
Abstract
The proinflammatory cytokine tumor necrosis factor (TNF) is known to be expressed in brain ischemia; however, its cellular and temporal appearance is not fully settled. In this study, nonradioactive in situ hybridization for murine TNF mRNA was performed on brain sections from adult C57×129 mice at 6 hours, 12 hours, 24 hours, 2 days, 5 days, or 10 days (six to eight mice per group) after induction of permanent focal cerebral ischemia. Cortical infarct volumes were estimated, and TNF mRNA-expressing cells were counted within the infarct and infarct border using Cast-Grid analysis. At 12 hours, a peak of 19.2 ± 5.1 TNF mRNA-expressing cells/mm2 was counted, contrasting two to three times lower values at 6 and 24 hours (6.4 ± 4.6 and 9.2 ± 3.4 cells/mm2, respectively) and <2 cells/ mm2 at 48 hours and later stages. The TNF mRNA-expressing cells were distributed along the entire rostrocaudal axis of the cortical infarcts and occasionally within the caudate putamen. At all time points, TNF mRNA colocalized with Mac-1-positive microglia/macrophages but not with Ly-6G (Gr-1)-positive polymorphonuclear leukocytes. Similarly, combined in situ hybridization for TNF mRNA and immunohistochemistry for glial fibrillary acidic protein at 12 and 24 hours revealed no TNF mRNA-expressing astrocytes at these time points. Translation of TNF mRNA into bioactive protein was demonstrated in the neocortex of C57Bl/6 mice subjected to permanent middle cerebral artery occlusion. In summary, this study points to a time-restricted microglial/macrophage production of TNF in focal cerebral ischemia in mice.
Keywords
The pleiotrophic cytokine tumor necrosis factor (TNF) is produced predominantly as a secreted protein and is reported to have both neurotoxic and neuroprotective effects in animal models of cerebral ischemia. Normal adult rats (Simmons and Willenborg, 1990) as well as rats undergoing focal cerebral ischemia (Barone et al., 1997) exhibit loss (or increased loss) of neurons when TNF is injected into the brain. Administration of TNF binding protein attenuates infarct volume in mice after establishment of permanent middle cerebral artery occlusion (pMCAO) up to 1 hour after occlusion (Nawashiro et al., 1997a) and after pMCAO in rats (Dawson et al., 1996; Barone et al., 1997) as well as after transient middle cerebral artery occlusion (MCAO) in both mice (Yang et al., 1998) and rats (Lavine et al., 1998). In contrast, treatment with small amounts of TNF induces ischemic tolerance in mice (Nawashiro et al., 1997b). Surprisingly, mice deficient in TNF develop larger infarct volumes than wild-type littermates 1 day after pMCAO induction (R. Gregersen et al., unpublished data), indicating that TNF also has neuroprotective properties. This is in line with data from Bruce et al. (1996) showing that mice deficient in the TNF p55 and p75 receptors exhibit larger infarcts than wild-type littermates at day 1 after transient MCAO.
Actions of TNF appear to depend on complicated signal transduction pathways that are initiated by the binding of TNF to its p55 and p75 receptors. Both receptors are expressed in differential levels on glial and nerve cells (Bruce et al., 1996; Tchelingerian et al., 1996; Dopp et al., 1997) as well as on infiltrating leukocytes (Hart et al., 1996) and are overexpressed in ischemic cortex after induction of pMCAO in rats (Botchkina et al., 1997). The p55 receptor contains cytoplasmic domains that may either induce apoptosis (Hsu et al. 1995) or induce downstream activation of nuclear factor-kappa B (Adam et al., 1995; Borset et al., 1996). Nuclear factor-kappa B activation has been demonstrated to be involved in modulation of voltage-dependent calcium channels and glutamate receptors (Furukawa and Mattson, 1998), which may explain the stabilizing effect of TNF on neuronal calcium metabolism (Cheng et al., 1994; Bruce et al., 1996). Nuclear factor-kappa B activation may also be implicated in TNF-mediated induction of antioxidant enzymes in neurons (Mattson et al., 1997). Binding of TNF to the p75 receptor may both transduce growth and cellular activation signals as well as activate intracellular cascades, leading to cell death (Rao et al., 1995).
Tumor necrosis factor induces activation of vascular endothelium (Barone et al., 1997) and adhesion molecule expression (Yang et al., 1998), which attract leukocytes to pass from the blood circulation into the ischemic brain. Within the brain, TNF might modulate the inflammatory processes by activation of microglia and macrophages (Colasanti et al., 1995; Winston et al., 1995; McLeish et al., 1998) and induction of respiratory burst in polymorphonuclear leukocytes (PMNs)(McLeish et al., 1998; Ottonello et al., 1998). Furthermore, TNF stimulates the production of inducible nitric oxide synthase and nitric oxide in astrocytes (Brodie et al., 1998) as well as prostaglandin E2 and collagenase production by human mononuclear cells (Dayer et al., 1985). Tumor necrosis factor has also been shown to increase the synthesis of and synergize with other inflammatory cytokines like interleukin-1β and interleukin-6, which are expressed in cerebral ischemia (Buttini et al., 1994; Wang et al., 1995; Saito et al., 1996). Among these, interleukin-1β is known to contribute to ischemia-induced nerve cell degeneration (Loddick and Rothwell, 1996; Stroemer and Rothwell, 1998).
The time course of TNF mRNA and protein expression has been established in ischemia by a number of standard molecular approaches. In hypoxia/ischemia in perinatal rats, reverse transcription-combined polymerase chain reaction showed a peak of TNF mRNA at 4 hours (Szaflarski et al., 1995); in t- and pMCAO in adult rats, Northern blotting showed maximal TNF mRNA expression at 3 and 12 hours, respectively (Liu et al., 1994; Wang et al., 1994); and in a model of transient global cerebral ischemia in gerbils, elevated levels of TNF were detected at 1 and 24 hours by enzyme-linked immunoassay techniques (Saito et al., 1996). However, the cellular sources of TNF in the ischemic brain still need to be defined. One of the few studies using in situ hybridization to locate TNF mRNA in brain ischemia points to microglia/macrophages as the cellular source in focal cerebral ischemia in rats (Buttini et al., 1996), whereas others have reported neuronal TNF mRNA expression in a model of traumatic hippocampal injury in mice (Tchelingerian et al., 1994). Similarly, immunohistochemical studies have suggested that TNF is localized to either ischemic neurons (Liu et al., 1994; Tchelingerian et al., 1996), astrocytes (Botchkina et al., 1997; Uno et al., 1997), brain vasculature (Botchkina et al., 1997), and/or microglia/macrophages (Liu et al., 1994; Botchkina et al., 1997; Uno et al., 1997) within the ischemic brain. This is in contrast to in vitro data pointing to activated microglia and secondarily activated astrocytes as major producers of TNF (Sawada et al., 1989; Lafortune et al., 1996). In inflammatory brain disease like fatal murine cerebral malaria and experimental allergic encephalomyelitis, TNF mRNA and protein are also associated primarily with microglia/macrophages and astrocytes (Renno et al., 1995; Villarroya et al., 1996; Medana et al., 1997).
In the present in situ hybridization study, we demonstrate by the use of enzyme-labeled oligodeoxynucleotide probes that TNF mRNA is expressed in a time-restricted manner from 6 hours and up to 2 days after surgery in cells located primarily at the edge of and within the infarct core in C57×129 mice. We also show that these TNF mRNA-expressing cells colocalize with Mac-1-positive microglia and macrophages and not Ly-6G (Gr-1)-positive PMNs, just as they do not coexpress the astrocytic marker glial fibrillary acidic protein (GFAP). Finally, we demonstrate by Western blotting analysis of brain homogenates from C57Bl/6 mice, which display the same TNF mRNA expression profile as C57×129 mice, that soluble and transmembranous TNF protein are induced in the brain after pMCAO, suggesting that microglial/macrophage TNF mRNA is translated into bioactive protein. In combination, our findings support the interpretation of microglia/macrophages as the major sources of TNF in pMCAO in mice.
MATERIALS AND METHODS
Experimental animals
The study was performed using age-matched, young adult, male C57×129 mice (bred in our own facility) and age-matched, young adult, male C57Bl/6 mice (Bomholtgaard A/S, Skensved, Denmark). All mice were given free access to food and water. Forty-six C57×129 mice, divided into six experimental groups with 6 to 8 mice per group, and 21 C57Bl/6 mice, divided into four experimental groups, were subjected to pMCAO. The groups of C57×129 mice were killed at either 6 or 12 hours or 1, 2, 5, or 10 days after surgery. One or two mice per group showing no histological signs of infarction, indicating unsuccessful MCAO, were included as sham-operated control mice. The C57Bl/6 mice were killed at 2 hours (n = 1), 12 hours (n = 7), 48 hours (n = 5), or 5 days (n = 5). All mice were housed and cared for in accordance with the protocols and guidelines approved by the Danish Animal Health Care Committee.
Focal ischemia
The left distal MCA was permanently occluded (Míller et al., 1995) under Hypnorm (fentanyl citrate 1.3 μg/10 g and fluanisone 40 μg/10 g subcutaneously; Janssen-Cilag, Denmark) and diazepam (20 μg/10 g subcutaneously; Dumex, Denmark) anesthesia. The mice were placed on an operating table at 37.5°C under a dissecting microscope, and an incision was made between the left lateral part of the orbit and the left ear. The parotid gland and the temporal muscle were pushed in the distal direction with a pair of anatomical forceps. A small craniotomy was made with a 0.8-mm burr above the anterior distal branch of the MCA using a high-speed microdrill. The inner layer of the skull was removed with fine forceps, and the dura and arachnoid were carefully opened. The MCA was bipolarly electrocauterized, applying forceps coupled to an electrosurgical unit(ICC50; Erbe, Germany). The muscle and soft tissue were replaced, and the skin was closed using 4-0 nylon suture. One milliliter of physiological 0.9% NaCl was given subcutaneously. The duration of anesthesia was 6 to 8 hours. Mice were kept for 20 hours in a recovery room at 28°C before they were transferred to the normal animal facility unit.
Decapitation
For histological processing, mice were decapitated after cervical dislocation, and brains were rapidly removed, frozen in CO2 snow, and stored at −80°C until sectioning.
Histological processing
The brains of C57×129 mice were cut into serial 16 μm (three to four mice per group) or 30 μm (four to five mice per group) coronal cryostat sections, and the brains of C57Bl/6 mice were cut into 30 μm serial slices (two mice per group). Parallel sections were Nissl stained; in situ hybridized for murine TNF mRNA, the “housekeeping” enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA, or myelin basic protein (MBP) mRNA; or reacted immunohistochemically for the microglial/macrophage Mac-1 antigen (Perry et al., 1985), the PMN Ly-6G (Gr-1) antigen (Fleming et al., 1993), or the isotype control (IgG2b) antibody for both the Mac-1 and the PMN stain. A number of sections were also subjected to combined in situ hybridization for TNF mRNA and immunohistochemistry for the astroglial marker GFAP (Bignami and Dahl, 1985).
In situ hybridization
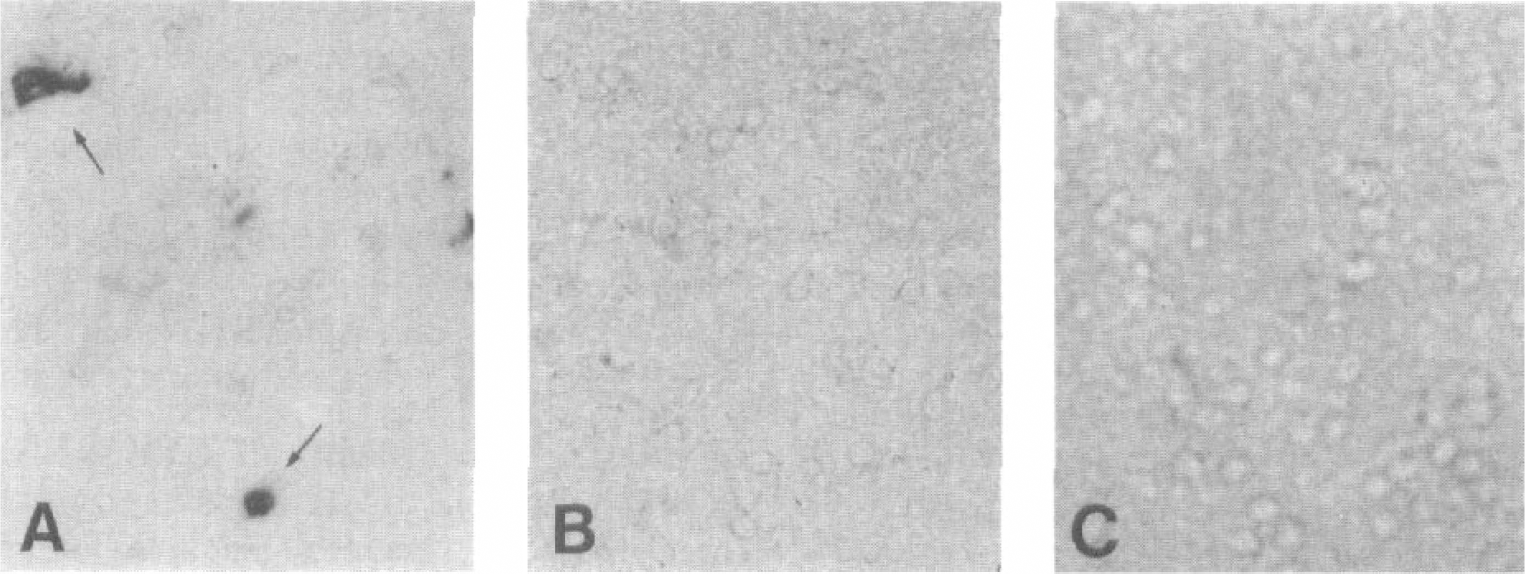
Test of the specificity of the tumor necrosis factor (TNF) mRNA hybridization signal.
Infarct volume estimation
Infarct areas or total infarct volumes were estimated on 30-μm Nissl-stained sections using the computer-assisted cast-grid microscope system (Olympus, Denmark) and Cavalieri principle for volume estimation (Moller et al., 1995; West et al., 1996). The number of points hitting the infarct (P) were counted on sections with a mean distance (t) of 0.36 mm. The sum of the infarct areas of different brain slices was calculated by multiplying the total number of counted points (ΣP) with the computer-given factor of area per point (Apoint). The total infarct volume (Vtotal) was calculated using the following formula: Vtotal = ΣP × Apoint × t. Volume estimation of the hemispheres or of the whole brain (without the cerebellum) was performed similarly to infarct volume assessment.
Density of TNF mRNA-expressing cells
Cells displaying a moderate to strong TNF mRNA hybridization signal in the infarct and bordering area were visualized by 500× amplification in the cast-grid microscope system. All TNF mRNA-expressing (TNF mRNA-positive) cells on ~10 hybridized sections within a distance of 360 μm were counted per animal using a 100% frame area stepping 290/290 in the XY position. The number of TNF mRNA-positive cells in the blank control sections or occasional sections with damaged infarcts was estimated by taking the mean count of the previous and subsequent sections. Cell density was calculated by dividing the total number of counted cells with the estimated infarct areas on parallel Nissl-stained sections. Cell counting was performed only on 30-μm sections.
Statistical analysis
Group mean ± SD of infarct volumes or cell density were compared by unpaired Student's t-test. Statistical significance was set at P < 0.05.
Western blotting for TNF
C57Bl/6 mice were given an overdose of pentobarbital (7278631; Den kgl. Veterinær-og Landbohøjskoles Apotek, Denmark) at 2 hours (n = 1), 12 hours (n = 5), 48 hours (n = 4), and 5 days (n = 4) after pMCAO surgery and perfused with 20 mL of ice-cold 0.1 mol/L phosphate-buffered saline (0.01 mol/L Na2HPO4, 2 × H2O, 2 mmol/L KH2PO4, 0.2 mol/L NaCl, and 3 mmol/L KCl) through the heart. The brains turned all completely white and were taken out after removal of the meninges. The ipsilateral and contralateral neocortices were dissected and homogenized in RIPA buffer [10 mmol/L sodium phosphate (pH 7.5; 1.7 mmol/L NaH2PO4, 1 × H2O, and 8.3 mmol/L Na2HPO4), 150 mmol/L NaCl, 1 mmol/L dithiothreitol (30077; Sigma-Aldrich, Denmark), 1% Nonidet P-40 (155942; ICN Biomedicals, U.S.A.), 1% sodium deoxycholate (5921; Sigma-Aldrich), 0.1% sodium dodecyl sulfate (1028685; Boehringer-Mannheim GmbH, Germany), and 1 mmol/L phenylmethylsulfonyl fluoride (6198; Sigma-Aldrich)]. Protein concentrations were estimated spectrophotometrically at 595 nm following 5-minute incubation with the Bio-Rad protein dye reagent (500-0006; Bio-Rad, Hercules, CA, U.S.A.). For the Western blotting assay, samples of 20 μg of protein brain homogenates were loaded and separated on a NuPAGE 10% Bis-Tris gel (NP0301; Novex, San Diego, CA, U.S.A.) along with a SeeBlue prestained standard (LC5625; Novex), 0.5 ng of the nonglycosylated 17-kDa murine recombinant TNF (T-7539; Sigma-Aldrich) as “positive” control for the N-linked glycosylated 19-kDa TNF band, and 1:10,000 dilution of normal rabbit serum (X0902; Dako) as “negative” control for the TNF-specific bands. The protein bands were transferred to a polyvinylidene difluoride membrane (IPUH00010; Millipore, Denmark), and unspecific binding was blocked by a 10-minute rinse in 2.5% dry milk (M248; Matas, Denmark) followed by 3 × 15-minute rinses in 0.2 mol/L TBS and 0.05% Tween-20 (pH 7.6; 2184; Merck) at room temperature. The membrane was then incubated with a 1:250 dilution of a rabbit anti-mouse TNF antibody (IP-400; Genzyme, Cambridge, MA, U.S.A.) at 4°C overnight. The membrane was rinsed 3 × 15 minutes in 0.2 mol/L TBS and 0.05% Tween-20 and incubated for 1 hour with an AP-conjugated antibody to rabbit Ig (1:230; A-3812; Sigma-Aldrich) for 1 hour at room temperature. Following 3 × 15-minute rinses in 0.2 mol/L TBS and 0.05% Tween-20 and a 10-minute rinse in Tris-HCl (pH 9.5), the AP signal was visualized by 5- to 7-minute exposure to NBT/BCIP (identical to the developer used for in situ hybridization).
RESULTS
Infarct development
The pMCAO caused unilateral cortical infarction within the parietal and insular cortices. The infarcts displayed characteristic Nissl-stainable shrunken neurons with hyperchromatic nuclei (not shown). Mean infarct volumes remained constant from 6 hours and up to 5 days after surgery (Fig. 2) and were ~25 mm3 or occupied ~8% of the total brain (minus cerebellum) volume. Infarct volumes at day 10 were significantly smaller due to resorption of the infarcted tissue (Fig. 2).
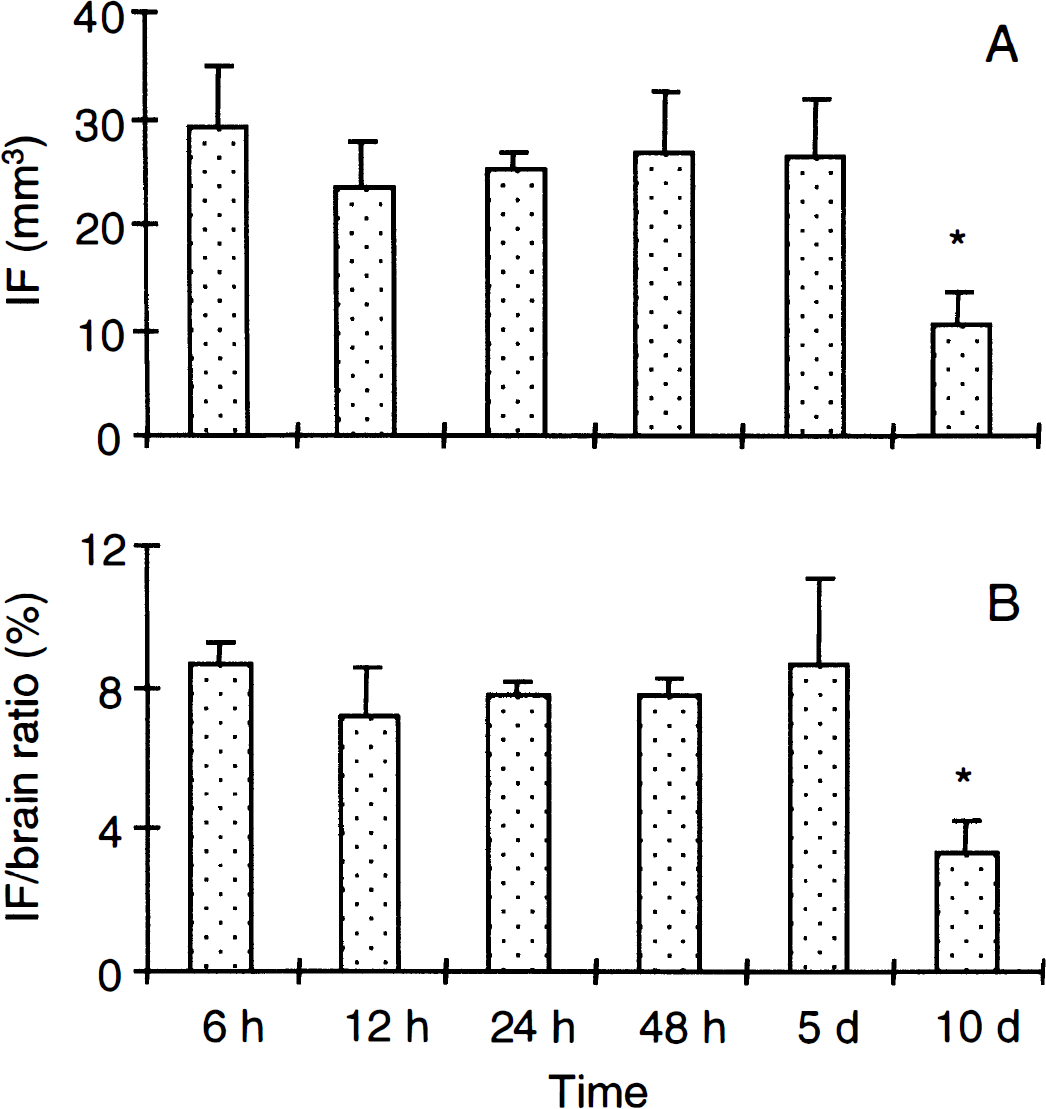
Infarct (IF) volume estimation.
Regional and temporal localization of TNF mRNA-expressing cells
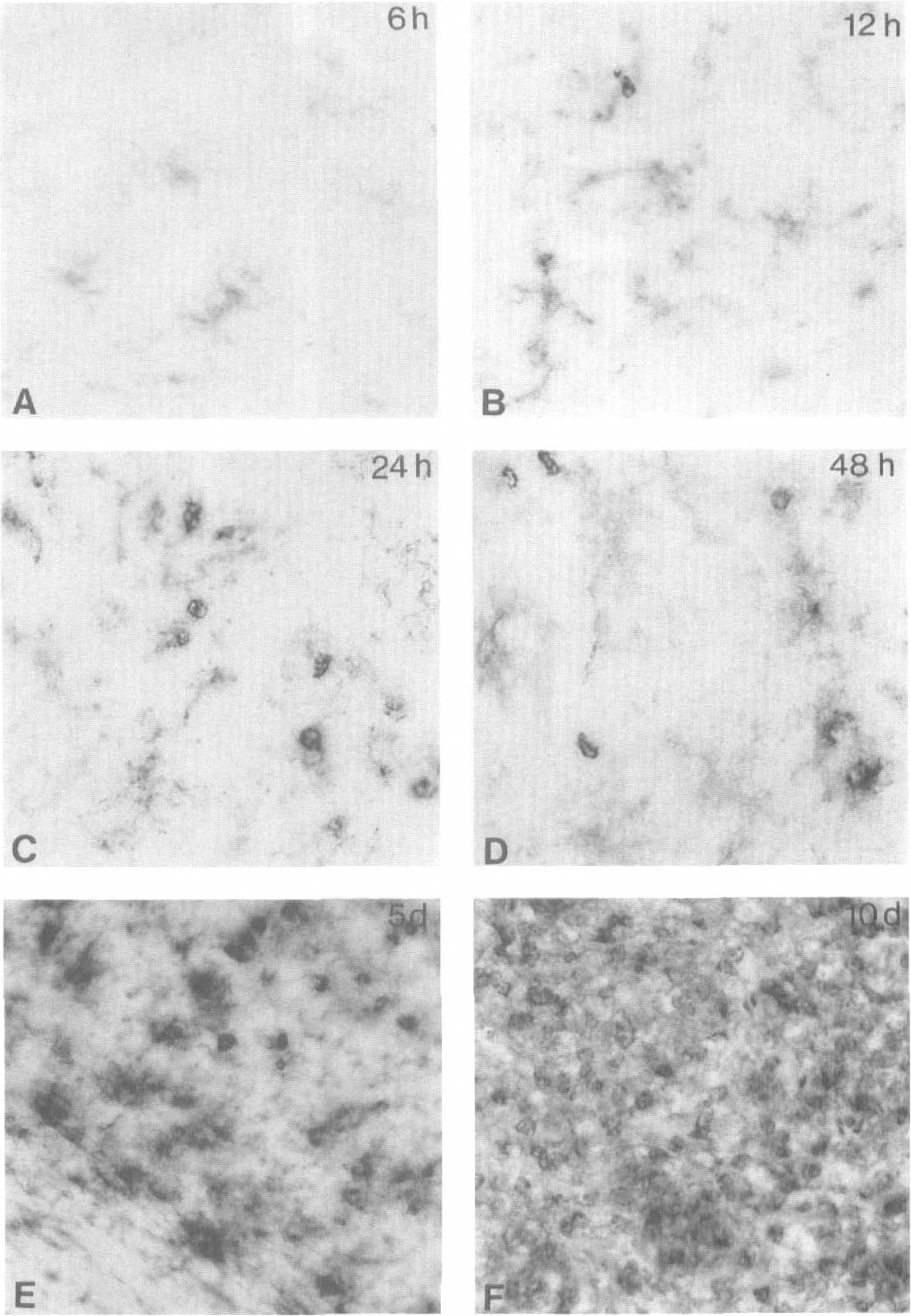
At 6 and 12 hours and 1 day after pMCAO, moderate to high levels of TNF mRNA were expressed in process-bearing microglia/macrophage-like cells located in variable densities at the margin of the infarcted areas and within the infarct core (Figs. 3 to 5). Brain sections from mice with 2-, 5-, or 10-day survival displayed no or minimal TNF mRNA signal (Fig. 3). Cast-grid analysis of the temporal density of TNF mRNA-positive cells in the infarct and at the infarct border showed peak density at 12 hours, with markedly lower density at 6 and 24 hours (Fig. 4). Strongly GFAP-immunoreactive astrocytes were present at the boundary of the infarct and in the corpus callosum and capsula externa (Fig. 5). TNF in situ hybridization combined with immunohistochemistry for astroglial GFAP gave no evidence of TNF mRNA expression by GFAP-immunoreactive astrocytes (Fig. 5).
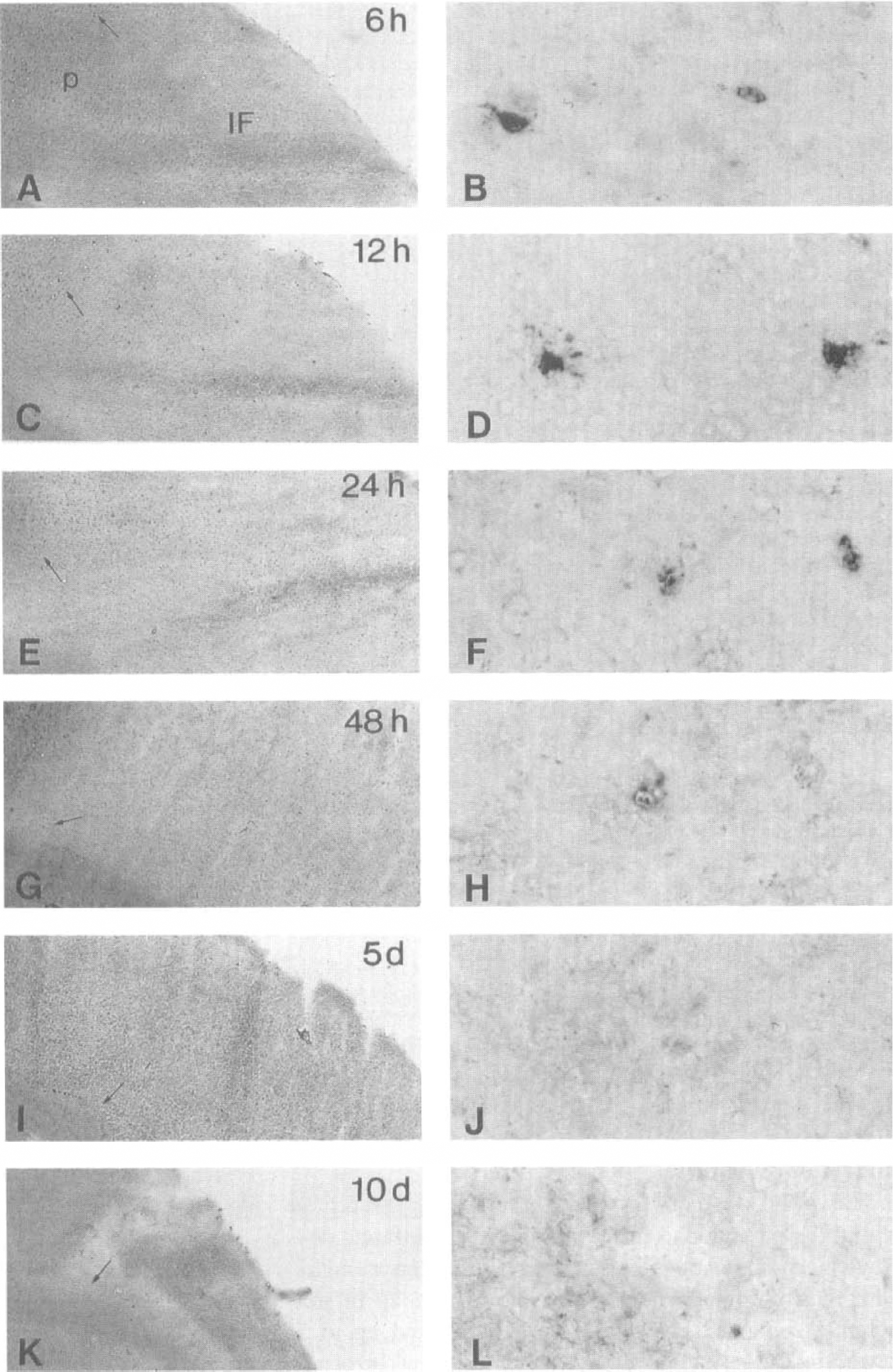
Time course and distribution of tumor necrosis factor (TNF) mRNA-positive cells within infarct (IF) and infarct border (P) at 6 hours (
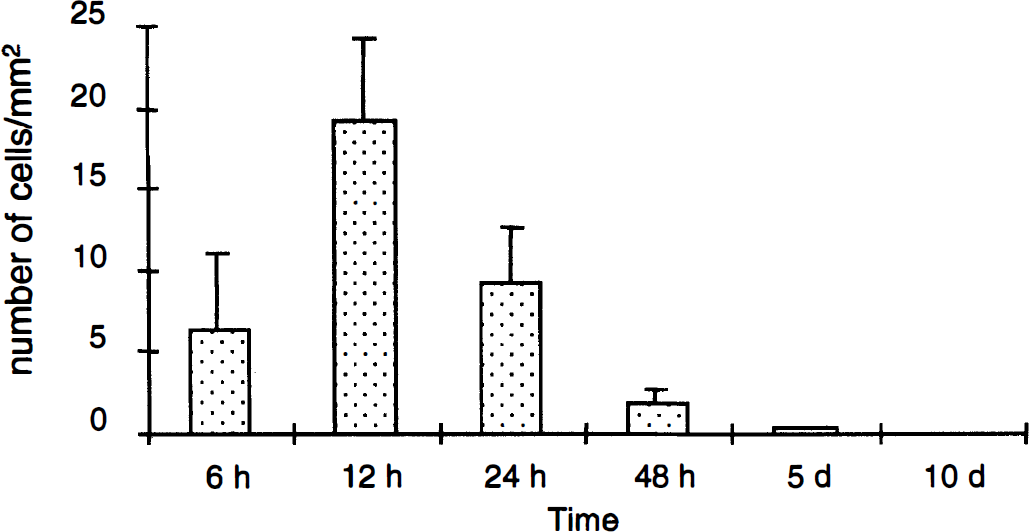
Estimation of the temporal density of tumor necrosis factor (TNF) mRNA-positive cells per square millimeter of infarct and infarct border after permanent middle cerebral artery occlusion surgery. Group means ± SD were as follows: 6 hours (6.4 ± 4.6/mm2; n = 3), 12 hours (19.2 ± 5.1/mm2; n = 4), 24 hours (9.2 ± 3.4/mm2; n = 4), 48 hours (1.3 ± 0.2 /mm2; n = 2), 5 days (0.3 ± 0.2/mm2; n = 5), and 10 days (0.0 ± 0.0/mm2; n = 6), revealing highest densities at 12 hours.
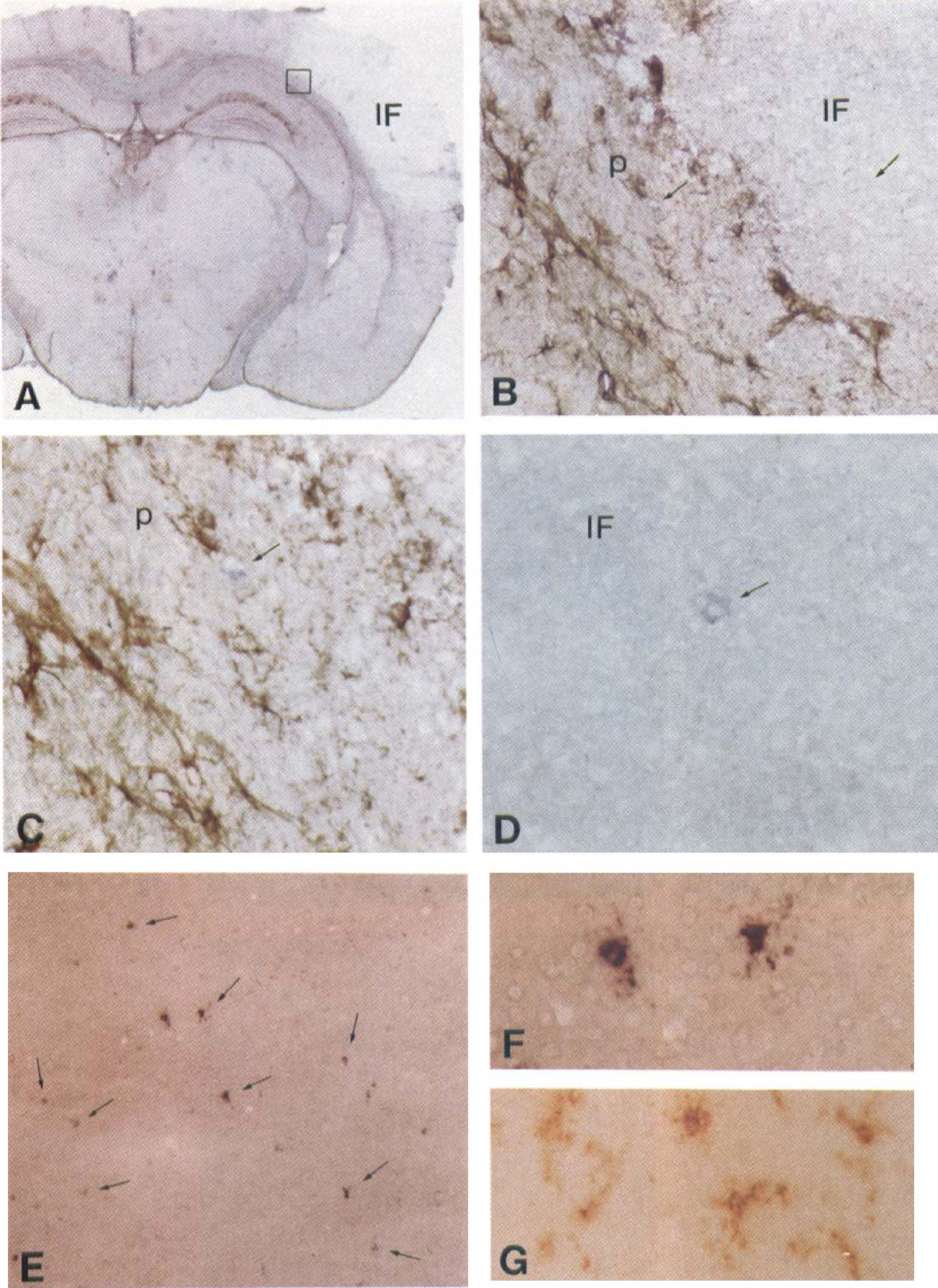
Permanent middle cerebral artery occlusion-induced cortical infarction at 12 hours. Shown is immunohistochemistry for astroglial glial fibrillary acidic protein (GFAP; brown signal) combined with in situ hybridization for tumor necrosis factor (TNF) mRNA (blue signal).
Mac-1 stain
At all time points, pMCAO-induced infarcts were demarcated by up-regulated levels of Mac-1 (Figs. 6 and 7). At 6 hours, microglial cells at the edge of the infarct were lightly activated, showing up-regulated levels of Mac-1 and slightly retracted processes. This microglial reaction had further increased at 12 hours, at which time typical microglial profiles no longer were visible within the core of the infarct. Also, at 12 hours and especially at 24 hours, infiltrating round Mac-1-positive macrophages appeared adjacent to vascular endothelium or meningeal tissue. These cells were distributed within cortical layers 1 to 3 at 12 to 24 hours, layers 3 to 4 at 48 hours, and cortical layers 4 to 6 of the infarct core (Fig. 6). “Bushy”-like microglia/macrophages were observed within the corpus callosum in both hemispheres and in the contralateral neocortex at days 5 and 10. At day 10, the partially resorbed infarct still contained intensely labeled Mac-1-positive macrophages (Fig. 7).
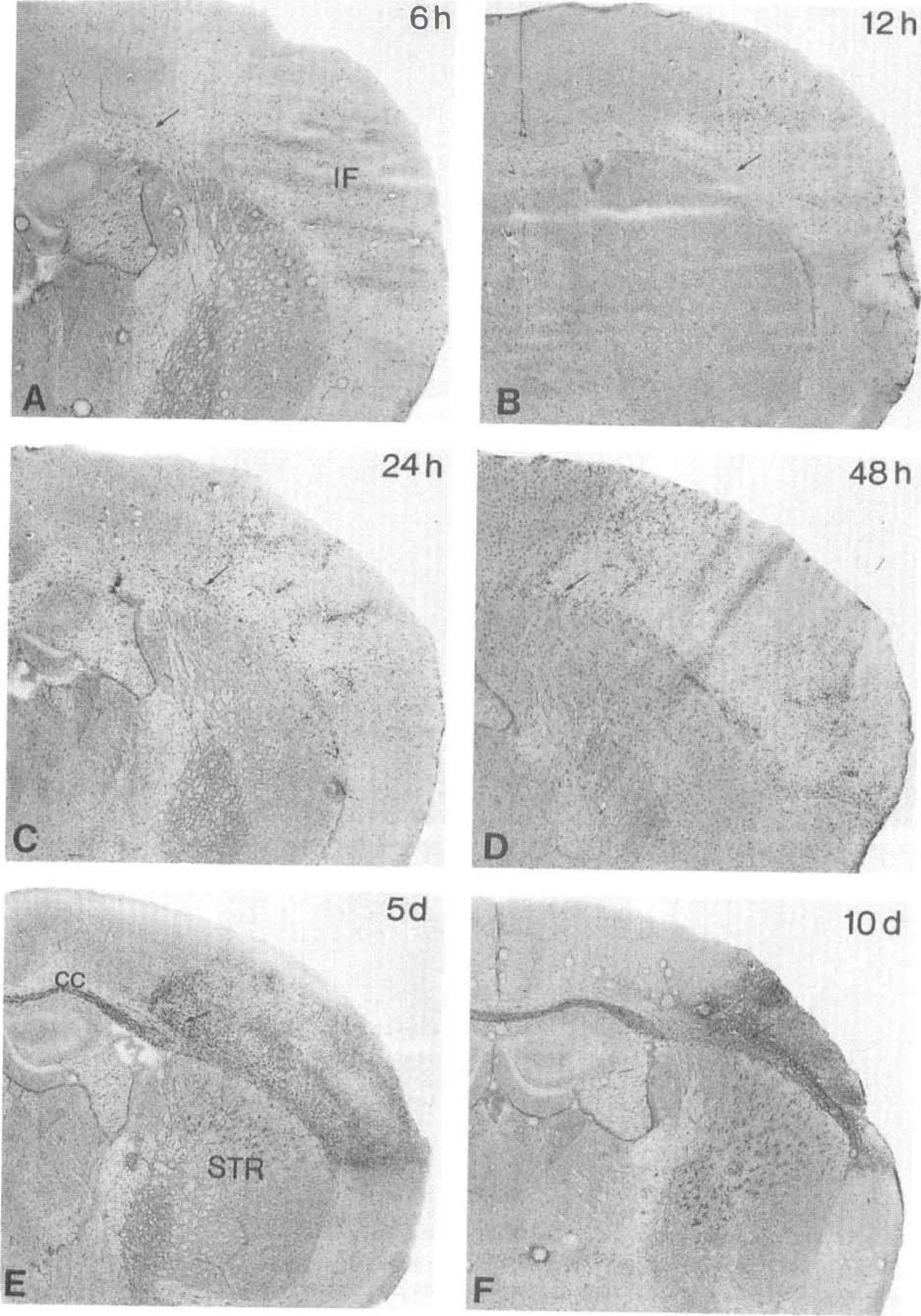
Mac-1-positive microglial/ macrophage responses after permanent middle cerebral artery occlusion at 6 hours
Mac-1-positive microglial/ macrophage responses at the infarct border at 6 hours
Polymorphonuclear leukocytes
From 24 to 48 hours after the onset of ischemia, PMNs extravasated from blood vessels located in the border zone of the infarct and in the corpus callosum (Fig. 8D). Whereas no PMNs were present at day 5, these cells reappeared at day 10, when they were abundant within and at the edge of the necrotic infarct and in the corpus callosum.
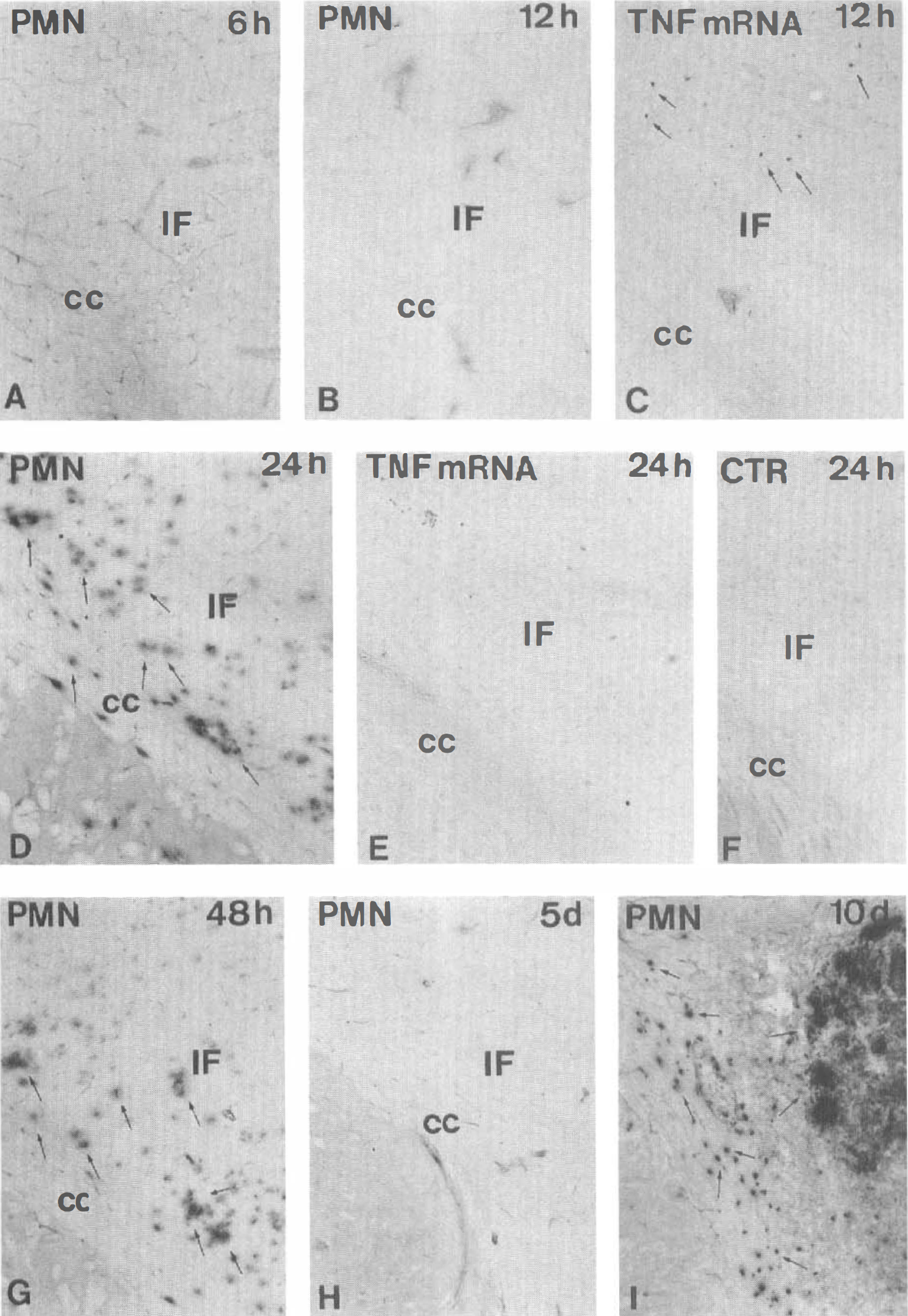
Polymorphonuclear leukocytes (PMNs) do not express tumor necrosis factor (TNF) mRNA in permanent middle cerebral artery occlusion. The PMNs are not present at 6 hours
TNF protein
Murine TNF soluble protein (19-kDa band) and the TNF transmembranous form (22-kDa band) were detected in homogenates of ipsilateral neocortex of C57Bl/6 mice at 2 hours, 12 hours, 48 hours, and 5 days after pMCAO surgery (Fig. 9) and in the contralatereal neocortex at 12 hours (Fig. 9). Furthermore, 14-kDa bands appeared, which were taken to represent degradation products of TNF. One control mouse (with craniotomy but no artery occlusion) showed almost undetectable TNF levels [or 14-, 19-, and 22-kDa bands] at 12 hours (Fig. 9). The “positive” control, murine recombinant TNF, gave in all cases a strong band at 17 kDa.
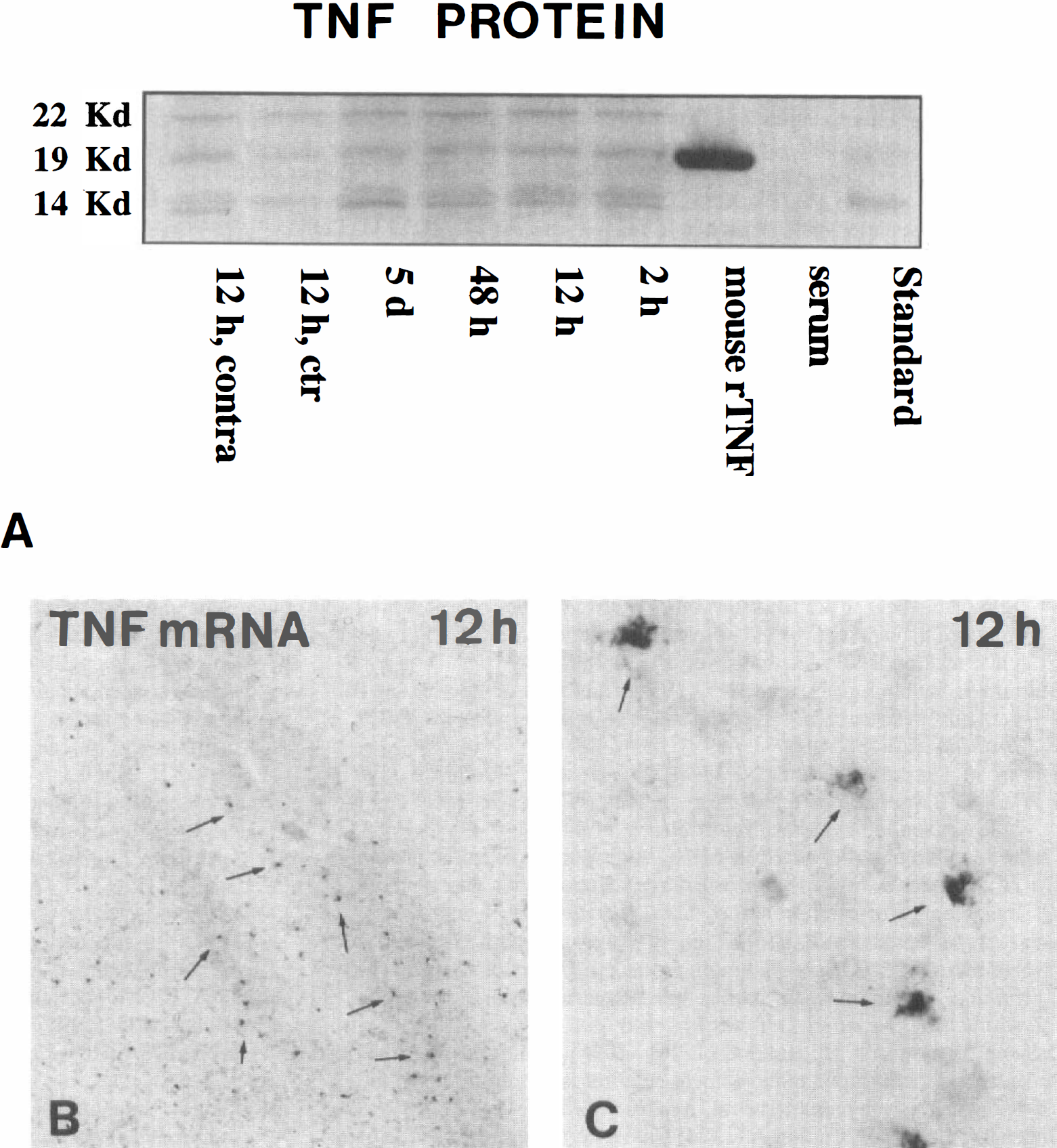
Western blot for tumor necrosis factor (TNF) protein
DISCUSSION
In the present study, we report on an early postsurgery induction of TNF protein in a murine model for focal cerebral ischemia. To determine the cellular origin of such TNF production in ischemia-induced neuropathology, a sensitive and reliable nonradioactive in situ hybridization method was established for visualization of TNF mRNA in brain tissue. This method revealed a distinct cellular TNF mRNA in situ hybridization signal within the infarct core and at the edge of the infarct from 6 to 48 hours after induction of pMCAO in C57×129 mice, with peak density of TNF mRNA-expressing cells at 12 hours. Importantly, neither astrocytes nor neurons were shown to express TNF mRNA. This was surprising as TNF protein has been reported to be produced by neurons in focal cerebral ischemia (Liu et al., 1994) and TNF mRNA to be expressed in neurons in adult mouse brain after traumatic hippocampal injury (Tchelingerian et al., 1996). The striking lack of PMNs at time points with high TNF mRNA expression and the absence of colocalization between PMNs and cellular TNF mRNA expression document that PMNs were not responsible for the observed TNF mRNA expression. In contrast, TNF mRNA expression colocalized with Mac-1-positive microglia/macrophages, thereby pointing to activated microglia and infiltrating blood-borne macrophages as the major sources of TNF from 6 to 48 hours after induction of pMCAO in mice. Estimated from the morphological characteristics, these Mac-1-positive cells were most probably infiltrating blood-borne macrophages within the infarct and activated microglial cells or a mixture of activated microglia and blood-borne macrophages at the infarct border. The early expression of TNF mRNA corresponds well with the impression of microglia/ macrophages to be robust and potent responder cells in the early stages of the pathological process after brain ischemia (Morioka et al., 1991; Finsen et al., 1996; Lehrmann et al., 1997). Furthermore, the absence of TNF mRNA-expressing cells at days 5 and 10 when the microglia/macrophage reaction reached its maximum suggests a finely tuned regulation of microglia/macrophage TNF gene expression and function in pMCAO.
Interestingly, we observed elevated levels of TNF protein at day 5, at which time the number of TNF mRNA-expressing cells was very low, in both the C57×129 hybrid mice as well as the C57Bl/6 mice. The observation of very low numbers of TNF mRNA-expressing cells in both strains of mice makes it unlikely that the discrepancy in TNF gene transcription and protein synthesis should be due to strain differences. A more plausible explanation might be that TNF at day 5 is expressed broadly by many cells, possibly also reactive astrocytes and neurons, but in such small levels per cell that our in situ hybridization technique is unable to visualize the transcriptionally active cells. Provided that sufficient numbers of cells are actively producing TNF, the Western blotting technique may, however, still show a strong signal under circumstances with a low rate of protein synthesis per cell. This explanation would also be in line with findings of Liu et al. (1994), who by Northern blotting analysis observed an almost identical temporal TNF mRNA expression profile in pMCAO in rats (Liu et al., 1994), except that TNF mRNA levels in that study were still significantly elevated in the ischemic cortex at day 5. Another explanation for the observation of TNF protein at day 5, when virtually no TNF mRNA-expressing cells can be observed, is that the transmembranous form of TNF may be present in microglia/macrophages and may continue to be cleaved from the membrane after down-regulation of TNF gene transcription. Importantly, from a functional point of view, the expression of TNF mRNA seems to follow the temporal profile of microglial/macrophage-derived interleukin-1β mRNA, which in rat pMCAO also peaks at 12 hours (Liu et al., 1993; Yang et al., 1998), thus opening the possibility of a synergistic effect of microglia/macrophage-produced TNF and interleukin-1β in the ischemia-induced pathology.
To our knowledge, this is the first study to describe the cellular localization of TNF mRNA in permanent focal cerebral ischemia in mice. Our presentation of equal infarct volumes from 6 hours and up to 5 days after the arterial occlusion points to the evolution of infarction within the first hours after the onset of permanent ischemia. This, taken together with the apparent role of TNF in ischemic neurodegeneration (Bruce et al., 1996; Dawson et al., 1996; Barone et al., 1997; Nawashiro et al., 1997a; Lavine et al., 1998; Yang et al., 1998) and the findings of elevated levels of TNF mRNA and protein very early after surgery in several ischemia models (Liu et al., 1994; Wang et al., 1994; Szaflarski et al., 1995; Saito et al., 1996; Buttini et al., 1996; Botchkina et al., 1997; Uno et al., 1997; present study), emphasizes the need for a further characterization of the cellular TNF mRNA expression within the first few hours after pMCAO.
CONCLUSION
We conclude that microglia and macrophages are major sources of TNF after pMCAO in mice. This observation points to a pivotal function of these cells in TNF-mediated neurotoxic and neuroprotective effects in cerebral ischemia.
Footnotes
Acknowledgments
The authors thank Arne Møller (NeuroSearch) for assistance with the pMCAO model and principles for infarct volume estimation and Trevor Owens for comments on the manuscript. The authors also thank Lene Jørgensen, Inger Margrethe Rasmussen, Dorte Frederiksen, Lily Poulsen, Grethe Jensen, and Albert Meier for excellent technical assistance.