
Abstract
We tested whether significant leukocyte infiltration occurs in a mouse model of permanent cerebral ischemia. C57BL6/J male mice underwent either permanent (3 or 24 hours) or transient (1 or 2 hours+22- to 23-hour reperfusion) middle cerebral artery occlusion (MCAO). Using flow cytometry, we observed ∼15,000 leukocytes (CD45+high cells) in the ischemic hemisphere as early as 3 hours after permanent MCAO (pMCAO), comprising ∼40% lymphoid cells and ∼60% myeloid cells. Neutrophils were the predominant cell type entering the brain, and were increased to ∼5,000 as early as 3 hours after pMCAO. Several cell types (monocytes, macrophages, B lymphocytes, CD8+ T lymphocytes, and natural killer cells) were also increased at 3 hours to levels sustained for 24 hours, whereas others (CD4+ T cells, natural killer T cells, and dendritic cells) were unchanged at 3 hours, but were increased by 24 hours after pMCAO. Immunohistochemical analysis revealed that leukocytes typically had entered and widely dispersed throughout the parenchyma of the infarct within 3 hours. Moreover, compared with pMCAO, there were ∼50% fewer infiltrating leukocytes at 24 hours after transient MCAO (tMCAO), independent of infarct size. Microglial cell numbers were bilaterally increased in both models. These findings indicate that a profound infiltration of inflammatory cells occurs in the brain early after focal ischemia, especially without reperfusion.
Keywords
INTRODUCTION
Stroke is a leading cause of death and disability worldwide, and accounts for ∼1 in every 18 deaths in the United States. 1 Recanalization of occluded vessels with intravenously administered recombinant tissue plasminogen activator within 4.5 hours of onset of stroke symptoms is currently the only approved acute stroke treatment. 2 However, the requirement for CT confirmation of an ischemic stroke together with the short therapeutic window limits its utility.
Approximately 10% to 15% of all ischemic brain infarcts affect large sections of one hemisphere, leading to ‘malignant’ media infarctions—a decisive factor for early mortality.3, 4 ‘Malignant middle cerebral artery (MCA) infarction’ describes a massive, space-occupying total or subtotal infarction in the territory of the MCA.4, 5 It is typically caused by an embolic occlusion of a distal internal carotid artery or proximal MCA segment. A very low recanalization rate (either spontaneous or therapeutic) of such occlusions results in ischemic lesions involving substantial parts of the affected MCA territory. Secondary to the initial damage, massive brain edema develops in the initial days after ischemia, leading to progressively elevated intracranial pressure. These events result in the involvement of formerly unaffected tissue and might even cause supratentorial herniation. In the subgroup of patients who develop malignant MCA infarction there is a mortality rate of 80%, compared with 5% to 45% in all patients with acute ischemic MCA territory stroke.4, 6 For treatment, decompressive hemicraniectomy surgery may be employed to normalize intracranial pressure and increase survival rate but it provides limited improvement in infarct volume and functional outcomes. 7 Opportunities to identify new directions that may enable treatment of acute stroke, especially in severe cases in which reperfusion has not occurred, must therefore be explored as a matter of urgency.
There is now strong evidence that inflammatory processes may contribute to secondary brain damage after ischemic stroke, and that this involves numerous types of immune cells infiltrating the postischemic brain with varying numerical, temporal, and spatial profiles after reperfusion. 8 These infiltrating cells include neutrophils, monocytes, macrophages, dendritic cells, T and B lymphocytes, and natural killer cells. 8 For example, clinical and experimental data suggest that neutrophils are a key immune cell to enter the brain after ischemia, and neutrophil number correlates with the severity of an ischemic lesion,9, 10 although there is some controversy as to whether this is an early event or is delayed by up to 3 days. 8 It is also remains controversial as to whether neutrophils contribute directly to secondary brain damage or are merely bystanders that mainly assist in promoting tissue repair and recovery.11, 12 Further, T lymphocytes are now understood to contribute to ischemic brain injury after reperfusion.13, 14 Thus, Rag1−/− mice that are deficient in lymphocytes appear to be partially protected from severe histochemical and functional outcomes after cerebral ischemia and reperfusion due to the absence of T lymphocytes. 15 By contrast, other infiltrating cells such as regulatory B cells may act to limit severity of stroke outcome after transient cerebral ischemia. 16 Hence, if the identities of culprit immune cell subpopulations and their time course of injury in specific stroke settings can be accurately determined, then it seems plausible that this information could be exploited to develop new interventional therapies to specifically block such effects within a rational therapeutic window of hours to days after stroke.
It is noteworthy that experimental assessment of inflammation and immune cell infiltration after stroke has typically been performed in models of cerebral ischemia with reperfusion. However, the involvement of such processes in brain injury occurring after severe permanent cerebral ischemia (i.e., ischemia with no reperfusion), such as in malignant MCA infarction, remains largely unknown. Indeed, it might be postulated that immune cell infiltration is minimal, or even absent, in the postischemic brain in the absence of reperfusion. Thus, here we have employed a flow cytometric approach to carefully assess the increases in multiple immune cell types in the postischemic mouse brain at an early (3 hours) and late (24 hours) time point in the first day after permanent MCA occlusion (pMCAO). We have compared those findings with the profile observed in other mice subjected to transient (1 hour) MCA occlusion (tMCAO) followed by 23-hour reperfusion. Our findings indicate that, despite the absence of reperfusion, immune cell infiltration occurs relatively quickly after the onset of ischemia, is sustained for at least 24 hours, and involves ∼2-fold more leukocytes than are present in the brain 24 hours after transient ischemia, independently of infarct size.
MATERIALS AND METHODS
Middle Cerebral Artery Occlusion
This study fully adheres to ARRIVE guidelines.
17
All experiments were approved by Monash University Animal Ethics Committee (Project MARP/2011/112) and conducted according to National Health and Research Council Australia guidelines for the care and use of animals in research. Male C57Bl6/J mice (22 to 32 g, 8 to 12 weeks,
Flow Cytometry
On each occasion when flow cytometry was utilized, we studied at least one poststroke mouse together with a time-matched sham-operated control mouse. Animals were euthanased at specified time points after ischemia (3or 24 hours after pMCAO; 24 hours after 1 or 2 hours of tMCAO) by isoflurane inhalation, followed by blood removal and decapitation. Brain, blood, and spleen were collected from each animal for flow cytometric analyses. Blood was collected by cardiac puncture and leukocytes were purified using red blood cell lysis buffer (155 mmol/L NH4Cl, 10 mmol/L KHCO3, and 3
The brain was removed from the skull and after removing the cerebellum, was separated into left (contralateral) and right (ischemic) hemispheres. In some experiments, brains were flushed with PBS before removal and cell counts were compared with those collected in parallel but without flushing (3-hour sham:
Antibodies used for flow cytometry and immunohistochemistry
Gating Strategy
Single cells were identified by forward scatter, and dead cells (7AAD+) were excluded. Cells were gated for CD45+high and CD45+med populations. CD45+high populations (leukocytes) were then divided into lymphoid cells, which include: B cells (B220+), T cells (CD49b+CD90−NK1.1−), thymocytes (CD49b−CD90+NK1.1−), natural killer (NK) cells (CD49b+CD90+NK1.1+) and natural killer T (NKT) cells (CD49b+CD90−NK1.1+); and myeloid cells (CD11b+). CD45+medCD11b+F4/80+ cells were considered as microglia. Two panels of antibodies were used, one of which was employed with each animal. Panel 1 enabled the counting of microglia and myeloid-derived leukocytes (i.e., CD11b+ cells; Supplementary Figure 1A), which included: neutrophils (Ly6G+), monocytes (Ly6C+), macrophages (F4/80+), and dendritic cells (CD11c+); whereas panel 2 divided lymphocytes into B cells (B220+), T cells (CD3+), NKT cells (NK1.1+CD3+), and NK cells (NK1.1+). T cells were then further subdivided into CD4+ T cells, CD8+ T cells, CD4−CD8− T cells, and CD4+CD25+ T cells (Supplementary Figure 1B). Fluorescence-minus-one was included as negative controls to define positive populations for F4/80, CD11c, B220, CD3, and CD25.
Immunohistochemistry
Animals (
Statistical Analysis
All data were analyzed using GraphPad Prism (GraphPad Software Inc., La Jolla, CA, USA) and presented as mean±standard error (s.e.m.). Data for pMCAO from flow cytometry were analyzed using two-way analysis of variance (ANOVA) with Bonferroni post tests for multiple comparison of different times and treatments. Data for tMCAO from flow cytometry and immunohistochemical studies were analyzed using one-way ANOVA with Bonferroni
RESULTS
Characteristics of Mouse Models of Stroke
All mice subjected to pMCAO or tMCAO received ∼80% reduction in cerebral blood flow in the region supplied by the MCA, as assessed by laser-Doppler flowmetry (Supplementary Figure 2A to D). After tMCAO (i.e., 1 or 2 hours of MCAO), regional blood flow increased to the preischemia level within 30 minutes after withdrawal of the filament (Supplementary Figure 2C and D). Eight animals were excluded due to death before the predetermined time of euthanasia (i.e., 24 hours after pMCAO or tMCAO; see the following text for details). Mortality was: 0% (0/15) among mice in the 3-hour pMCAO group, 12% (3/25) in the 24-hour pMCAO group, 23% (3/13) in the 1-hour tMCAO group, and 25% (2/8) in the 2-hour tMCAO group. There were no mortalities among sham-operated control mice. In the surviving animals, pMCAO produced a large infarct throughout the cortex and the striatum, which was evident at 3 hours (Supplementary Figure 2E), and which occupied most of the ipsilateral hemisphere by 24 hours (Supplementary Figure 2F). By comparison, infarct volume was smaller and restricted mainly to the striatum at 23 hours after 1 hour of tMCAO (Supplementary Figure 2G), whereas 2-hour tMCAO plus 22-hour reperfusion produced an infarct that was comparable in size to the pMCAO groups.
Immune Cell Numbers in the Brain After Permanent Middle Cerebral Artery Occlusion
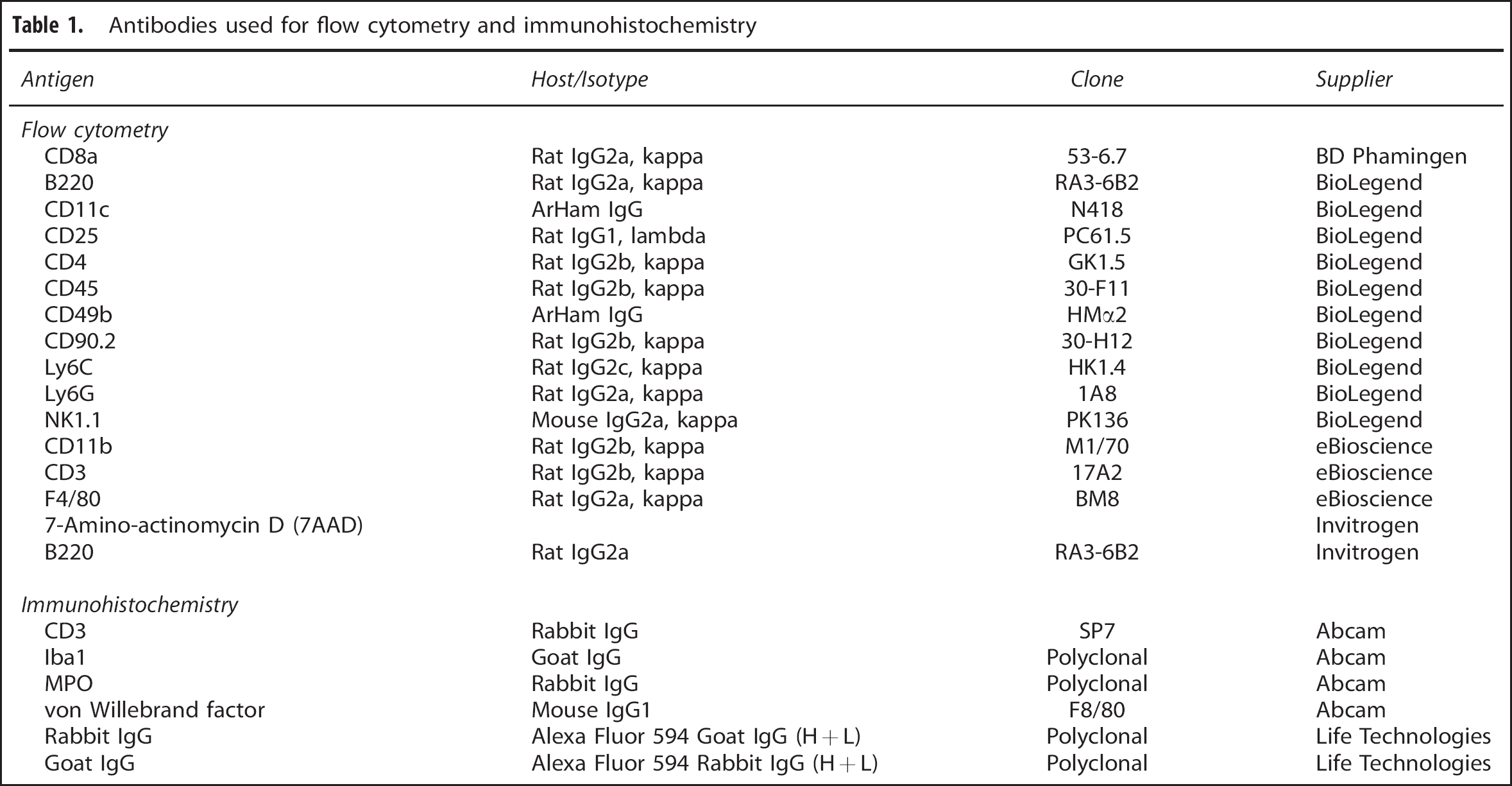
There were a total of ∼15,000 leukocytes present in the ischemic hemisphere at both 3and 24 hours after pMCAO, which was 3- to 5-fold more than in the contralateral (nonischemic) hemisphere or in sham controls (Figures 1A and 4). Of these, lymphoid cells (CD11b−) represented 30% to 40%, whereas myeloid cells (CD11b+) represented 60% to 70% (Figure 1) of total leukocytes.
Flow cytometric quantification of leukocytes in the brain, expressed as total, myeloid, or lymphoid cells, at 3 or 24 hours after permanent middle cerebral artery occlusion (pMCAO) (
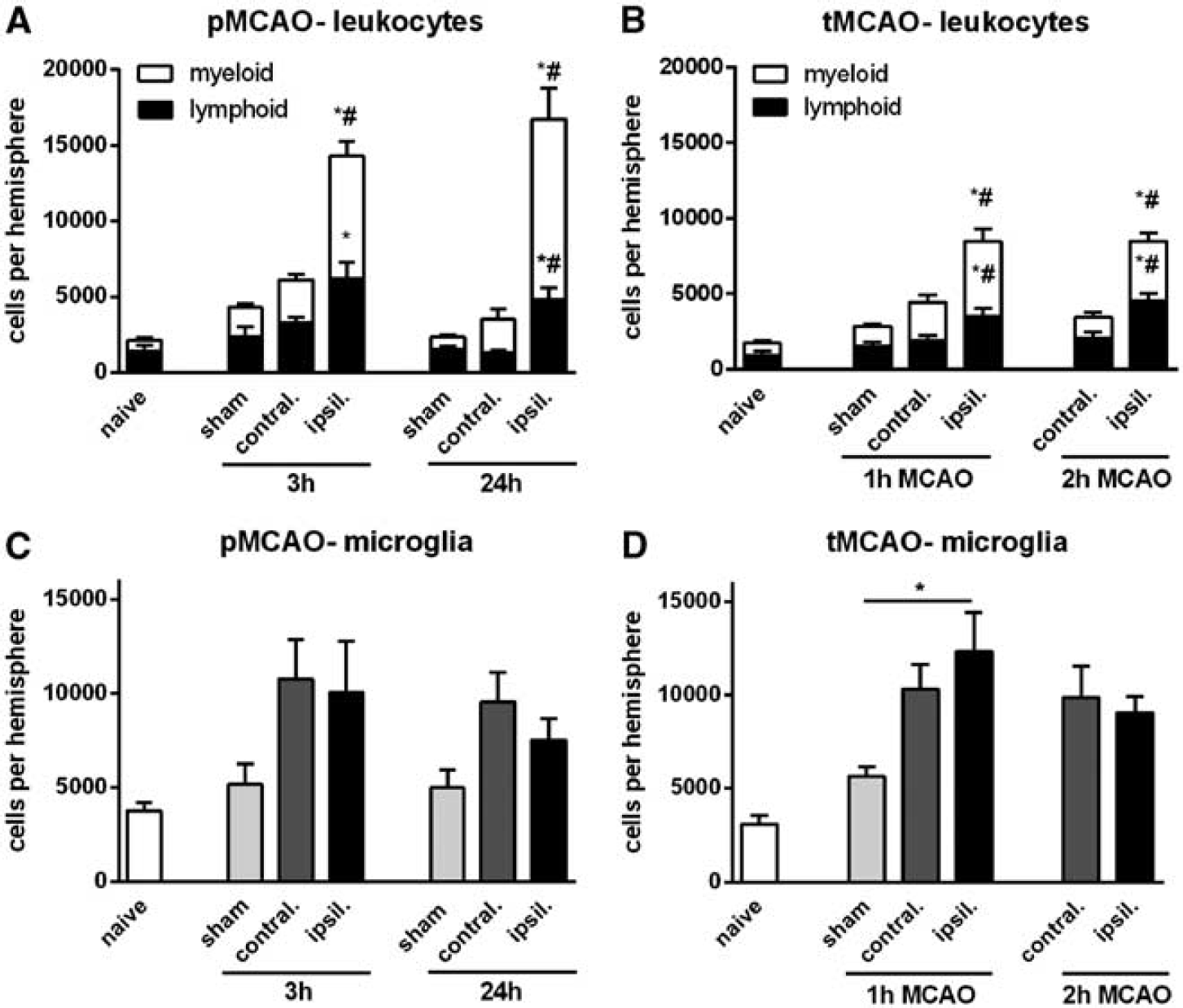
Myeloid cells (CD11b+) were increased by ∼4-fold in the ischemic brain at 3 and 24 hours after pMCAO (Figure 1A). Neutrophils (Ly6G+) comprised >70% of myeloid cells (and up to 60% of all infiltrating leukocytes) after pMCAO, with ∼5,000 cells at 3 hours and ∼9,000 cells at 24 hours (Figures 2A and 4). A 2- to 4-fold increase in macrophages and monocytes (F4/80+ or Ly6C+, respectively) was observed at both 3 and 24 hours (Figures 2D and 2E). Dendritic cells (CD11b+CD11c+) were not increased at 3 hours but were ∼6-fold more numerous at 24 hours (Figure 2F). Numbers of resident brain microglia (CD45+medCD11b+F4/80+) tended to be increased ∼2-fold after pMCAO (Figure 1C).
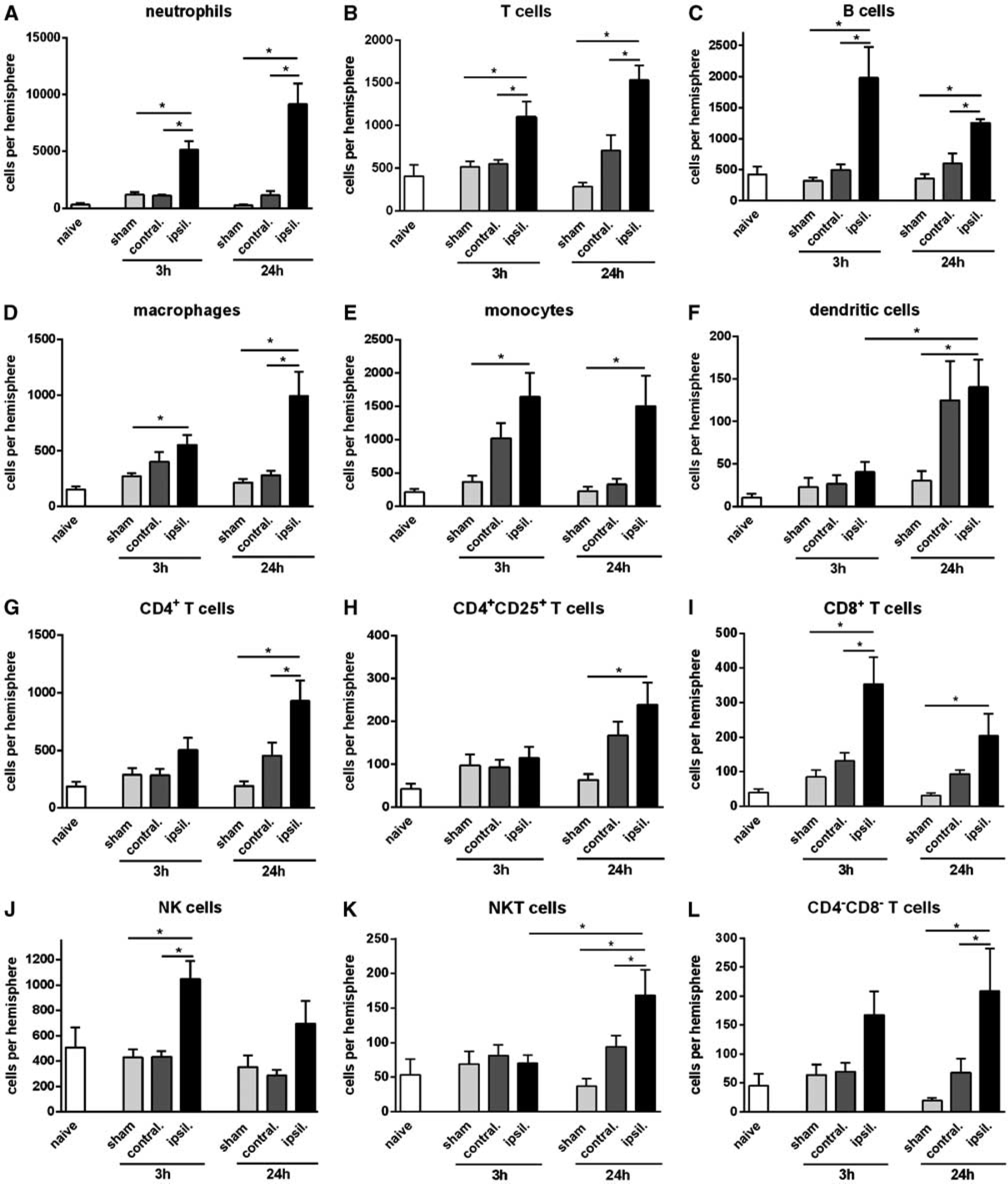
Quantification of leukocyte subtypes in brain at 3 or 24 hours after permanent middle cerebral artery occlusion, compared with naïve and time-matched sham controls (
Lymphoid cells were increased by ∼2-fold at 3 and 24 hours after pMCAO (Figure 1A). B cells (B220+) were increased by 3- to 4-fold (Figure 2C). T lymphocytes (CD3+) were increased by 2- to 3-fold at 3 and 24 hours (Figure 2B). Of these, CD4+ T cells had not increased significantly by 3 hours, but were 4- to 5-fold more numerous at 24 hours (Figure 2G). CD4+CD25+ T cells comprised ∼30% of CD4+ cells, and followed the same profile as total CD4+ cells (Figure 2H). By contrast, CD8+ T cells were increased by 3- to 4-fold at 3 and 24 hours (Figure 2I). NK cells (NK1.1+CD3−; Figure 2J) and CD8−CD4− T cells (i.e., also CD3+; Figure 2L) followed by a similar profile to CD8+ cells, whereas NKT cells (NK1.1+CD3+) were more like CD4+ cells in that their numbers were not increased until 24 hours after pMCAO (Figure 2K).
Immune Cell Numbers in the Brain After Transient Middle Cerebral Artery Occlusion
There were ∼8,000 leukocytes (CD45+high cells) present in the ischemic hemisphere at 24 hours after either 1 or 2 hours of tMCAO, which was 2- to 3-fold more than in the contralateral (nonischemic) hemisphere or in sham controls (Figure 1B). Of these, lymphoid cells (CD11b−) and myeloid cells (CD11b+) each represented ∼50% of total leukocytes (Figures 1B and 4). Fewer leukocytes in brains after tMCAO versus pMCAO was largely due to lower numbers of myeloid cells (Figures 1A and 1B).
Myeloid cells (CD11b+) were increased in the ischemic brain by ∼3-fold at 24 hours after tMCAO (Figure 1B). Neutrophils (Ly6G+) were increased by 5- to 6-fold at 24 hours (Figure 3A) and comprised ∼60% of myeloid cells (and ∼40% of all infiltrating leukocytes) after tMCAO (Figures 3A and 4). An ∼3-fold increase in macrophages and monocytes (F4/80+ or Ly6C+, respectively) was observed at 24 hours after tMCAO (Figures 3D and 3E). Dendritic cells (CD11b+CD11c+) tended to increase by >10-fold at 24 hours tMCAO (Figure 3F), but represented only ∼2% of all infiltrating leukocytes (Figure 4). Numbers of resident brain microglia (CD45+medCD11b+F4/80+) increased by 3- to 4-fold after tMCAO in both ischemic and contralateral hemispheres (Figure 1D).
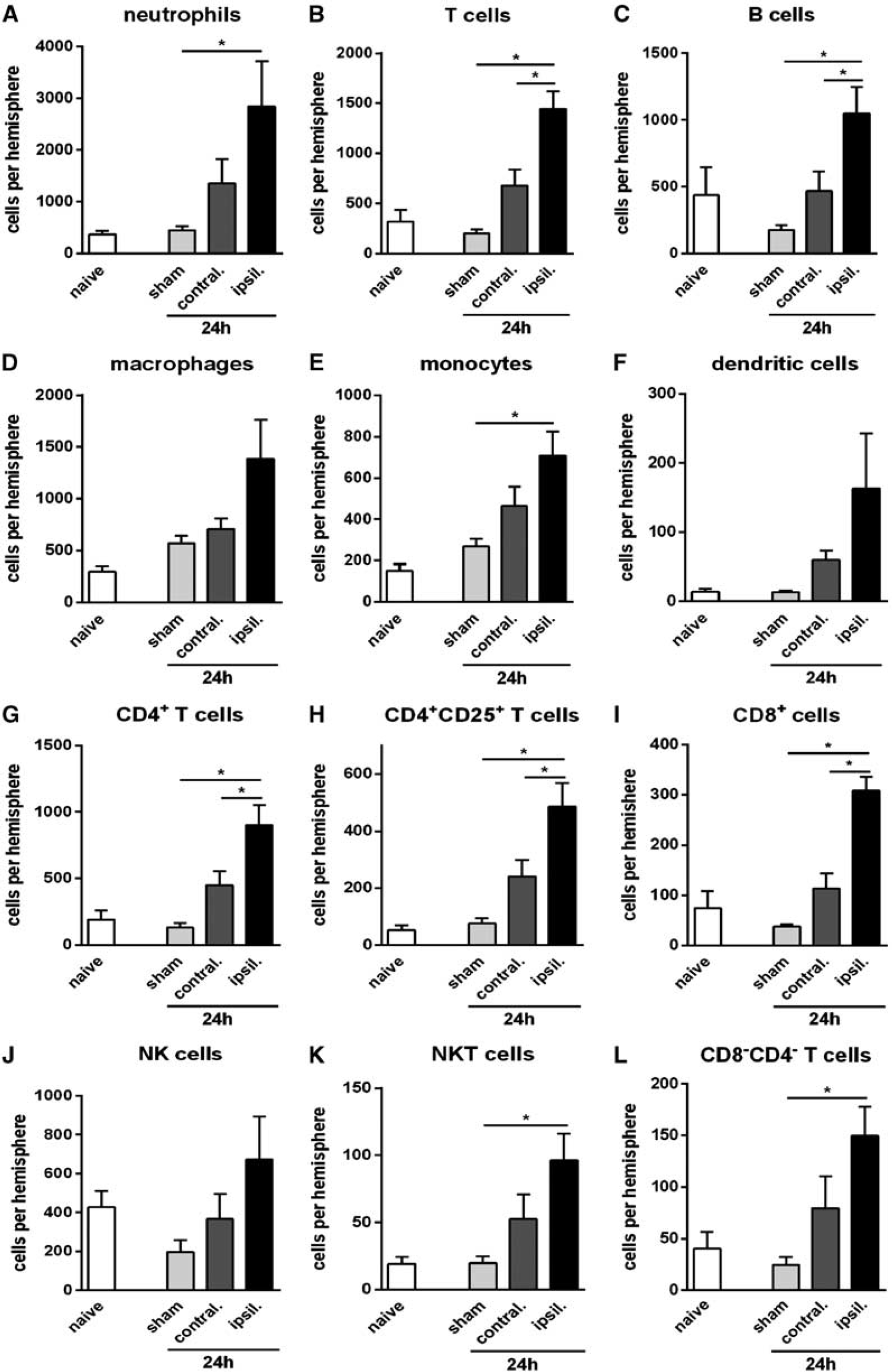
Quantification of leukocyte subtypes in brain at 24 hours after transient middle cerebral artery occlusion, compared with naïve and time-matched sham controls (
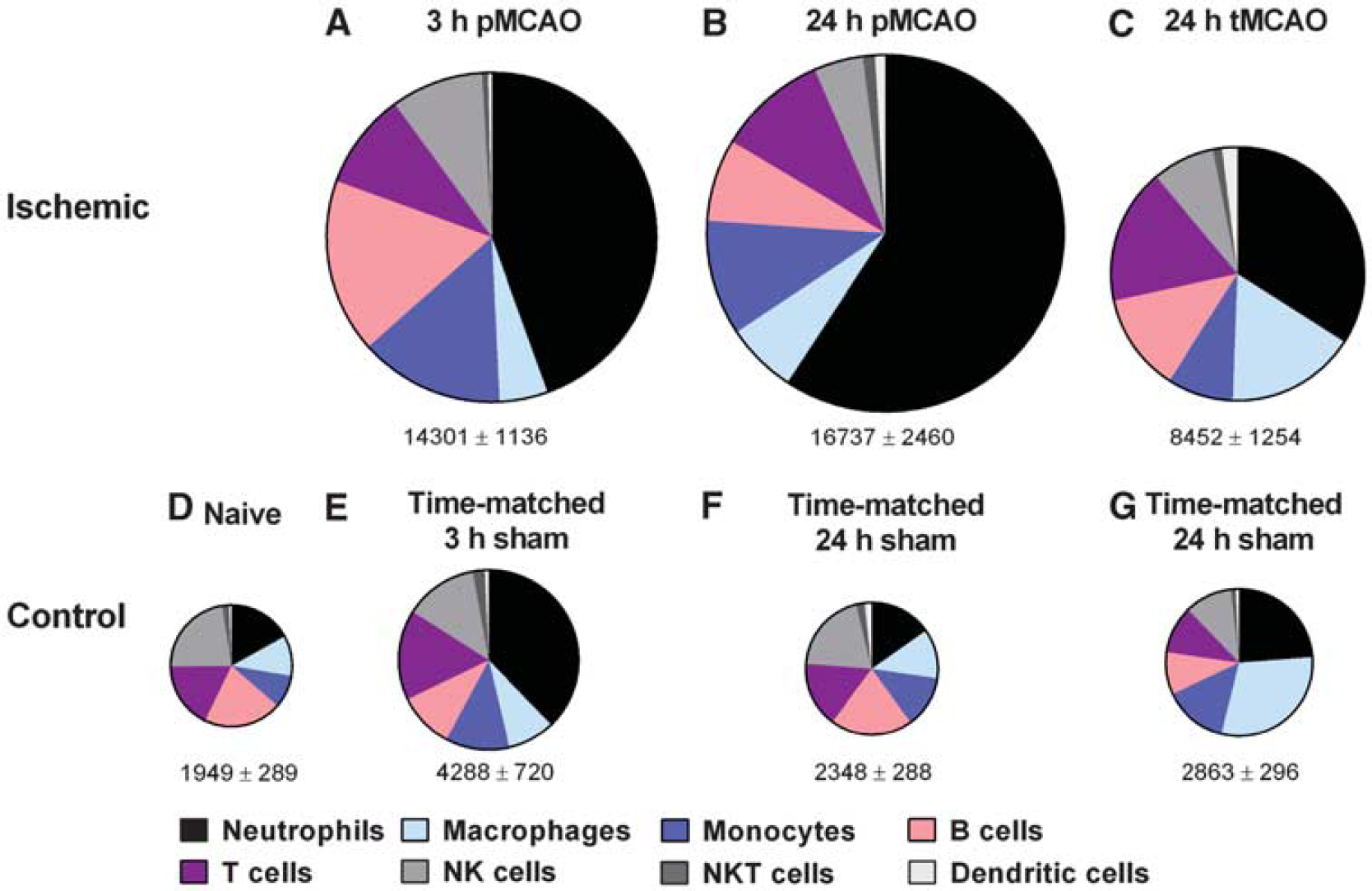
Pie charts summarizing number and composition of immune cells in ischemic hemispheres of mouse brains at 3 hours (
Lymphoid cells were increased by 2- to 3-fold at 24 hours after tMCAO (Figure 1B). Approximately 40% of these were B cells (B220+; Figures 3C and 4), which were increased by ∼2-fold (Figure 3C). T cells (CD3+) were increased by 3- to 5-fold at 24 hours (Figure 3B). Of these, CD4+ T cells were ∼4-fold more numerous (Figure 3G). CD4+CD25+ cells comprised >50% of CD4+ cells, and followed the same profile as total CD4+ cells (Figure 3H). CD8+ T cells and CD8−CD4− T cells were also increased by 3-fold at 24-hour tMCAO (Figures 3I and 3L). NK cells (NK1.1+CD3−; Figure 3J) were increased by ∼2-fold whereas NKT cells (NK1.1+CD3+) were increased by ∼4-fold at 24 hours after pMCAO (Figure 3K).
Immune Cell Numbers in Blood and Spleen After Permanent Middle Cerebral Artery Occlusion
No significant changes in the number of any circulating immune cells were found at 3 hours after pMCAO. However, at 24 hours after pMCAO the proportion of leukocytes that were neutrophils increased by ∼3-fold. By contrast, lymphoid cells were reduced from ∼80% to ∼30% of total leukocytes at 24 hours after pMCAO (Supplementary Figure 3). No changes were observed in numbers of any immune cells in spleen at either 3 or 24 hours after pMCAO (Supplementary Figure 4). Similarly, spleen weights were not significantly different at 3 hours (103.2±8.5 mg) or at 24 hours (82.2±5.7 mg) after pMCAO, compared with time-matched sham controls (89.5±3.8 mg and 75.2±5.1 mg, respectively;
Localization of Leukocytes After Permanent Middle Cerebral Artery Occlusion
We utilized immunohistochemistry, via an anti-MPO antibody, to localize neutrophils—the most prominent type of infiltrating leukocyte—in relation to the infarcted region within 3 hours after pMCAO (Figures 5 and 6A). It is possible that some more weakly stained MPO+ cells were instead macrophages. Cell density of MPO+ was markedly higher (∼7-fold; Figure 5) in the infarct compared with noninfarct regions from the ischemic hemisphere or the contralateral hemisphere. Myeloperoxidase cells appeared to be evenly distributed within the infarct, with no notable difference between infarct border areas (within 1 mm from the infarct border), and remote areas (>1 mm within the infarct border) (Figure 5). Most of the infiltrating leukocytes within the infarct were found to be present outside the vasculature (Figure 6). There was no evidence of hemorrhagic transformation (e.g., areas of erythrocytes in the parenchyma) in any of the brain sections examined after pMCAO.
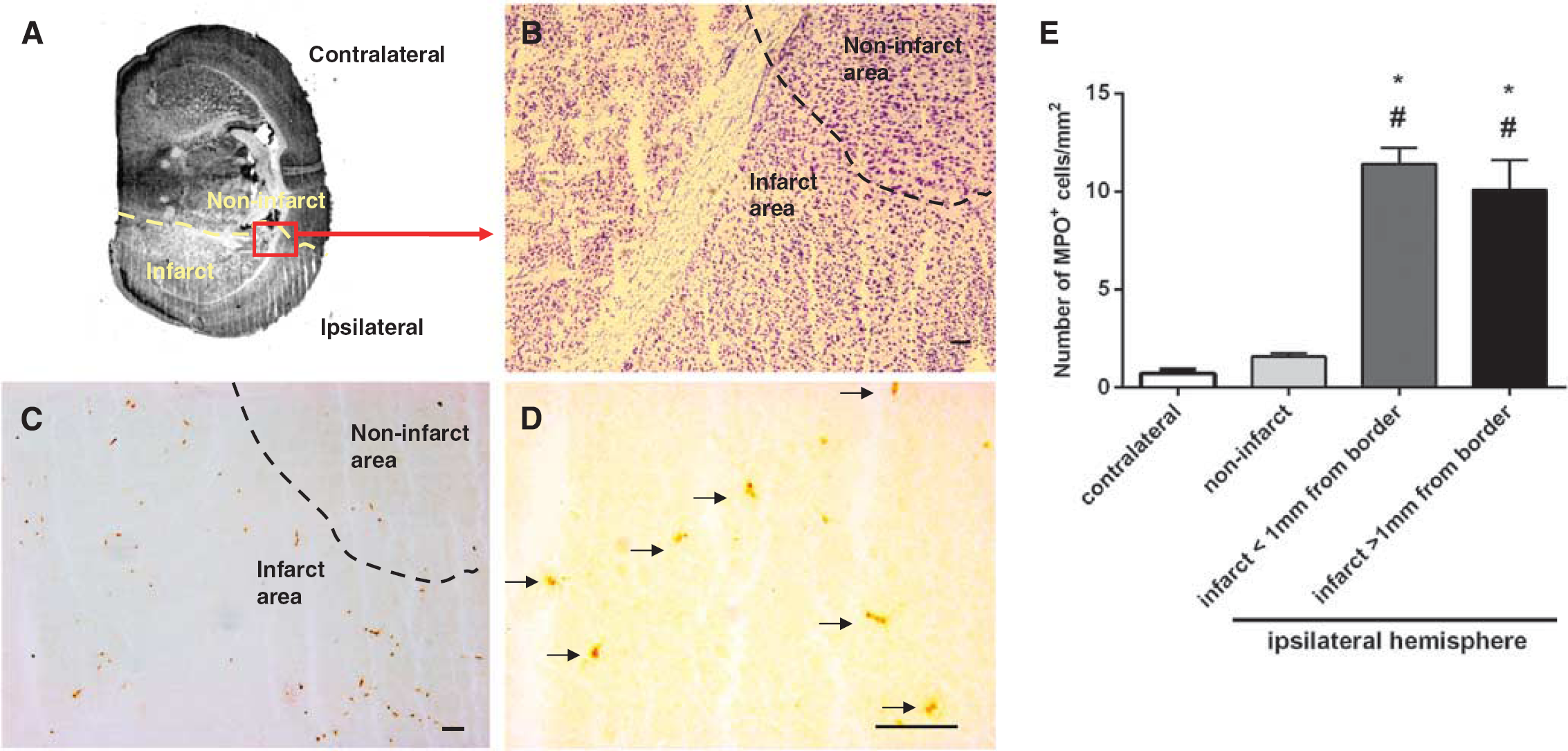
Representative photomicrographs showing localization of myeloperoxidase (MPO+) cells in mouse brain at 3 hour after permanent middle cerebral artery occlusion (pMCAO). (
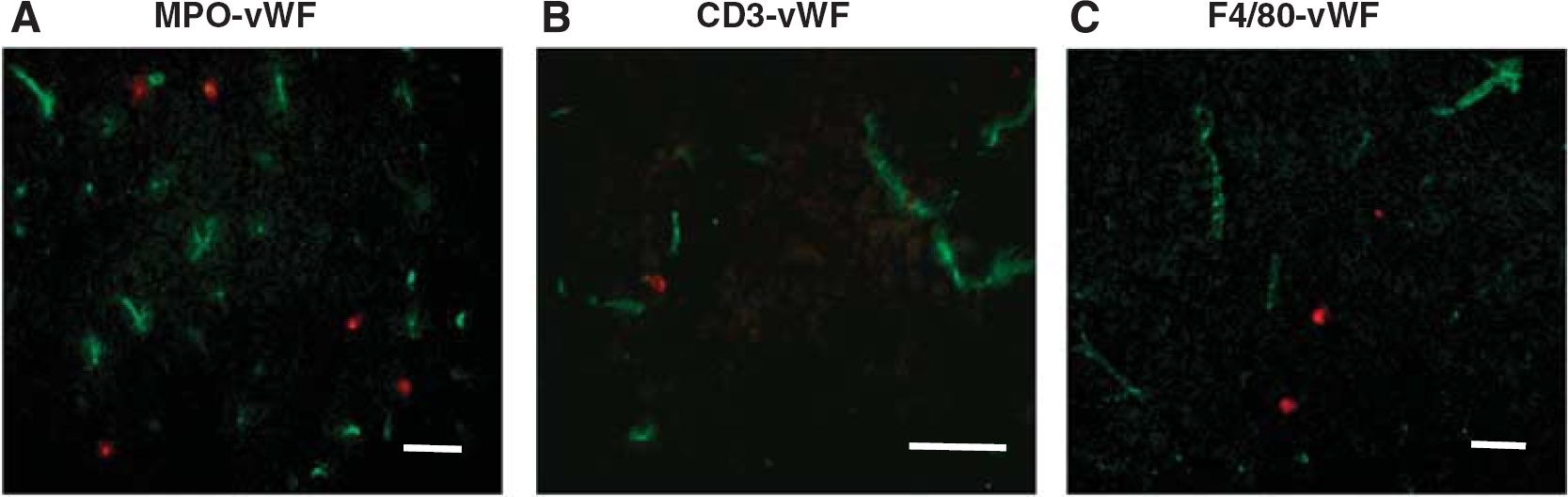
Neutrophils, T cells, and macrophages are infiltrated into the ischemic infarct at 3 hours after permanent middle cerebral artery occlusion (pMCAO). Representative overlayed images of myeloperoxidase (MPO+) cells (
Effect of Flushing with Phosphate-Buffered Saline Before Brain Removal
Consistent with the immunohistochemical data in Figure 6, the quantity of infiltrating leukocytes remaining within in the 24-hour pMCAO brain, as estimated by flow cytometry, was reduced by only ∼10% to 20% by flushing the brain with saline before removal (
Transient Effect of Sham Surgery on Number of Intravascular Leukocytes
We noted that sham surgery appeared to cause a small, transient increase in leukocyte numbers in the brain at 3 hours but which had disappeared by 24 hour (see Figures 4D–4G). To explore whether these cells might be present in the intravascular space, we performed a further 12 experiments using flow cytometry in either naïve or saline-flushed versus nonflushed brains from sham-operated mice taken 3 hours after surgery. The data indicate that the increased leukocyte numbers can be accounted for by a greater presence of neutrophils, and this can be reduced by saline flushing (Supplementary Figure 6).
DISCUSSION
Inflammation appears to have an important role in the brain injury that results from cerebral ischemia.13, 18 It has been assumed that the process of postischemic brain inflammation is facilitated by reperfusion but is limited in the absence of reperfusion. Our study has examined the profile of immune cell infiltration at early time points after pMCAO in mice, a model of human MCA infarction lacking cerebral reperfusion. We discovered that marked leukocyte infiltration occurs in the brain within 3 hours after pMCAO and is maintained for at least 24 hours. Specifically, infiltrating leukocyte populations that are already present in the ischemic hemisphere in stable numbers as early as 3 hours include neutrophils in particular, but also CD8+ T cells, B cells, NK cells, monocytes, and macrophages. Infiltration of CD4+ T cells, NKT cells, and dendritic cells is slower, and occurs later than 3 hours. We hypothesized that brain infiltration of leukocytes after cerebral ischemia would be substantially greater after tMCAO in comparison with pMCAO, due to the augmented brain perfusion occurring after removing a temporary (1 or 2 hours) occlusion of the MCA. Surprisingly, we found that over 24 hours after tMCAO, numbers of most leukocyte populations infiltrating the ischemic brain were less than those seen after pMCAO, including substantially fewer (∼50%) neutrophils.
We found that neutrophils were the most numerous cell type (representing approximately one half) infiltrating the brain throughout the first 24 hours after pMCAO, most of which were present within 3 hours of pMCAO. Previous studies of experimental stroke in rats using histochemical approaches identified neutrophils to be one of the earliest cell types infiltrating the ischemic brain, contributing to inflammation as early as 12 hours after pMCAO. 19 Furthermore, pharmacological inhibition (at 1.5 hours) or genetic deletion of neutrophil elastase was reported to reduce infarct volume in mice after tMCAO but not pMCAO, suggesting that reperfusion may be necessary to allow neutrophils to access and elicit damage to vulnerable ischemic tissue. 20 However, here our comprehensive analyses of whole ischemic hemispheres utilising flow cytometry have revealed that neutrophils are ∼3-fold as numerous in the ischemic hemisphere at 24 hours after pMCAO compared with 1 or 2 hours of tMCAO, despite a comparable infarct volume after 2-hour tMCAO and the pMCAO models. Further histochemical analysis indicated that leukocytes, particularly neutrophils, T cells, and macrophages, were mostly present within the parenchyma of the infarcted zone after pMCAO. This indicates that neutrophil infiltration readily occurs throughout the infarcted tissue after cerebral ischemia and is apparently not dependent on blood perfusion being restored. Studies in poststroke human brain have also suggested that neutrophil accumulation is positively correlated with severity of lesion and neurologic deficit 9 or rate of infarct expansion. 21 Given that in the present study, continuing occlusion of the MCA was evidently no barrier to the effective infiltration of the ischemic tissue by neutrophils over 24 hours, and that pMCAO in our model generates an infarct that is ∼2-fold larger than 1 hour of tMCAO, 14 it seemed to be plausible that the degree of neutrophil infiltration into the ischemic hemisphere was quantitatively dependent on the infarct volume of each model. However, our additional studies after 2 hours of tMCAO plus 22 hours of reperfusion indicated that numbers of infiltrating leukocytes were not strictly related to infarct volume but were perhaps more related to whether reperfusion occurred. Surprisingly, reperfusion resulted in a markedly lower number of immune cells infiltrating the parenchyma. This may suggest that the severity of ongoing hypoxia in the infarct core, and not simply the size of the infarct, is a major stimulus for recruitment and retainment of immune cells in the brain after stroke.
Whether neutrophil accumulation causes secondary brain damage, and can be effectively blocked, after cerebral ischemia in animals and/or humans remains controversial. In addition to the findings of Stowe
We observed increased numbers of monocytes and macrophages in the ischemic hemisphere after pMCAO, both of which had increased substantially by 3 hours. Monocytes were ∼2-fold more prevalent at 24 hours after pMCAO compared with tMCAO, whereas the opposite trend was true for macrophages. Denes
We found a substantial increase in T lymphocytes in the ischemic hemisphere as early as 3 hours after pMCAO, which is earlier than most others have reported to occur after tMCAO.8, 14 Moreover, a differential temporal profile was observed among subpopulations of T cells. CD8+ cells were markedly increased by 3 hours and tended to be less numerous at 24 hours after pMCAO. Numbers of CD4−CD8− T cells, which are involved in the downregulation of immune responses, 26 were similar to those of CD8+ cells in both models. By contrast, CD4+ cells were not significantly increased until 24 hours after pMCAO. Similar numbers of both CD8+ and CD4+ cells were present at 24 hours in the two models. Infiltrating NKT cells displayed a similar temporal profile as CD4+ T cells in both models, but were only ∼10% as numerous. Natural killer T cells recognize self and foreign lipid antigens presented by CD1d, including bacterial glycolipids, and are reported to have important roles in mechanisms of immune modulation after stroke, especially in stroke-induced immunosuppression.27, 28 T lymphocytes have previously been reported to be present in postischemic brain tissue by 24 hours after tMCAO,8, 14 and there is evidence that both CD8+ and CD4+ T cells contribute to postischemic injury in mice. 29 CD4+CD25+Forkhead box P3+(FoxP3+) regulatory T (Treg) cells have been reported to have a protective, immunomodulatory role in the brain over several days after stroke,30, 31 but a detrimental role during more acute conditions. 32 We found no increase in CD4+CD25+ T cells, which includes activated CD4+ T cells and Tregs, at 3 hours after pMCAO, but a marked increase in these cells in the ischemic hemisphere 24 hours after either pMCAO or tMCAO. A 10-fold increase in Treg cells was recently reported at 14 days after tMCAO. 33
We found that numbers of B cells were increased by 2- to 3-fold over sham levels at 24 hours after either pMCAO or tMCAO, and these cells were even more numerous earlier at 3 hours after pMCAO. B cells can function as antigen-presenting cells to activate cytotoxic (CD8+) T cells, but there is recent data to suggest that a regulatory B-cell subpopulation may modulate outcome in experimental stroke. B cell-deficient mice had larger infarct volumes, higher mortality and an increased number of activated T cells, macrophages, microglia, and neutrophils, compared with wild-type control mice after tMCAO.16, 34 IL-10-secreting regulatory B cells have been identified as a major regulatory B-cell subtype,16, 34 however, little is known about the initial temporal profile of B-cell infiltration in the brain after ischemia. Previous flow cytometric studies have identified increases in B cells at 3 days after 0.5 hour tMCAO 8 and 5 days after pMCAO by electrocoagulation. 35 Clarification of the functions of different B-cell subtypes will be important for the possible development of B cell-related therapies for acute stroke.
We observed an ∼4-fold increase in dendritic cells at 24 hours after pMCAO. A previous immunohistochemical study reported increased levels of dendritic cells in the ischemic lesion after pMCAO. 36 Dendritic cells have also been shown to be increased until day 6, predominantly around the infarcted brain tissue after photochemically induced focal ischemia in mice. 37 Dendritic cells are involved in antigen presentation during immune cell activation. They can also induce self-tolerance via release of different cytokines, which have an important role in adaptive responses of immune regulation. 38 The precise role(s) of dendritic cells in outcome after cerebral ischemia is yet to be established.
We found that at 3 hours after sham surgery, the number of total leukocytes, especially neutrophils, were more numerous in the brain than at 24 hours after sham surgery (see Figures 4E–4G). Flow cytometry data indicated that these increased leukocyte numbers could be reduced by saline flushing. This suggests that general anesthesia with ketamine/xylazine may result in a mild and temporary neutrophil-related inflammation within the cerebral vasculature of mice.
Overall, we have described the infiltration profile of several leukocyte subtypes during the first 24 hours after pMCAO—a model of human malignant MCA infarction—as well as at 24 hours after tMCAO in mice. One limitation of the study could be that the brains were not flushed with saline before tissue collection, so that the data do not strictly exclude cells remaining in the vasculature and not extravasated. However, we do not believe that such a phenomenon contributed significantly to the interpretation of our data because: (1) in a subset of experiments we found using flow cytometry that flushing had only a modest effect on the number of CD45+ cells present in the ischemic hemisphere and (2) immunohistochemical analyses of unflushed brains indicated that most infiltrating leukocytes had extravasated and were localized outside blood vessels and extensively dispersed throughout the infarcted tissue. Moreover, we chose to focus on data from unflushed brains since any such leukocytes adherent to the cerebral endothelium after stroke are likely to be relevant to postischemic brain inflammatory mechanisms and are therefore appropriate to be included in any quantitation of cell numbers. Finally, we cannot exclude the possibility that increased expression of CD45, together with CD11c or F4/80, by microglia after stroke was in part falsely ascribed to dendritic cells or macrophages, respectively.
In summary, these comparative data describing the infiltration profiles of numerous leukocyte subtypes into the postischemic brain in the absence or presence of reperfusion should assist in our understanding of early inflammatory changes in brain after ischemic stroke. Our data are particularly relevant for the severe setting of malignant MCA infarction, where we found a surprisingly large degree of leukocyte infiltration to occur very early after focal ischemia in the absence of reperfusion. So far, no established medical therapies for malignant MCA infarction have reached clinical practice. The major clinical problem in malignant MCA infarction is the development of massive brain edema, which then leads to increased intracranial pressure and subsequently evolving secondary tissue damage. Due to the lack of pharmacological strategies, decompressive surgery of the rapidly evolving brain edema has been proposed for patients with space-occupying hemispheric infarction. 39 Inflammatory mechanisms are well accepted to contribute to the development of brain edema in ischemic tissue damage. 40 Our observation of massive immune cell infiltration in the setting of permanent brain ischemia suggests that pharmacological strategies targeting inflammatory mechanisms might be a more promising therapeutic option than expected to reduce brain edema in patients with malignant MCA infarction. These new medical treatments could be applied in combination with established surgical approaches. Future studies should evaluate therapies to interfere with leukocyte infiltration early after stroke, regardless of whether reperfusion is possible.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.