
Abstract
Caspase-3 has been identified as a key protease in the execution of apoptosis and appears to be an important downstream event after hypoxia-ischemia in the immature brain. The efficacy of a pan-caspase inhibitor, boc-aspartyl-(Ome)-fluoromethyl-ketone (BAF), was tested in a model of unilateral focal ischemia with reperfusion in 7-day-old rats. The BAF inhibitor was given intraperitoneally 5 minutes before reperfusion via the carotid artery. This procedure reduced the activity of caspase-3 by 79% but did not induce a significant reduction in infarct volume (23.8 ± 7.5% versus 30.1 ± 6.4%). Animals were distributed in two populations. One population exhibited an infarct, whereas the other appeared to be fully protected. BAF-treated animals exhibiting an infarct mostly displayed necrotic cell death, whereas apoptotic nuclei were observed in untreated or vehicle-treated animals. Repeated dose of BAF (5 minutes before and 9 hours after reperfusion) did not also provide benefit after neonatal ischemia, although a general trend to reduce lesion was observed (20.5 ± 3.7% versus 34.4 ± 5.9%). These findings raise critical questions about the use of peptide ketone apoptotic inhibitors in improving histopathologic outcomes after neonatal stroke.
Cerebral infarction during the perinatal period remains a major cause of mortality and morbidity in newborn infants (Volpe, 1987), although obstetric and neonatal care has improved over the past decades. Survivors of perinatal asphyxia have moderate to severe brain injury (Berger and Garnier, 1999) for which there currently is no promising therapy.
The pathophysiology of the lesion after hypoxiaischemia (HI) has been extensively studied in animal models, and these models have shown that multiple mechanisms act to lead to neuronal death after the injury. In the immature brain, HI lesions result in a decrease of mitochondrial respiration (Rosenberg, 1986) and secondary energy failure leading to cell necrosis. Nevertheless, it has been clearly shown that a large number of neurons die by the apoptotic mitochondrial pathway (Blomgren et al., 2001; Han et al., 2000; Zhu et al., 2000). In addition, we recently reported that the mitochondrial pathway was expressed in neurons, the primarily affected cells, but also within the astrocytes, which constituted a dense network delineating the infarct, following neonatal stroke (Benjelloun et al., 2003). Transient HI induces not only acute brain damage but also delayed neuronal death in selected regions in human neonates after HI encephalopathy (Edwards et al., 1997; Stadelmann et al., 2001). Because apoptotic cell death occurs in a delayed fashion, opportunities for neuroprotective treatment targeting specific upstream regulators of apoptotic cascade may exist.
A number of specific therapeutic agents have been tested in animal models for their ability to reduce neuronal damage associated with HI injury. Among the possible molecular targets, caspases (a family of enzymes implicated in the apoptotic cascade) have elicited a particular interest. Caspase-3, a key enzyme of this family, appears to be an important downstream event after HI in the immature brain, but a minor event in adult brain (Gill et al., 2002; Wang et al., 2001; Yakovlev et al., 2001; Han et al., 2002). Mice deleted for the caspase-3 (Le et al., 2002) and caspase-1 (Liu et al., 1999) gene were reported to have reduced infarct after neonatal HI when compared to wild-type animals. Intracerebroventricular or systemic treatment with BAF [boc-aspartyl-(Ome)fluoromethyl-ketone], a membrane-permeable general caspase inhibitor, has been shown to provide benefit over a prolonged therapeutic window in the neonatal rodent model of HI, whereas a caspase-1 inhibitor did not confer protection (Cheng et al., 1998).
Therefore, the effect of BAF administration after transient unilateral focal ischemia with reperfusion in the developing rat brain (Renolleau et al., 1998) was assessed with biochemical (caspase assay) and histologic (infarct volume, cell count and cell death assessment) methods.
MATERIALS AND METHODS
Animals
All animal experiments were conducted according to the French and European Community guidelines for the care and use of experimental animals (licence number 01352). Wistar female rats in the proestrus period (R. Janvier, Le Genest-St-Isle, France), as shown by daily vaginal smears, were housed together with males for 24 hours, and then maintained during gestation in separate cages under standard laboratory conditions on a 12/12-hour light/dark cycle with food and water available ad libitum. The litter was adjusted to eight or nine pups on the day of delivery.
Perinatal ischemia
Ischemia was performed in 7-day-old rats (17 to 21 g, n = 137) of both sexes, as previously described (Renolleau et al., 1998). Rat pups were anesthetized with an intraperitoneal injection of chloral hydrate (350 mg/kg). Anesthetized rats were positioned on their back and a median incision was made in the neck to expose the left common carotid artery (CCA). Rats were then placed on the right side and an oblique skin incision was made between the ear and the eye. After excision of the temporal muscle, the cranial bone was removed from the frontal suture to a level below the zygomatic arch. Then, the left middle cerebral artery, exposed just after its appearance over the rhinal fissure, was coagulated at the inferior level of the cerebral vein. After this procedure, a clip was placed to occlude the left CCA. Rats were then placed in an incubator to avoid hypothermia. After 50 minutes, the clip was removed and carotid blood flow restoration was verified with the aid of a microscope. Neck and cranial skin incisions were then closed. During the surgical procedure, body temperature was maintained at 37°C to 38°C. Pups were transferred in an incubator (32°C) until recovery and then returned to their mothers.
Therapeutic protocol
To examine the effect of a pan-caspase inhibitor, BAF (Calbiochem, VWR International S.A.S., Fontenay sous Bois, France) at a dose of 2 μg/10 g weight (in 100 μL) was administered intraperitoneally 5 minutes before the clip was removed on the left CCA (BAF-treated group). Control animals received an equivalent volume of 0.9% saline containing 10% dimethyl sulfoxide (DMSO), the vehicle required to solubilize the peptidergic caspase inhibitor (vehicle-treated group). Eighty-six animals were used in these experiments (n = 16, n = 7 or 8, and n = 5 in each group for infarct volume, caspase activity, and cell counts on histological sections, respectively). In a second set of experiments, BAF, at the same concentration, was administered as a single injection (5 minutes before reperfusion) or as a double injection (5 minutes before and 9 hours after the clip was removed) and compared to untreated ischemic animals (n = 17 each group). In rat pups subjected to ischemia, mortality rates during ischemia or before killing did not differ between control, BAF-treated, and vehicle-treated groups in any of the experiments (<4%).
Measurement of infarct volume
Rats were killed 48 hours after reperfusion and brains were fixed 2 days in 4% buffered formaldehyde. Fifty-micrometer coronal brain sections were cut on a cryostat and collected on gelatin-coated slides. Sixteen sections from anterior striatum to posterior hippocampus (corresponding to plates 9 to 27 in Paxinos's rat brain atlas) were selected and taken at equally spaced 0.5-mm intervals. Infarct size was determined on cresyl violetstained sections using an image analyzer (IMSTAR, Paris, France), as previously described (Ducrocq et al., 2000). The data are expressed as mean percentage volume ± SEM.
Tissue preparation
For biochemical studies, rat pups were killed at 24 hours after reperfusion and the brains were rapidly removed. An observer who was blinded to the treatment visually scored the infarct lesion (pale zone on the cortex surface). The size of the lesion was graded from 0 to 3, where 0 indicated no observable lesion and 1, 2, and 3 indicated small, medium, and large infarcts, respectively. The brains were then dissected on a cold plate. Cortical tissue from both lesioned and unlesioned hemispheres were harvested and stored at −70°C. The volume of the harvested tissue corresponded to the parietal cortex, including the ischemic core and penumbra (Benjelloun et al., 2003).
For histologic analysis, quantification of cell density and apoptotic and necrotic nuclei density, ischemic rats, and ischemic rats treated with BAF or vehicle (n = 5 in each group), at 24 hours after reperfusion, were anesthetized with 350-mg/kg chloral hydrate intraperitoneally and perfused through the left ventricle with heparinized saline followed by 0.12-mol/L phosphate buffer (pH 7.4) containing 4% paraformaldehyde. Brains were then processed for paraffin embedding. Seven-micrometer coronal brain sections were cut and stained with cresyl-violet. The recently developed fluorescent tracer, Fluoro-Jade B (Histo-Chem, Jefferson, AR, U.S.A.), was also used to stain both the cell bodies and processes of degenerating neurons (Schmued and Hopkins, 2000) and to distinguish the core and penumbra zones (Butler et al., 2002).
DEVD-AFC cleaving caspase activity
Caspase-3 activity was performed as previously described (Benjelloun et al., 2003). Briefly, tissues were homogenized in a lysis buffer containing 10-mmol/L N-Tris(hydroxymethyl)methyl-2-amino Ethane Sulfonic acid (TES) (pH = 7.4), 1-mmol/L Ethylene diamine tetra acetic acid (EDTA), 250-mmol/L sucrose, 0.1% ethanol (EtOH), 0.2-mmol/L phenylmethylsulfonyl fluoride (PMSF), and 1-μmol/L leupeptin for 10 minutes at 4°C. Cell lysates were then centrifuged at 10,000g for 10 minutes. Supernatants were extracted and assayed for activity on synthetic substrate linked to 7-amino-4-trifluoromethykl-coumaryl (AFC, Biomol, Tebu, France). Ac-DEVD-AFC (Asp-Glu-Val-Asp) substrate (20-μmol/L), diluted in PBS, was added and incubated at 37°C. Caspase activity was measured for 1 hour using a fluorimeter (Bioteck, France) at 400 nm excitation and 505 nm emission wave lengths. The protease inhibitor DVED-CHO (5 μmol/L; Alexis, Qbiogen, Illkirch, France) was incubated with alternate samples to inhibit caspase-3–like activity. Arbitrary fluorescent units were converted into picomoles of AFC release per minute and milligrams of protein using a standard curve of free AFC (Aldrich, Sigma, France). Enzyme activity is expressed as mean ± SEM.
Assessment of cell damage
The number of cells was determined in the infragranular part of the parietal cortex (core and penumbra, layers III-V) at a 40× magnification in at least five standardized microscopic fields of 45,000 μm2 each, defined by a morphometric grid. Control, BAF-treated, and vehicle-treated ischemic brains were analyzed after 24 hours of reperfusion. For each selected field, only cells with their nuclei present in the focal plane were counted. Values represent the number of cells per square millimeter.
Apoptotic cell death was determined on sections stained with Hoechst 33342 (Sigma). Criteria for the assessment of apoptosis were nuclear condensation and fragmentation and flocculent chromatin for necrosis. Characteristic nuclei were scored under fluorescence microscopy by counting concerned nuclei in at least three distinct areas of 100 nuclei.
Statistical analysis
Assuming a β risk of 0.2 and an α risk of 0.05, it was estimated that 16 animals in each group were needed to detect a 50% infarction volume reduction between two groups. Data were drawn from our previous study (Ducroq et al., 2000). Because three groups of animals are compared in the experiments, these values are only informative. A predetermined list with blocks of six animals was used to randomized the animals among the three groups. An investigator blind to the treatment condition performed all measurements. Nonparametric statistics were used because infarct volume data were not normally distributed. The Kruskal-Wallis test was used for comparison of multiple groups. The significance of differences between groups (caspase-3 activity, cell number) was determined using an analysis of variance followed by probing for the source of the difference using the Fischer PLSD test. All data are expressed as the group mean ± SEM values with a P < 0 .05 significance level.
RESULTS
Infarct volume in treated animals
Because our previous data indicated significant caspase-3–like activity at 24 hours of recovery (Benjelloun et al., 2003), we examined whether the caspase inhibitor BAF could provide neuroprotection when administrated in our ischemic model. One dose of BAF (2 μg/mL) or vehicle was administrated intraperitoneally 5 minutes before the clip was removed. Three groups of ischemic animals were studied: untreated rats, rats treated with BAF diluted in 10% DMSO, or rats treated with the vehicle. Brains were then analyzed 48 hours later, a time point at which the infarct was stabilized (Ducrocq et al., 2000) without significant edema (no more than 1.5%). Ischemia induced an infarct volume of 74.9 ± 15.6 mm3, which represents a 30.1 ± 6.4% (Fig. 1A) damage in the lesioned ipsilateral hemisphere. Infarct volumes appeared normally distributed (between 7% and 63%), except one animal for which the infarct involved the entire ipsilateral hemisphere (Fig. 1B). A single dose of BAF, given 5 minutes before reperfusion, did not significantly reduce the infarct volume (23.8 ± 7.5%), as shown in Fig. 1A. However, of the 16 animals treated with BAF, eight showed no infarct whereas the eight others exhibited an infarct with a volume between 6% and 95% (mean, 44.6 ± 9.8%). These results suggested that BAF-treated animals could be divided in two populations, one with no apparent infarct and the other with an infarct, which appeared to be aggravated. Administration of the vehicle by the same route had no effect (mean volume, 21.5 ± 6.2%). Of the 15 animals studied, five had no infarct. Of the nine remaining animals, seven presented a normal distribution with a percentage of infarct volume between 17 and 25, and the two others exhibited a larger infarct (73%). Comparison of infarct area at different rostrocaudal levels in BAF-treated animals exhibiting a cortical infarct revealed that area values were significantly higher in sections six to eight (Fig. 2), corresponding to plates 23 to 27 in a rat brain atlas (Paxinos, 1988), as compared to ischemic control rats.
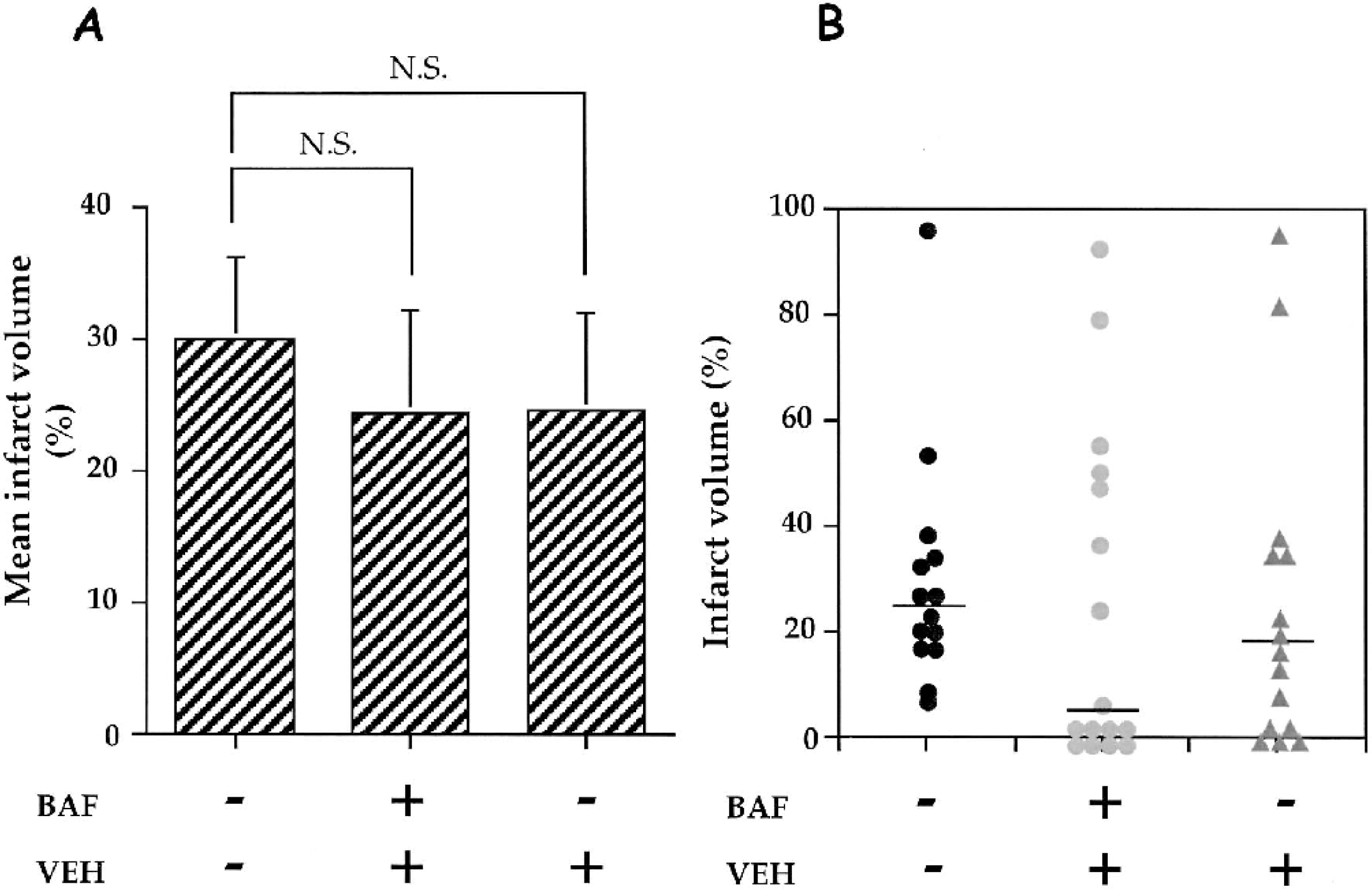
Effect of BAF on infarct volume measured 48 hours after ischemia. The drug was given 5 minutes before clip removal on the left common carotid artery and consisted of a single intraperitoneal injection of BAF (2 μg/mL in 10% DMSO, n = 16) or vehicle (10% DMSO, n = 15). Control ischemic rats (n = 14) were also studied. Data are mean ± SEM (bar). (
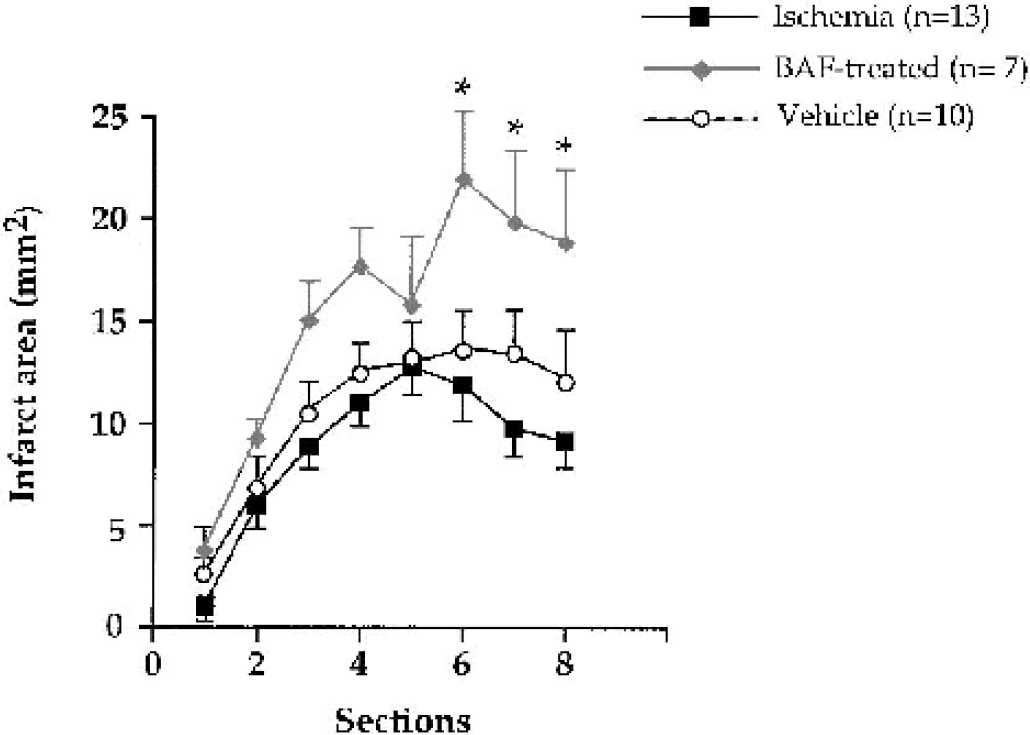
Rostrocaudal distribution of infarct areas plotted as a function of anterior to posterior distance in untreated, BAF-treated and vehicle-treated ischemic rat pups. *P < 0.05 versus control animals. Only BAF- and vehicle-treated ischemic animals exhibiting a significant cortical infarct are represented. Again, the ischemic control animal with a 95% infarct volume was not included.
Caspase-3–like activity in treated animals
To confirm that BAF administration was correlated with its ability to inhibit caspase activity, we tested whether BAF was able to inhibit caspase-3–like activity in vivo. P7 rat pups were subjected to ischemia and received a single injection of either BAF or vehicle. Twenty-four hours later, brains were removed, scored for the infarct lesion, and ipsilateral cortical tissue was harvested and assayed for its ability to cleave ac-DEVD-AFC substrate. As shown in Table 1, the distribution of the infarct lesion in ischemic pups belongs score between 1 and 3 (with a maximum at score 2), whereas contralateral hemisphere displayed no infarct (score 0). In contrast, infarct lesion in BAF- and vehicle-treated ischemic animals were distributed in the 4 different scores. There was a 10-fold and an eightfold increase in activity in vehicle-treated and untreated ischemic samples, respectively, as compared to contralateral tissues. Conversely, BAF treatment induced a significant reduction in caspase-3–like activity (79.3% compared to vehicle-treated tissue).
Caspase-3–like activity in cortical samples 24 hours after ischemia

Crude cytosolic fractions from control (contralateral tissue), untreated (ischemic) and BAF- or vehicle-treated ischemic brains were assayed for their ability to cleave ac-DEVD-AFC substrate and activity was expressed in picomoles of AFC released per min and per milligram of protein (mean ± SEM, three independent experiments with seven or eight animals in each group). The number of animals for each size of the infarct (lesion scoring, see Materials and Methods is indicated.
P < 0.05 versus contralateral.
P < 0.05 versus vehicle treated.
Assessment of cell death in treated animals
Because our results showed that the BAF-treated animals could be separated into two groups (protected and not protected), we investigated cell death by neuronal density in the core and penumbra of the cortical infarct at 24 hours of recovery by comparing nonprotected BAF-treated animals with untreated and vehicle-treated animals (those exhibiting an infarct). In ischemic animals, cell density decreased by 39% and 20% in the core and penumbra respectively, compared to the unlesioned contralateral cortex (Table 2). Similar results were observed in vehicle-treated animals with a cortical infarct. In contrast, cortical cell density decreased by 61% (P < 0.01 versus contralateral and P < 0.05 versus ischemic brains) and 38% (P < 0.05 versus contralateral) in the core and penumbra, respectively, in BAF-treated animals, which did not show neuroprotection (Table 2). Of the five animals studied, one had no cell loss and could be considered as a protected animal similar to those seen in the first experiment (Fig. 3B). In contrast, the four other animals exhibited a larger infarct (Fig. 3C) compared to a control ischemic rat pup (Fig. 3A). With Hoechst staining, chromatin fragmentation into apoptotic bodies (white arrows in Fig. 3E) was seen in the core of the infarct of ischemic control pups whereas flocculent chromatin (Fig. 3F), suggesting necrosis, was detected in BAF-treated animals. Quantification of apoptotic and necrotic nuclei in the ischemic core (Table 3) showed that percentage of the former was high in ischemic and vehicle-treated animals but less in BAF-treated animals, in which most of cell death was necrosis. In addition, the Fluoro-Jade B fluorescent marker also showed early cellular degeneration (not easily detectable by violet-cresyl staining) in the hippocampus of BAF-treated animals (Fig. 3I) but not in control ischemic rats.
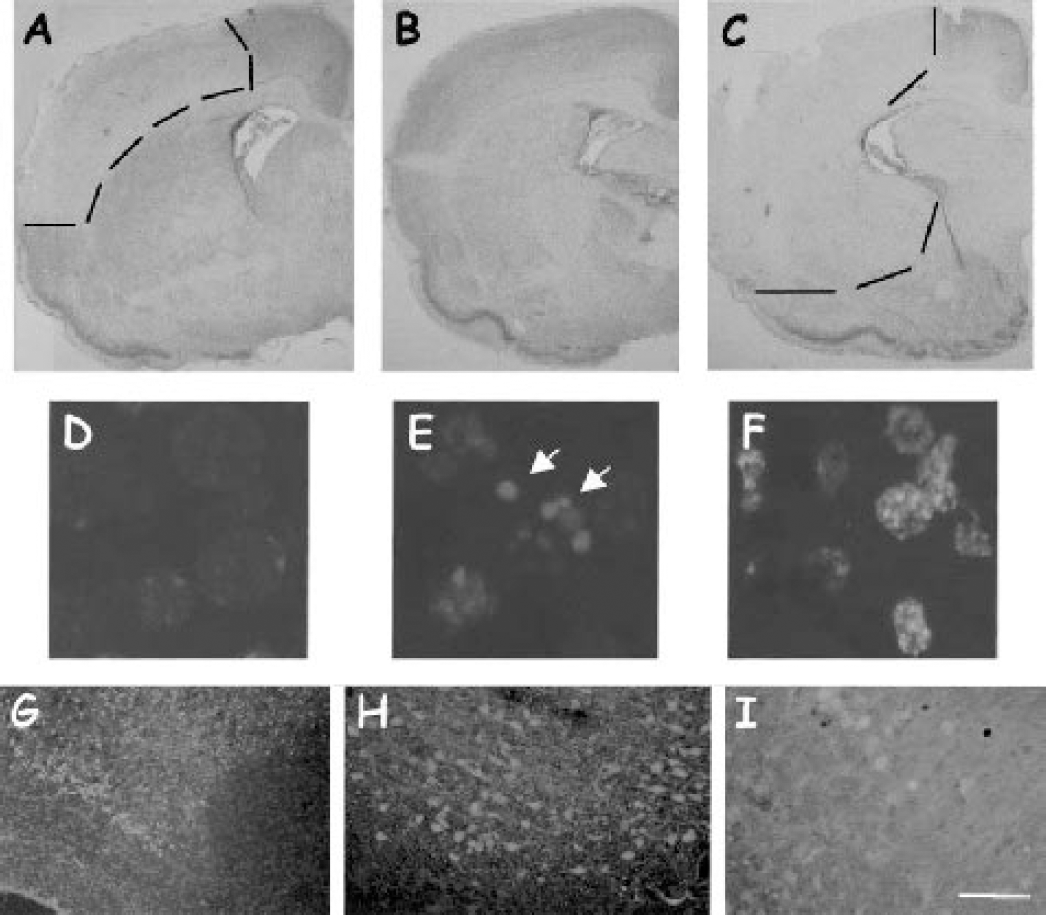
Brain damage and cell death 24 hours after neonatal ischemia as shown with cresyl-violet, Hoechst, and Fluoro-Jade B staining. Coronal sections at the level of the anterior commissure were obtained from ischemic (
Quantification of cell number in the core and penumbra 24 hours after ischemia
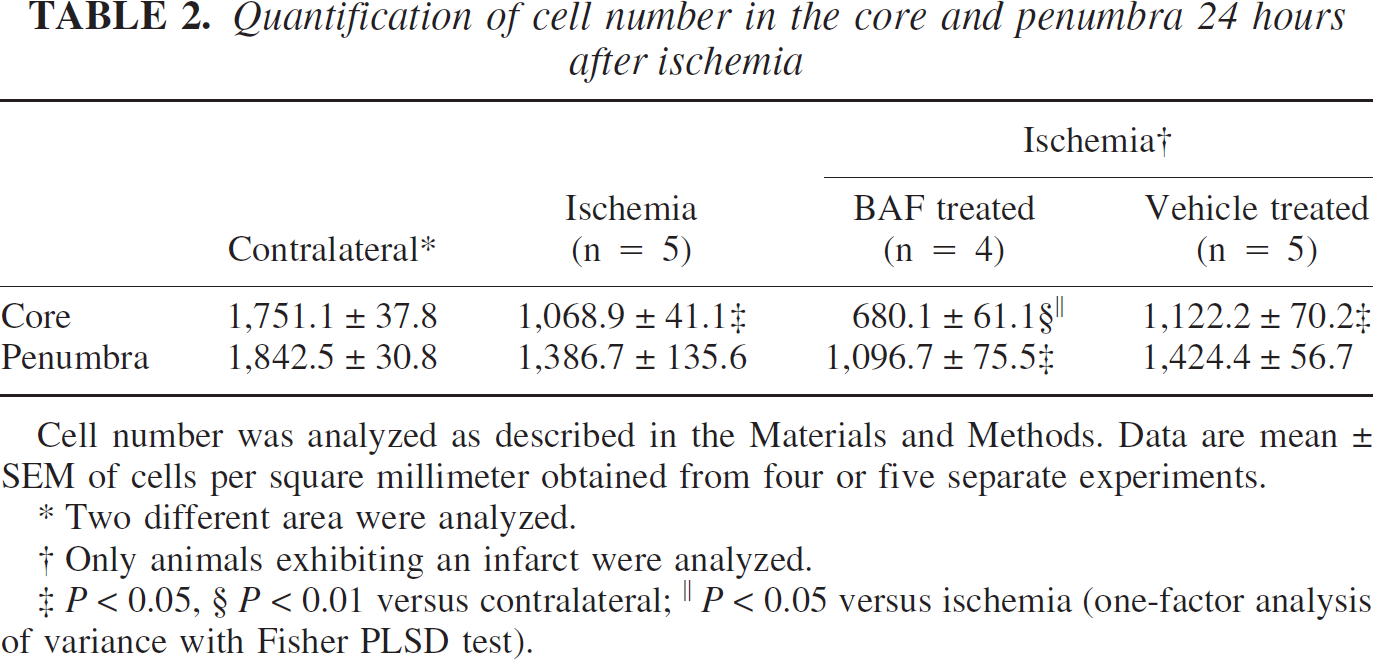
Cell number was analyzed as described in the Materials and Methods. Data are mean ± SEM of cells per square millimeter obtained from four or five separate experiments.
Two different area were analyzed.
Only animals exhibiting an infarct were analyzed.
P < 0.05, § P < 0.01 versus contralateral; ∥ P < 0.05 versus ischemia (one-factor analysis of variance with Fisher PLSD test).
Quantification of apoptotic and necrotic nuclei in the core 24 hours after ischemia

Number of apoptotic and necrotic nuclei (as determined after Hoechst 33342 staining, see Materials and Methods). Data are mean percentage ± SEM obtained for three triplicate counts in four or five experiments.
Only animals exhibiting an infarct were analyzed.
Infarct volume after a double dose of BAF
As described above, a single dose of BAF given 5 minutes before reperfusion did not significantly reduce the infarct volume (25.0 ± 5.8% versus 34.4 ± 5.9%; Fig. 4A). In another set of experiments, a second dose given 9 hours after the first dose also did not significantly reduce the infarct volume (20.5 ± 3.7%; Fig. 4A); however, a general trend toward reduced lesion size was observed (P = 0.0512, n = 17). As described above, treated rat pups (single and double dose) could be divided into two populations: a few (five and three animals, respectively) with no apparent lesion and the others (12 and 14 animals, respectively) with no protection. However, the values were more normally distributed in the group that received two doses of BAF than in that which received a single dose (Fig. 4B).
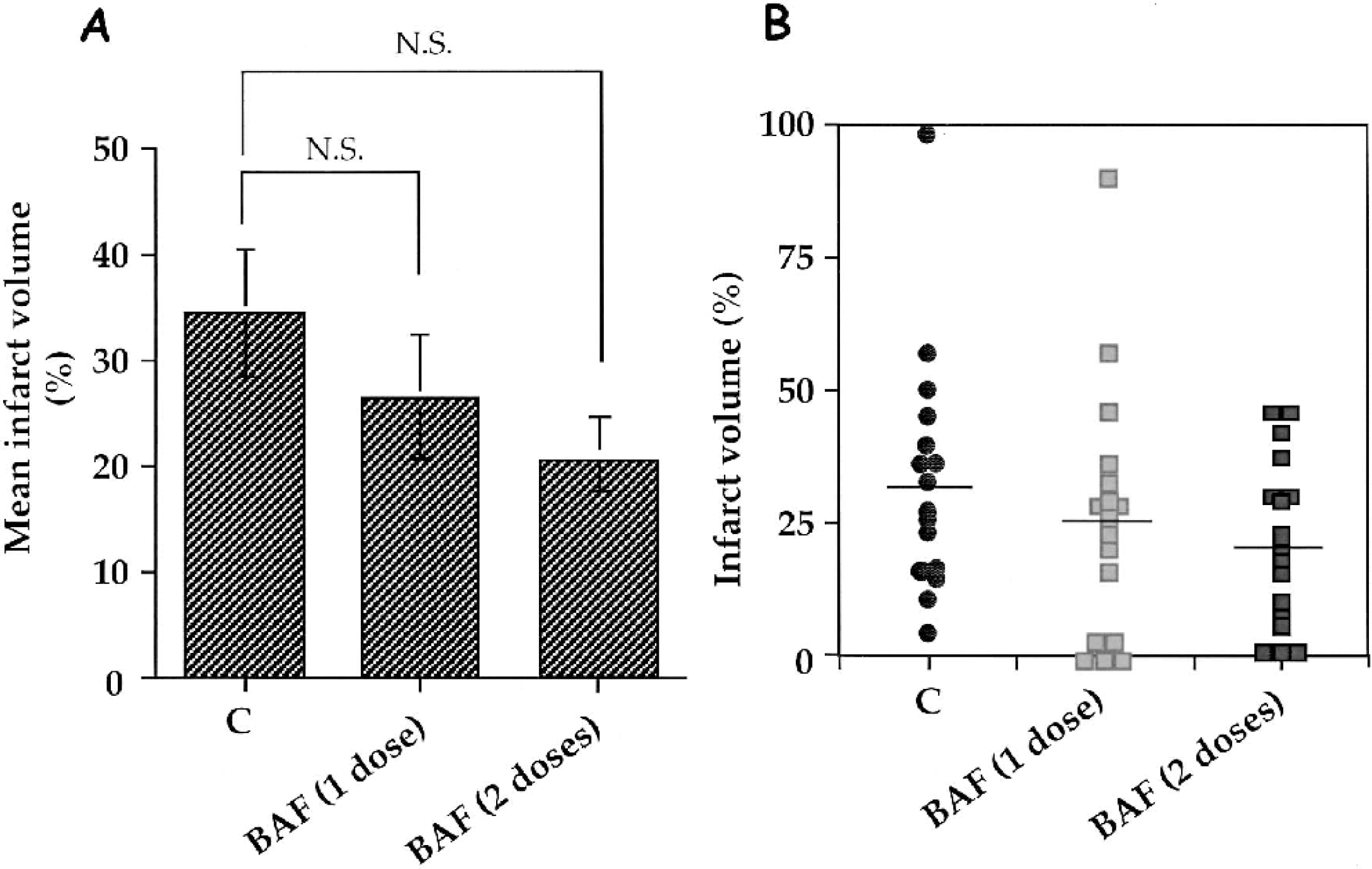
Effect of BAF (single versus double dose) on infarct volume measured 48 hours after ischemia. The drug (at the same concentration than above) was given 5 minutes before (single dose, n = 16) and 5 minutes before and 9 hours after (n = 17) clip removal on the left common carotid artery and compared to untreated animals (n = 16). Data are mean ± SEM (bar). (
DISCUSSION
In this study, we assessed the neuroprotective effects of caspase inhibitor administration against damage induced by ischemia in the P7 rat brain. Our results show that the pan-caspase inhibitor BAF, administered systemically, did not reduce the cortical infarct, although caspase-3–like activity was significantly inhibited. The discrepancy between our findings and those reported by Cheng et al. (1998) are discussed.
Historically, the involvement of caspases in ischemia-induced cell death was inferred from studies showing that z-VADfmk, BAF, or z-DEVDfmk, which are broadspectrum caspase-inhibitors, were neuroprotective in adult animal models of focal and global ischemia (reviewed in Loetscher et al., 2001). However, the efficacy of these inhibitors in focal ischemia was less clear in earlier studies (Gillardon et al., 1999, Li et al., 2000). After ischemia in adult rodents, the lesion evolves over 24 hours, although 80% of the damage has already occurred by 4 hours. The relative contributions of necrosis and apoptosis to neuronal death appear to differ between the mature and developing brain, with necrosis prevailing in the former and apoptosis in the latter (Gill et al., 2002). In neonatal HI models (using the Rice-Vannucci model), there is evidence that cerebral ischemia leads to delayed cell death with DNA damage (Ferrer et al., 1994; Sidhu et al., 1997), and apoptotic mechanisms of cell death (Nakajima et al., 2000; Northington et al., 2001a,b). After transient unilateral focal ischemia with reperfusion in the P7 rat pup (Renolleau et al., 1998), DNA fragmentation (Charriaut-Marlangue et al., 1999), morphologic features characteristic of apoptosis (Aggoun-Zouaoui et al., 2000), and activation of the mitochondrial pathway (Benjelloun et al., 2003) were reported. The proteolytic activity of caspase-3 on the ac-DEVD-AFC substrate was shown to increase between 3 and 24 hours, then gradually decrease (Benjelloun et al., 2003). This prompted us to investigate the effect of the pan-caspase inhibitor BAF early during reperfusion. Surpringsingly, half of the animals did not show a neuroprotection whereas the other half did. As we performed two separate experiments with one dose of BAF, the sum of data using the same number of animals as in the Cheng et al. (1998) study gives a mean of 23.7 ± 4.6% (BAF-treated animals, n = 32), which is not statistically different from the mean of ischemic control animals (32.1 ± 4.2%, n = 33). Again we observed 40% (13 of 32) of animals with a clear neuroprotection and 60% (19 of 32) with no protection. The process of neuroprotection or death in this bimodal distribution may involve one or more molecule(s) having a dual role in cellular damage. One of the most studied is the nuclear enzyme poly-(ADP-ribose) polymerase (PARP-1), which is involved in DNA repair and appears to be a key component of molecular mechanisms leading to cell death or survival after an ischemic insult (Ha and Snyder, 2000; Scovassi and Poirier, 1999). However, PARP overactivation can deplete tissue stores of ATP and NAD+ and may lead to necrotic cell death by energy failure (Ha and Snyder, 1999). PARP inactivation by caspase-3–mediated cleavage has been shown to preserve energy stores necessary for apoptotic events (Nicholson et al., 1995). PARP is then at the parting of the ways of major biochemical pathways regulating DNA repair or cell death. We recently reported that cell death after neonatal stroke implicated an early and transient PARP activation (Joly et al., 2003) and that PARP inhibitor reduced PARP activation and ischemic damage (Ducrocq et al., 2000). Therefore, the energy depletion state after inhibition of PARP cleavage by the BAF inhibitor, depending on blood reflow, might be more or less important. This allows either DNA repair leading to neuroprotection or, in contrast, overactivation of PARP leading to increased necrotic cell death in unprotected animals, as shown in the current study. Very recently, Zhu et al. (2003) used the Rice-Vannucci model but with a shorter hypoxia duration and reported that BAF did not reduce infarct volume, brain injury scoring, and DNA damage 72 hours after ischemia. Furthermore, these authors showed that BAF treatment did not affect aptosis-inducing factor (AIF) release from mitochondria or the frequency of positive nuclear AIF, an apoptosis-inducing factor that triggers apoptosis in a caspase-independent manner. AIF-mediated cell death may be also an important mechanism of HI-induced neuronal loss in the immature brain. Finally, it is difficult to compare data from these three studies (Cheng et al., 1998; Zhu et al., 2003, and the present study) in terms of the HI model, severity of the insult, and mechanisms such as apoptosis and/or necrosis (Leist and Jäättelä, 2001; Northington et al., 2001a,b) involved in cellular damage. A combination of moderate systemic hypothermia at 29°C and BAF produced a strong neuroprotective effect against neonatal HI in the CA1 region of the hippocampus (Adachi et al., 2001). Negative experimental findings have also been reported in other systems. The caspase inhibitor z-VADfmk, administrated by a variety of routes (local, subarachnoidal, intravenous, and intraperitoneal), did not inhibit apoptotic processes (Ozawa et al., 2002) or improve neurologic deficits and tissue damage after spinal cord injury (Lapchak et al., 2003).
BAF treatment significantly reduced caspase-3–like activity (in two previous studies and in the current study) and caspase-2, and produced a smaller decrease in caspase-9 activity (Zhu et al., 2003). It is interesting to note that the vehicle alone (20% and 40% DMSO in previous studies, respectively, and 10% in our study) was able to increase caspase-3–like activity and to produce an 18% significant (Zhu et al., 2003) and a 28.8% (nonsignificant in our study) reduction in infarct volume. Taken together, these data suggest that caspase-3 activity induced by DMSO can counterbalance the neuroprotective effect induced by the BAF inhibitor in some animals, leading to an inefficient neuroprotection.
As previously reported (Renolleau et al., 1998), association of transient homolateral carotid artery and permanent middle cerebral artery proximal occlusion leads to low cerebral blood flow, but anastomoses may allow a secondary recirculation phase. However, we know very little about this secondary recirculation phase, and blood reflow (slow or fast) after transient CCA occlusion may vary among animals owing to variation in numbers or efficacy of anastomoses. Secondary invading inflammation with granulocytic cells were not involved (McRae et al., 1995; Towfighi et al., 1991, 1995) or were involved (Benjelloun et al., 1999; Coeroli et al., 1998) after neonatal HI in the Rice-Vannucci and Renolleau models, respectively. Polymorphonuclear cells were observed in a greater numbers in the BAF-treated animals than in untreated animals 24 hours after ischemia (data not shown). Therefore, these differences may lead to the involvement of various brain damage pathways (oxidative stress, nitric oxide production) and may also account for different levels of neuroprotection.
In summary, the efficacy of the permeant multicaspase inhibitor BAF is less clear in our neonatal stroke model as compared to a previous report (Cheng et al., 1998), but is in agreement with that of Zhu et al. (2003). However, there was a general trend toward reduced cellular damage, and because caspase-3 activation is a major actor of cell death in the developing brain, further investigations are required to establish the specifics of treatment of HI brain injury with caspase inhibitors.
Footnotes
Acknowledgements
The authors thank B. Palmier (Laboratoire de Pharmacologie) and P. Bouquet (UMR-CNRS) for their excellent technical assistance, and Ann Lohof (UMR-CNRS) for editorial assistance.