Induction of VCAM-1 in remote organs, distant to that directly affected by inflammation, has been previously reported. Indeed, selective upregulation (5.5-fold) of VCAM-1 was found in the cerebral endothelium of colitic rats and mice, which was not followed by leukocyte infiltration to the brain, in spite of the level of VCAM-1 induction in brain vasculature being correlated with that induced in the colon and with the severity of colon inflammatory changes (Sans et al, 2001). Likewise, here we failed to detect any CD45-positive cells in the heart, but they were found in the ischemic area of brain. The interaction of endothelial VCAM-1 with its receptor in leukocytes, α4-integrin, is involved in the firm adhesion of leukocytes to the endothelial surface, but it does not trigger transendothelial migration of T-cells through the blood-brain barrier (Laschinger and Engelhardt, 2000). These findings show that endothelial VCAM-1 induction is not sufficient to enable tissue infiltration of circulating leukocytes, while it is likely that induction of additional adhesion molecules or leukocyte-activating factors in the endothelium are needed (Sans et al, 2001).
In brief, the present results show that VCAM-1 expression is upregulated in the cerebral microvasculature of the ischemic rat brain, with a peak of around 4-fold at 24 h. In addition, the results show a small but significant induction of VCAM-1 in the contralateral hemisphere and in a remote organ, the heart, indicating that the inflammatory process propagates beyond the frame of the injured brain tissue. However, blocking VCAM-1 is not protective in transient focal cerebral ischemia in rats and mice.
Footnotes
Acknowledgements
We thank Ms Noelia Montoya and Ms Eugenia Gómez for skillful technical assistance.