
Abstract
These studies have addressed the role of caspase-3 activation in neuronal death after cerebral ischemia in different animal models. The authors were unable to show activation of procaspase-3 measured as an induction of DEVDase (Asp-Glu-Val-Asp) activity after focal or transient forebrain ischemia in rats. DEVDase activity could not be induced in the cytosolic fraction of the brain tissue obtained from these animals by exogenous cytochrome c/dATP and Ca2+. However, the addition of granzyme B to these cytosolic fractions resulted in a significant activation of DEVDase, confirming that the conditions were permissive to analyze proteolytic cleavage of the DEVD-AMC (7-amino-4-methyl-coumarin) substrate. Consistent with these findings, zVal-Ala-Asp-fluoromethylketone administered after focal ischemia did not have a neuroprotective effect. In contrast to these findings, a large increase in DEVDase activity was detected in a model of hypoxic-ischemia in postnatal-day-7 rats. Furthermore, in postnatal-day-7 animals treated with MK-801, in which it has been suggested that excessive apoptosis is induced, the authors were unable to detect activation of DEVDase activity but were able to induce it in vitro by the addition of cytochrome c/dATP and Ca2+ to the cytosolic fraction. Analysis of cytochrome c distribution did not provide definitive evidence for selective cytochrome c release in the permanent focal ischemia model, whereas in the transient model a small but consistent amount of cytochrome c was found in the cytosolic fraction. However, in both models the majority of cytochrome c remained associated with the mitochondrial fraction. In conclusion, the authors were unable to substantiate a role of mitochondrially derived cytochrome c and procaspase-3 activation in ischemia-induced cell death in adult brain, but did see a clear induction of caspase-3 in neonatal hypoxia.
There has been much debate in recent years about the role of apoptosis in the etiology of neuronal cell loss after acute neurodegeneration in the adult brain. Apoptosis is a form of cell death characterized by specific ultrastructural changes that serve to eliminate dying cells in proliferating or differentiating cell populations, without eliciting a gross inflammatory response (Kerr et al., 1972). Thus, apoptosis plays a critical developmental role in neonates (Oppenheim, 1991) and is also referred to as programmed cell death.
During the past few years, ischemic cell death and apoptotic cell death have been reported to share common features (Charriaut-Marlangue et al., 1996; Van Lookeren-Campagne and Gill, 1996; Macmanus and Linnik, 1997). These findings were based largely on the detection of DNA breaks using TUNEL (terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling) staining. However, there is now substantial evidence that TUNEL can stain both apoptotic and necrotic cells (Enright et al., 1994; Grasl-Kraupp et al., 1995). Therefore, the gold standard for identifying apoptosis remains electron microscopic analysis (Kerr et al., 1972). Furthermore, in an animal model of focal cerebral ischemia involving permanent middle cerebral artery (MCA) occlusion, we showed that the morphologic changes did not exhibit characteristics of apoptosis (Van Lookeren-Campagne and Gill, 1996). These observations have been extended to animal models of transient forebrain ischemia in which detailed electron microscopy and DNA labeling failed to reveal signs of apoptosis (Petito et al., 1997; Colbourne et al., 1999).
Caspases are cysteine proteases that play a central role in apoptotic cell death (Alnemri, 1997; Thornberry and Lazebnik, 1998). The caspase protease family has at least 14 members representing the mammalian homologs of CED3, a gene essential to programmed cell death in Caenorhabditis elegans. Caspase-3 shares the highest sequence homology to CED3 and is thought to play a critical role in mammalian apoptosis (Alnemri, 1997). One of the pathways in the initiation of the caspase cascade is the translocation of cytochrome c from the mitochondria to the cytoplasm (Yang et al., 1997; Kluck et al., 1997), where it interacts with apoptosis protease-activating factor-1 (Apaf1) in the apoptosome complex (Green and Reed, 1998). Procaspase-9 becomes activated and goes on to cleave procaspase-3, initiating the proteolytic cascade involved in apoptosis. The caspase-3-, caspase-9-, and Apaf1-knockout mice showed a similar phenotype with abnormal brain development (Kuida et al., 1996; 1998; Hakem et al., 1998; Cecconi et al., 1998; Yoshida et al., 1998). These findings lend further support to the hypothesis that caspase-3 plays an effector role in neuronal cell death during normal development of the brain. Thus, attention has been focused on the role of caspase-3 in neurodegenerative disorders such as stroke, Alzheimer disease, and Huntington disease.
The involvement of caspases in ischemia-induced cell death were inferred from studies showing that zVAD-fmk (z-Val-Ala-DL-Asp-fluoromethylketone), a broad-spectrum caspase inhibitor, was neuroprotective in animal models of focal and global ischemia (Loddick et al., 1996; Hara et al., 1997; Endres et al., 1998; Chen et al., 1998; Namura et al., 1998). Furthermore, cerebral ischemia studies also reported an increase in caspase-3 messenger RNA after focal or global ischemia (Schulz et al., 1999; Ni et al., 1997, 1998). However, procaspase-3 has to be activated by proteolytic cleavage before it can become an effector of apoptosis. We have investigated activation of caspases using an enzymatic assay in various animal models of focal and global ischemia in neonatal and adult brain. Because release of cytochrome c from mitochondria plays a critical role in the activation of procaspase-3, we investigated the release of cytochrome c after focal ischemia. Finally, the neuroprotective effects of zVAD-fmk have been studied in a model of focal ischemia, which replicates the pathological changes seen after stroke in humans. This work has previously been presented in abstract form (Gill et al., 1999).
MATERIALS AND METHODS
Reagents
Acetyl-Asp-Glu-Val-Asp-7-amino-4-methyl-coumarin (Ac-DEVD-AMC) was obtained from Alexis Corporation (Laeufelfingen, Switzerland) and purified granzyme B was purchased from Calbiochem-Novabiochem Corporation (La Jolla, CA, U.S.A.). Mouse anticytochrome c antibody (clone 7H8.2C12) was purchased from RDI (Flanders, NJ, U.S.A.) and the rabbit anticaspase-3 antibody H-277 (sc-7148) and antimouse and antirabbit horseradish peroxidase conjugates were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). Granzyme B was purchased from Calbiochem-Novabiochem Corporation (Juro, Switzerland). zVAD-fmk was prepared in the form of the ethyl ester as a 1:1 mixture of diastereoisomers (z-Val-Ala-(D)-Asp(OEt)-fluoromethylketone and z-Val-Ala-(L)-Asp(OEt)-fluorometh-yl-ketone) using a modified procedure described previously (Revesz et al., 1994).
Surgical procedures
Male Sprague-Dawley (330-350 g) and Wistar (300 g) rats were used for permanent focal and transient forebrain ischemia, respectively. Male C57BL/6 mice (20–25 g) were used for transient forebrain ischemia. All animals were maintained on a 12-hour light/dark cycle with food and water ad libitum until required for surgical procedures. All animal experiments were conducted according to the guidelines of the Swiss Federal Animal Protection Act.
Permanent focal ischemia in rats
The animals were anesthetized with a mixture of 4% isoflurane in 30% oxygen and 70% air, and were then intubated and ventilated with a mixture of 3% isoflurane in 30% oxygen and 70% air. The left middle cerebral artery (MCA) was permanently occluded as described previously (Gill et al., 1992). The MABP and blood gases were maintained at normal levels during the entire surgical procedure and for up to 1 hour after MCA occlusion. The temperature of the animals was maintained at 37 ± 0.2°C for the entire surgical procedure and for up to 48 hours during the survival period. Animals (n = 2 per time point) were decapitated at 4, 8, and 24 hours after permanent MCA occlusion. The ipsilateral, frontal, and parietal cortices and the striatum were dissected from the brain and mitochondrial and cytosolic fractions were prepared. The equivalent areas from the contralateral side were used as controls. All dissections were carried out on ice and the tissues were collected and processed immediately.
Transient forebrain ischemia in rats
Adult male Wistar rats (280–340 g) were subjected to transient forebrain ischemia induced by occlusion of both common carotid arteries (CCAs) and combined with systemic hypotension by withdrawal of blood, as detailed elsewhere (Van Lookeren-Campagne and Gill, 1998). Briefly, rats were fasted overnight before the surgery, but had free access to water. Animals were anesthetized with 4% isoflurane in 70% air and 30% oxygen, intubated, and mechanically ventilated with a mixture of 1.5% isoflurane in 70% air and 30% oxygen. The femoral artery was cannulated to enable monitoring of physiologic variables, such as MABP and blood gases. A catheter was placed in the inferior vena cava through the external jugular vein for withdrawal and readministration of shed blood. The EEG was continuously recorded using two silver-needle electrodes inserted in to the temporalis muscles. The core body temperature was maintained at 37°C during the surgical procedures. Ischemia was produced by withdrawing blood via the jugular vein to produce a MABP of 50 mm Hg, then the CCAs were occluded using microsurgical clips (n = 4). After 11 minutes, the ischemic period was rapidly terminated by removing the clips from the CCAs and rapidly reinfusing the shed blood. Reperfusion of the CCAs was confirmed by visual inspection. Sham-operated animals underwent the same surgical procedure but without occlusion of the CCAs and hypotension (n = 2). Body temperature was monitored continuously for the next 24 hours using telemetry (Data Sciences International, Frankfurt, Germany) and maintained at 37.5°C. The animals were decapitated 24 hours later. The brain was removed and the hippocampal CA1 region, somatosensory cortex, striatum, and cerebellum were dissected, and cytosolic and crude mitochondrial fractions were prepared. Sham-operated animals were used as controls.
Transient focal ischemia in mice
Male C57BL/6 mice were anesthetized using a mixture of 3% isoflurane in 30% oxygen and 70% air. Anesthesia was subsequently maintained with 2% isoflurane in 30% oxygen and 70% air using a facial mask. Transient focal ischemia was induced as described by Hara et al., (1997). Briefly, the internal carotid artery was exposed and an 8/0 silicone-coated suture was introduced into the internal carotid artery via the common carotid artery and advanced to occlude the origin of the MCA. The suture was secured in place and the mice allowed to recover from the anesthesia for 2 hours, then they were reanesthetized and the suture was removed to allow reperfusion of the MCA. The animals were decapitated 30 minutes after reperfusion and the brains were processed for analysis of caspase activity. The contralateral hemisphere served as the control tissue.
Perinatal hypoxic-ischemia
Unilateral hypoxic-ischemia was induced in 7-day-old Wistar rats of both sexes. The pups were anesthetized with halothane (3.0% for induction and 1.0%–1.5% for maintenance) in a mixture of nitrous oxide and oxygen (1:1), and the duration of anesthesia was less than 10 minutes. The left common carotid artery was cut between double ligatures of prolene sutures (6–0). After the surgical procedure, the wounds were covered with a local anesthetic and the pups were allowed to recover for 1 to 2 hours. The litters were then placed in a chamber perfused with a humidified gas mixture (7.70 ± 0.01% oxygen in nitrogen) for 70 minutes. The temperature in the gas chamber was maintained at 36°C. After hypoxic exposure, the pups were returned to their biological dams until time of death. Control animals were operated and ligated but not subjected to hypoxia. The Ethical Committee of Göteborg approved all animal experiments.
MK-801 administration to neonatal animals
Intraperitoneal MK-801 (0.5 mg/kg) was administered to 7-day-old male Wistar rats at 0, 8, and 20 hours as described by Ikonomidou et al. (1999). Control animals received the vehicle at the same times (n = 4 per group). The animals were decapitated 24 hours after the first dose of MK-801 or vehicle, the brains were rapidly removed, and various brain regions (cortex, caudate, and hippocampus) were dissected on ice. The samples were processed to yield mitochondrial and cytosolic fractions for the analysis of caspase activity.
Preparation of cytosolic and crude mitochondrial fractions
Dissected brain areas were suspended in isolation buffer (10-mmol/L Trishydrochloride pH 7.3, 0.32-mol/L sucrose, 1-mmol/L K-ethylenediaminetetracetic acid [EDTA]) at 100 to 300 mg tissue/mL and homogenized in a Dounce (Fischer Scientific, Wohlen, Switzerland) homogenizer with 12 to 15 up-and-down strokes of a tight-fitting glass pestle. The homogenate was centrifuged at 1,330 g for 3 minutes, and the resulting postnuclear supernatant was centrifuged at 10,000 g for 10 minutes. The pellet was washed with isolation buffer and resuspended in approximately half of the original volume to yield the crude mitochondrial fraction. The cytosolic fraction was obtained by centrifuging the postmitochondrial supernatant at 100,000 g for 20 minutes to remove particulate material. Protein concentrations were determined by the Bradford dye-binding procedure (Bio-Rad Laboratories, Munich, Germany) using bovine serum albumin as a standard. In the neonatal hypoxia–ischemia model, the animals were decapitated and the brains were rapidly dissected on a bed of ice, weighed, quickly frozen in isopentane and dry ice, and stored at −80°C. Cortical tissue rostral to the hippocampus (=50 mg), was dissected out from each hemisphere at −10°C. The tissue was homogenized by sonication in 10 volumes of ice-cold 50-mmol/L Trishydrochloride pH 7.3 containing 5-mmol/L EDTA, aliquoted, and stored at −80°C until measurement of caspase-3 activation. Caspase activity was measured in the homogenate and in the soluble fraction obtained after centrifugation at 20,000 g for 15 minutes.
Caspase-3 activity measurements
The brain subcellular fractions (20 μL) were added to 80 μL caspase-3 assay buffer (100-mmol/L HEPES pH 7.0, 0.5-mmol/L EDTA, 5-mmol/L dithiothreitol, 20% glycerol, 0.1% CHAPS (3-[(3-cholamidopropyl)-dimethyl-ammonio]-1-proponsulfonate), 25-μmol/L Ac-DEVD-AMC) in a white mi-crotiterplate (Life Technologies, Basel, Switzerland). The fluorescence of released AMC (excitation, 380 nm; emission, 460 nm) was measured every 20 minutes for up to 3 hours at ambient temperature in a Perkin Elmer (Ueberlingen, Germany) LS50B instrument. The enzymatic activity was calculated from the linear increase of fluorescence as a function of time and converted to pmol AMC produced per milligram cytosolic or mitochondrial protein per hour using an AMC standard curve.
Experimental groups for the neuroprotection study
For the neuroprotection experiments, a cannula was implanted in the left ventricle 24 hours before MCA occlusion in anesthetized animals. The left MCA was permanently occluded in anesthetized animals, as described previously. After surgery, the animals were allowed to recover from the anesthetic and 80 ng zVAD-fmk (n = 13) was administered intracerebroventricularly 5, 60, and 120 minutes after MCA occlusion. Vehicle was administered to control animals (2μL over 2 minutes, n = 12) intracerebroventricularly at the same time points. Additionally one group of animals was administered 1.5 mg/kg MK-801 intraperitoneally 5 minutes after MCA occlusion (n = 9). The animals were allowed to survive for 48 hours, after which time they were anesthetized and the brains were removed and prepared for histologic evaluation of the lesion volume.
Quantitative measurement of the volume of ischemic damage
The brains were embedded in paraffin, sectioned, and the volume of infarction was evaluated at the level of the cortex and caudate nucleus in 13 slices taken through the brain. The volume of infarction was measured using unbiased stereological tools (Olympus, Copenhagen, Denmark) in terms of the volume of hemispheric, cortical, and striatal damage.
Statistical analysis
The differences in the volume of ischemic damage for each brain region (hemisphere, cortex and caudate) for the various groups of animals was tested using analysis of variance with Bonferroni correction (BMDP version 1.1, Statistical Software, Cork, Ireland). All data are presented as mean ± SD for N animals.
RESULTS
Permanent focal ischemia in rats
Caspase activity was measured in the ipsilateral frontal and parietal cortices and striatum at various times after permanent MCA occlusion. The respective areas from the contralateral side of the brain served as controls. A constitutive caspase-3–like or DEVDase activity was found in the cytosolic and mitochondrial fractions derived from the various brain areas (Fig. 1A). However, there was no significant increase of caspase-3 activity in the ischemic penumbra and core areas as analyzed at 4, 8, and 24 hours after MCA occlusion. The DEVDase activity in the corresponding contralateral hemisphere also remained unchanged in the cytosolic and mitochondrial fraction. Interestingly, a marked decline of the constitutive DEVDase activity was observed in the ipsilateral parietal cortex and striatum at 8 hours that was most pronounced at 24 hours and probably reflected the ongoing ischemic damage in these areas at these time points. We could not show an upregulation of procaspase-3 in any of the brain regions measured 24 hours after focal ischemia using an antibody against procaspase-3 (Fig. 1B). Although the antibody detected a faint band at 34 kd (i.e., the position of procaspase-3), there was no apparent increase of this signal in the samples from the ipsilateral side. Whether this band indeed represents trace amounts of procaspase-3 in the adult rat brain, or whether it is caused by an unspecific crossreactivity of the antibody (similar to the reaction with some higher molecular weight bands) remains unresolved. The antibody clearly identified procaspase-3 in the cytosol of neonatal rat brain and showed downregulation of procaspase-3 expression to nearly undetectable levels in adult rat brain (Fig. 1C). The same antibody was also used to show an age-related decrease in procaspase-3 expression in murine brain extracts (Fig. 1D). Furthermore, addition of granzyme B resulted in activation of procaspase-3 and the active fragment (p17 subunit) could clearly be detected on the immunoblot with the same anticaspase-3 antibody (Fig. 1D). This band also decreased in an age-dependent manner and corresponded to the decrease in procaspase-3 expression.
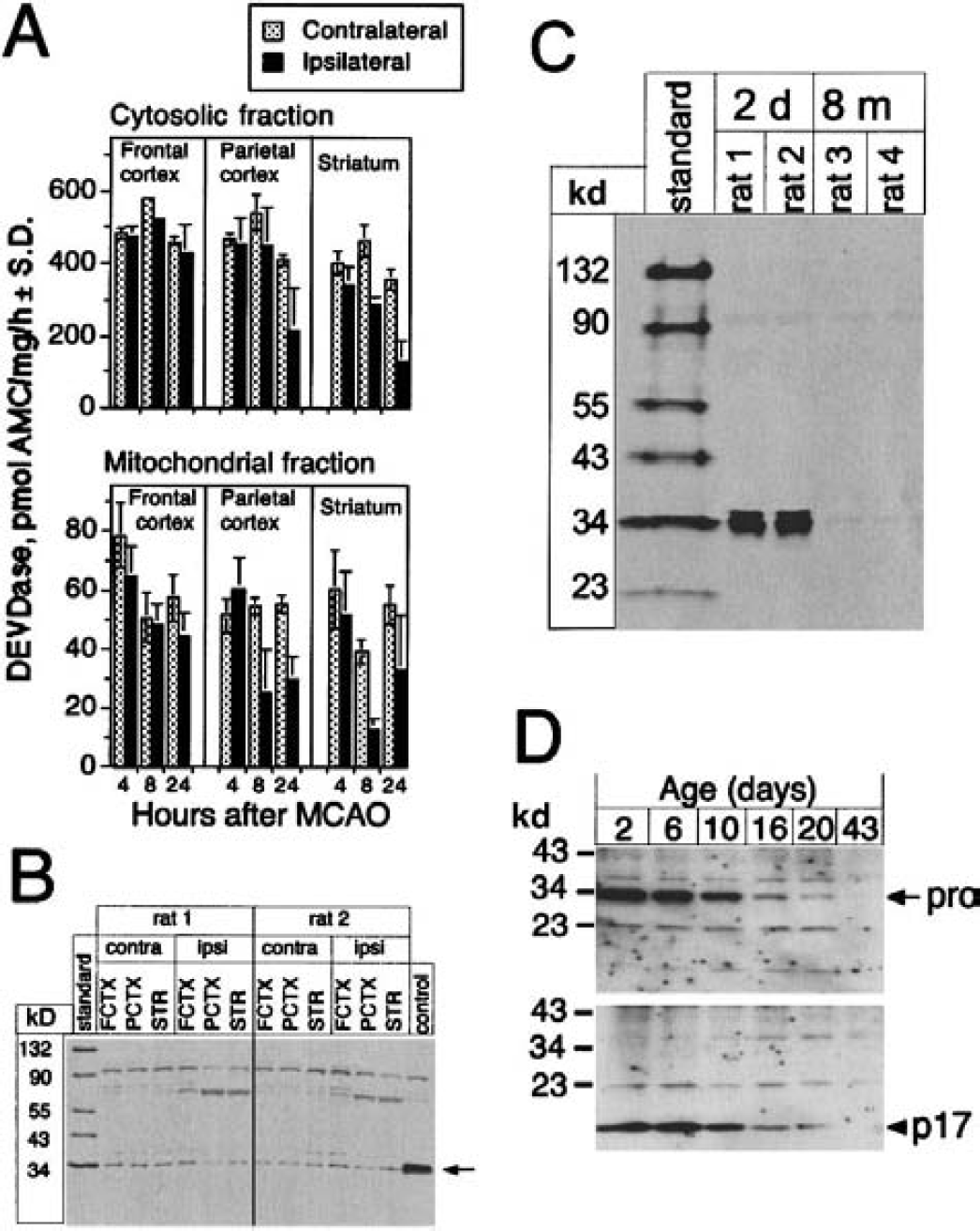
DEVDase activity and procaspase-3 immunoblot of various brain areas after permanent middle cerebral artery occlusion (MCAO) in rats. (Pro)caspase-3 immunoblot of neonatal and adult rat and mouse brain.
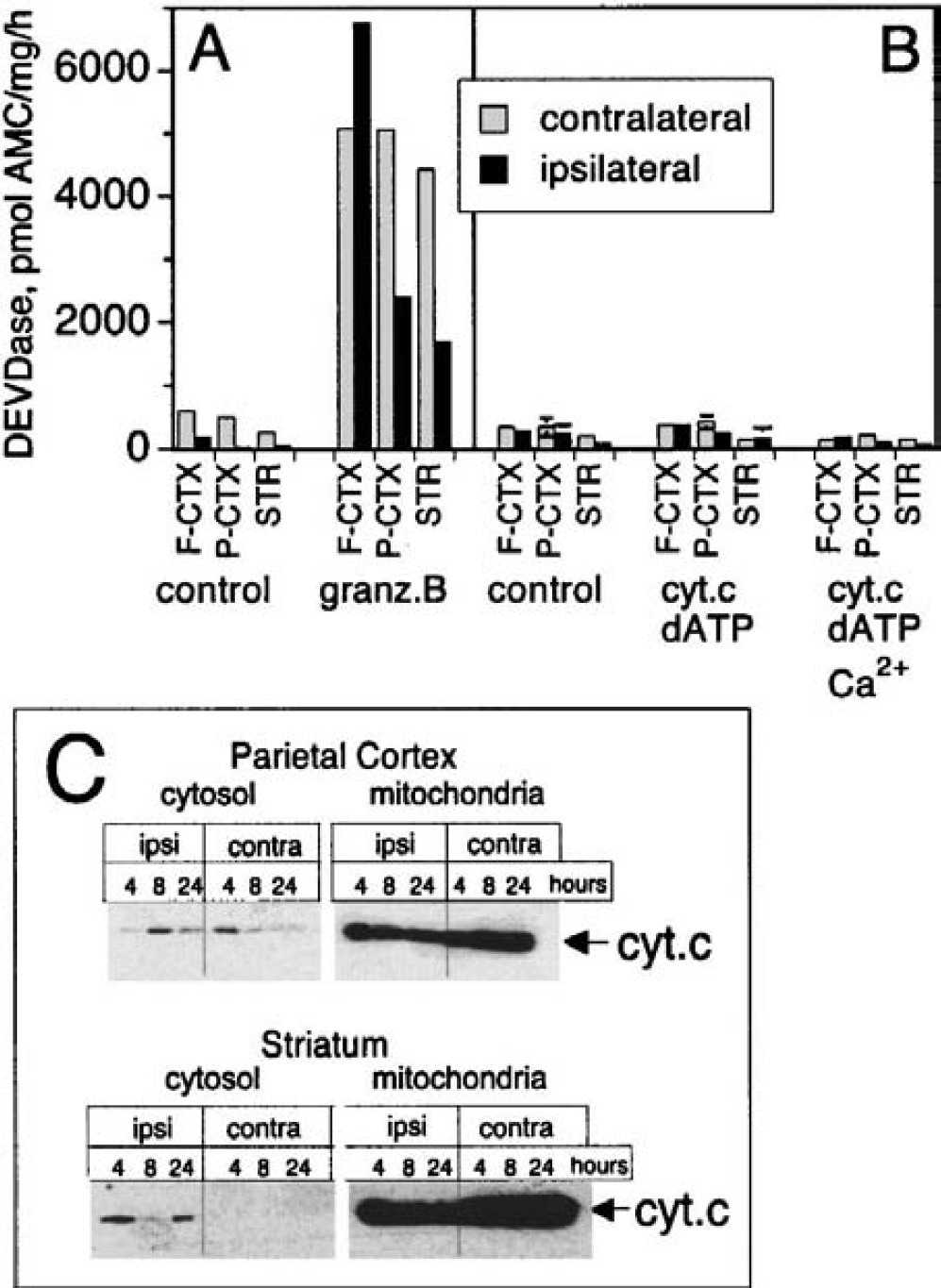
To show that the chosen conditions allowed measurement of caspase activity in rat brain extracts (Talanian et al., 1997; Yang et al., 1998), we added granzyme B, a known activator of caspase-3, −7, and −10, to the cytosolic fraction derived from the different tissues collected 24 hours after permanent MCA occlusion (Fig. 2A). This experiment clearly revealed a granzyme B–inducible DEVDase activity that was present in the various cytosolic fractions from both the ipsilateral and contralateral sides of the brain. However, the DEVDase activity seen after incubation with granzyme B was unlikely to represent caspase-3 because addition of cytochrome c and dATP with or without Ca2+ did not result in a similar induction of DEVDase activity (Fig. 2B). Using an identical experimental design with cytosolic extracts from neuronal and nonneuronal cell lines and neonatal brain, we observed a marked activation of procaspase-3 by exogenous cytochrome c/dATP that was enhanced several-fold in the presence of Ca2+ ions (Niederhauser and Loetscher, unpublished data, 2000). We also analyzed the cellular distribution of cytochrome c in the various brain regions 4, 8, or 24 hours after focal ischemia by Western blotting (Fig. 2C). Although we could see small amounts of cytosolic cytochrome c in some samples, there was no clear correlation of mitochondrial cytochrome c release and severity of the ischemia (i.e., no time-dependent increase of soluble cytochrome c between 4 and 24 hours after MCA occlusion). However, it appeared that the samples from the ipsilateral sides in general contained slightly more cytosolic cytochrome c than those from the corresponding contralateral sides, particularly in the striatum (Figure 2C). Nevertheless, in all cases the cytosolic cytochrome c represented only a minor fraction and the large majority of cytochrome c cosegregated with mitochondria.
In vitro activation of DEVDase and distribution of cytochrome c after permanent middle cerebral artery (MCA) occlusion in rats. Cytosolic fractions prepared from frontal (FCTX) and parietal cortex (PCTX) and striatum (STR) after 24-hour MCA occlusion were treated with granzyme B
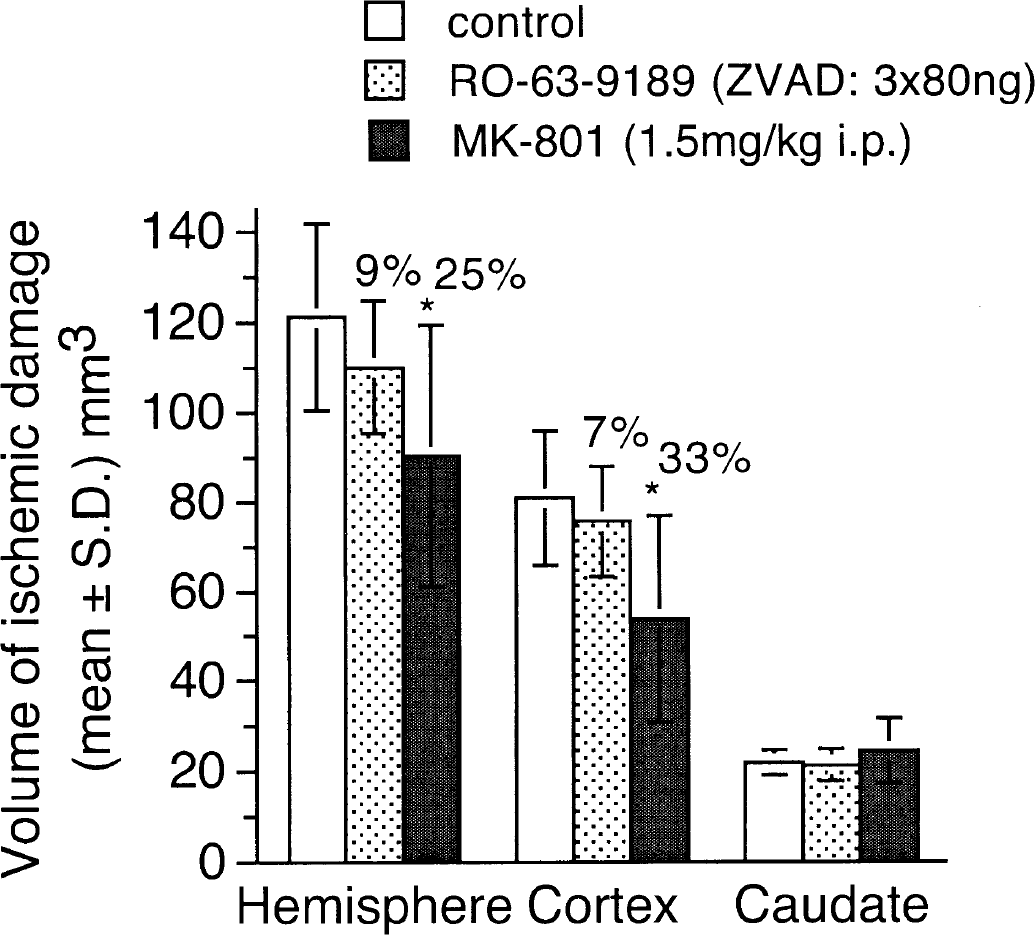
The results of the neuroprotection experiment are shown in Fig. 3. There was no significant protective effect of zVAD-fmk administered at a total dose of 240 ng against the volumes of hemispheric and cortical infarction when compared with the control group of animals. In the same experiment, MK-801, which was used as a positive control, produced a significant (P < 0.05) reduction in the volumes of hemispheric and cortical infarction. There was no significant reduction in the volume of striatal damage because in this model the lenticulostriate artery was occluded and this is the main branch serving the striatum.
Determination of the ischemic damage after permanent middle cerebral artery (MCA) occlusion in rats. At 5, 60, and 120 minutes after permanent MCA occlusion, zVAD-fmk (80 ng at each time point) was administered intracerebroventricularly (n = 13). The control group received vehicle only (n = 12). Another group of animals was administered 1.5 mg/kg MK-801 intraperitoneally 5 minutes after MCA occlusion (n = 9). At 48 hours after treatment the brains, were prepared for histologic evaluation of the lesion volume. Values are mean ± SD. Asterisk indicates statistically significant differences (P < 0.05) according to analysis of variance data analysis and Bonferroni correction.
Transient focal ischemia in mice
Transient focal ischemia in the mouse for 2 hours followed by 30 minutes of reperfusion also failed to produce an increase of DEVDase activity in the cytosolic or mitochondrial fraction (Fig. 4A). As seen in the permanent MCA occlusion model in rats, DEVDase activity could be measured after addition of granzyme B to the cytosolic fraction, again indicating that the experimental conditions were permissive to analyze caspaselike enzymatic activities (Fig. 4B). Supplementation of the cytosol with exogenous cytochrome c/dATP in the absence or presence of Ca2+ did not reveal any DEVDase activity that would be indicative for procaspase-3 activation. In this model of ischemia, however, a consistent small fraction of cytochrome c was observed in the cytosol prepared from the ipsilateral hemisphere, whereas no cytosolic cytochrome c was detected in the contralateral hemisphere (Fig. 4C).
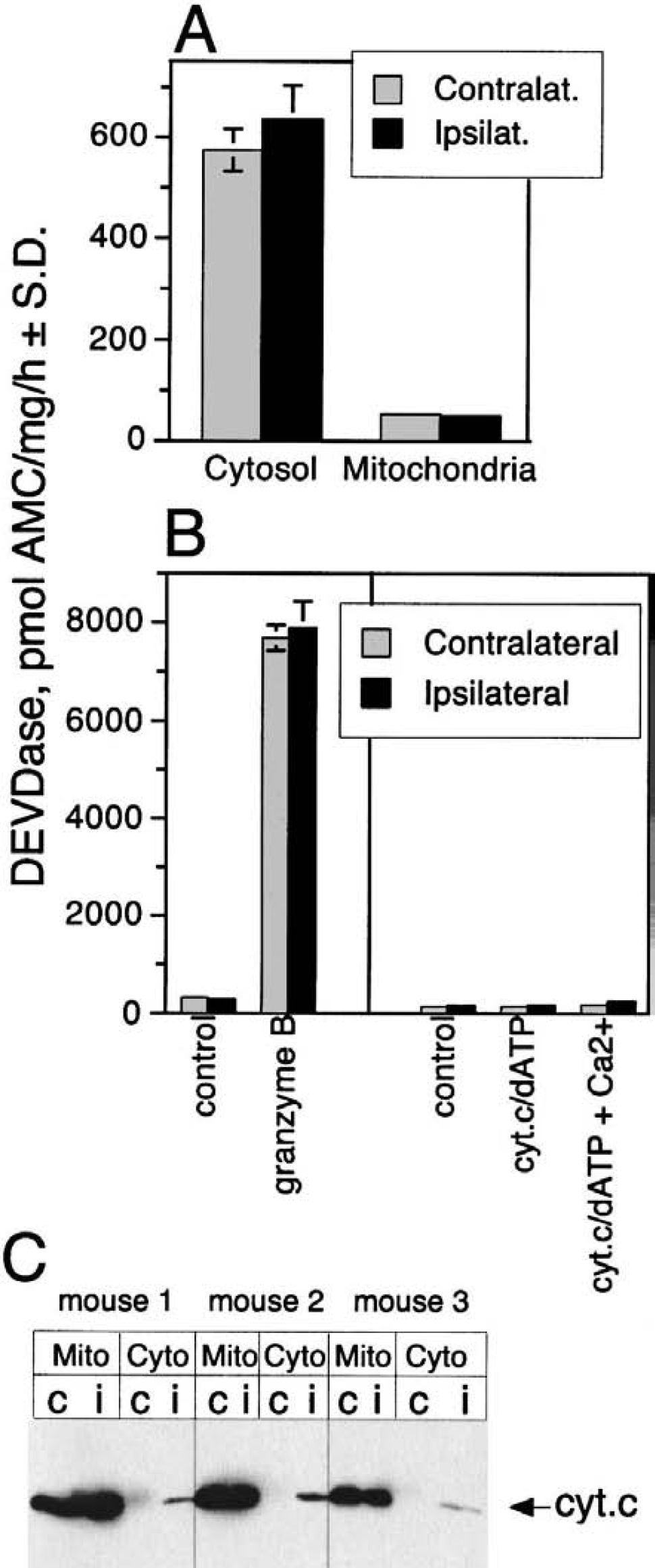
DEVDase activity and distribution of cytochrome c after transient focal ischemia in mice. Animals were subjected to middle cerebral artery (MCA) filament occlusion for 2 hours, and the brain hemispheres were dissected after 30-minutes reperfusion.
Transient forebrain ischemia in rats
The potential role of caspase activation was also measured in a model of transient forebrain ischemia induced by occlusion of both CCAs and concomitant hypotension for 11 minutes. This protocol resulted in delayed neuronal damage in the hippocampal CA1 neurons and some morphologic changes in the striatum and cortex (Van Lookeren-Campagne and Gill, 1998). DEVDase activity was measured 24 hours after transient forebrain ischemia and, similar to the other two ischemia models described previously, we did not see any induction of a caspaselike activity in any of the brain regions examined (Fig. 5).
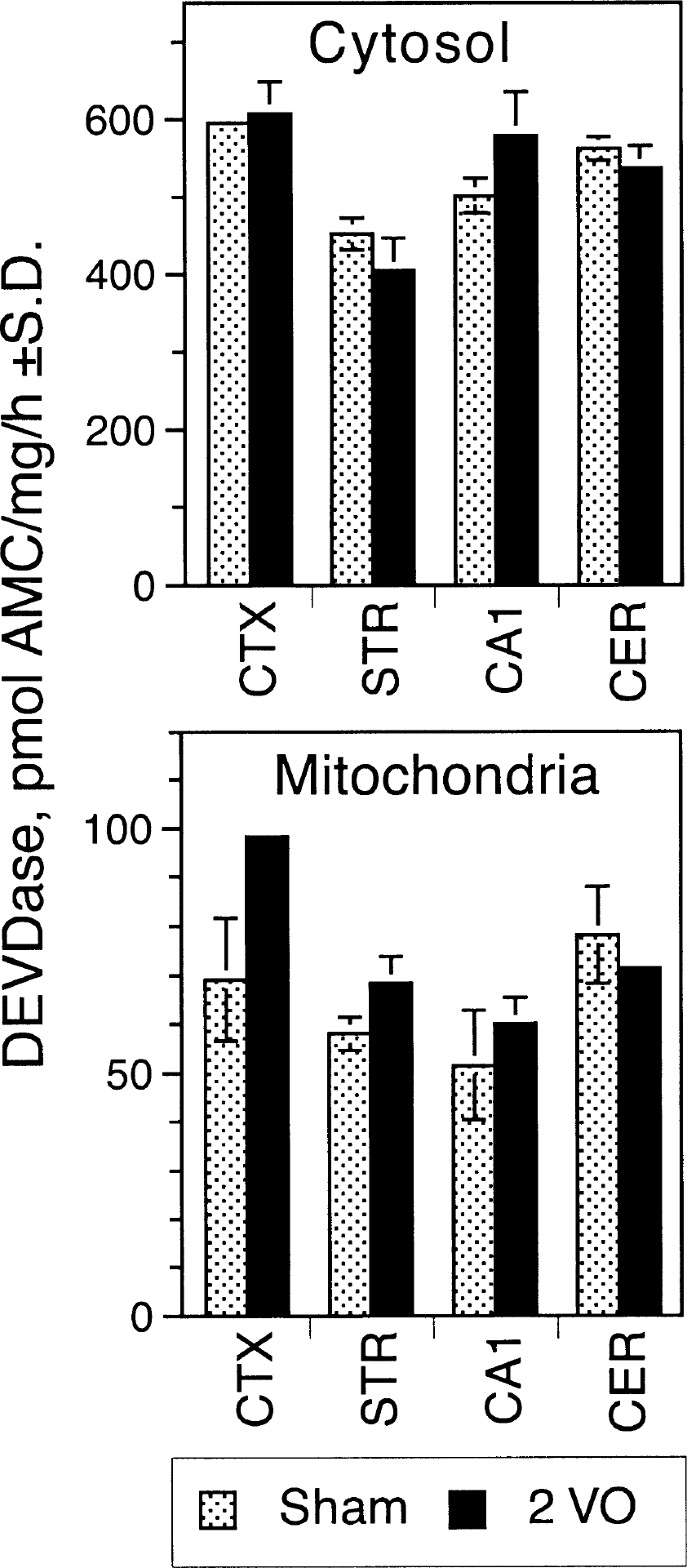
DEVDase activity in various brain regions after transient forebrain ischemia in rats. Rats were subjected to transient forebrain ischemia by occlusion of both common carotid arteries combined with systemic hypotension as described. The ischemic period was terminated and reperfusion initiated after 11 minutes. Twenty-four hours later, the brain was removed and the different brain regions were dissected. Mitochondrial and cytosolic fractions were prepared and analyzed for DEVDase activity. The average values (duplicate determinations) of two animals ± SD are shown.
Neonatal model of hypoxic ischemia
The hypoxia–ischemia model in immature rats results in neuronal damage in the ipsilateral cortex, whereas the contralateral side is left undamaged (Rice et al., 1981; Andiné et al., 1990). Again, we studied the potential involvement of caspase-3 in the hypoxia–ischemia model in immature rats by enzymatically measuring DEVDase activity in total brain homogenates and soluble brain extracts prepared from the ipsilateral and contralateral hemispheres. There was some constitutive DEVDase activity seen in the sham-operated 7-day-old animals (Fig. 6) with no difference between left and right hemispheres. However, there was a dramatic increase of DEVDase activity in the ipsilateral cortex compared with the contralateral cortex in the hypoxic ischemia animals. Increased enzymatic activity could be seen in both the homogenate and soluble fraction. The induction of DEVDase activity was evident as early as 1 hour after the hypoxic exposure and peaked at approximately 24 hours (Puka-Sundvall et al., 2000; Zhu et al., 2000). Thus, activation of caspase(s), most likely procaspase-3, does appear to play an important role in neuronal degeneration seen after hypoxic ischemia in neonates.
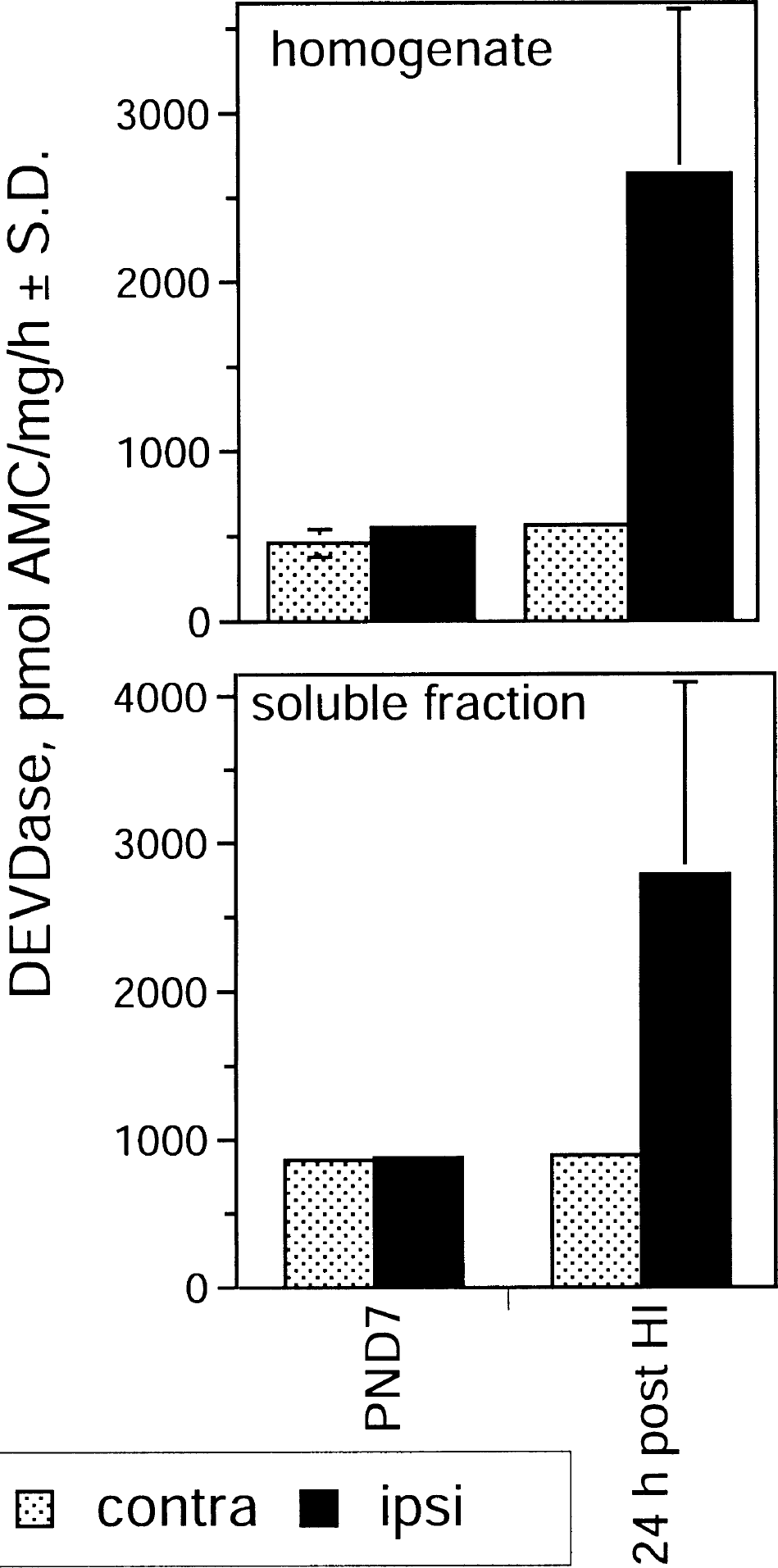
Induction of DEVDase activity in a hypoxic-ischemia model in neonatal rats. Postnatal-day-7 (PND7) rats were subjected to unilateral hypoxic ischemia by cutting the left common carotid artery followed by hypoxic exposure for 70 minutes. After 24 hours, the brains were removed and cortical tissue from both hemispheres was dissected. DEVDase activity was measured in the homogenate and in the soluble fraction after centrifugation. PND7 represent operated control animals not subjected to hypoxic exposure. Seven animals were used in each group and average values ± SD are shown. HI, hypoxic ischemia.
MK-801 administration to neonates
When MK-801 (0.5mg/kg) was administered intraperitoneally to 7-day-old neonatal rats at 0, 8, and 20 hours, only sporadic cells staining positive for TUNEL were seen in both these and control animals (data not shown). We have used this model to test whether activation of caspase(s) contributed to the observed phenotype 24 hours after initiation of the dosing of MK-801 or vehicle, the cortex, caudate and hippocampus were dissected and processed to measure DEVDase activity. As shown in Figs. 7A and 7B, we did not see an increase of DEVDase activity in the samples prepared from the MK-801– treated animals in the cytosolic or mitochondrial fraction. However, addition of exogenous cytochrome c and dATP to the cytosolic fraction induced a clear increase of DEVDase activity in the cortex and hippocampus, but there was no significant difference between vehicle- and MK-801–treated animals (Fig. 7C). The effect of cytochrome c/dATP was much more pronounced in the presence of Ca2+ in all three brain regions studied, though the change seen in the cortex was of the largest magnitude. Cytochrome c/dATP has been described to specifically activate procaspase-3, involving an interaction with Apaf1 and caspase-9 in the so-called apoptosome. Therefore, it is likely that the cytochrome c/dATP-induced DEVDase activity in the neonatal brain samples indeed represents active caspase-3.
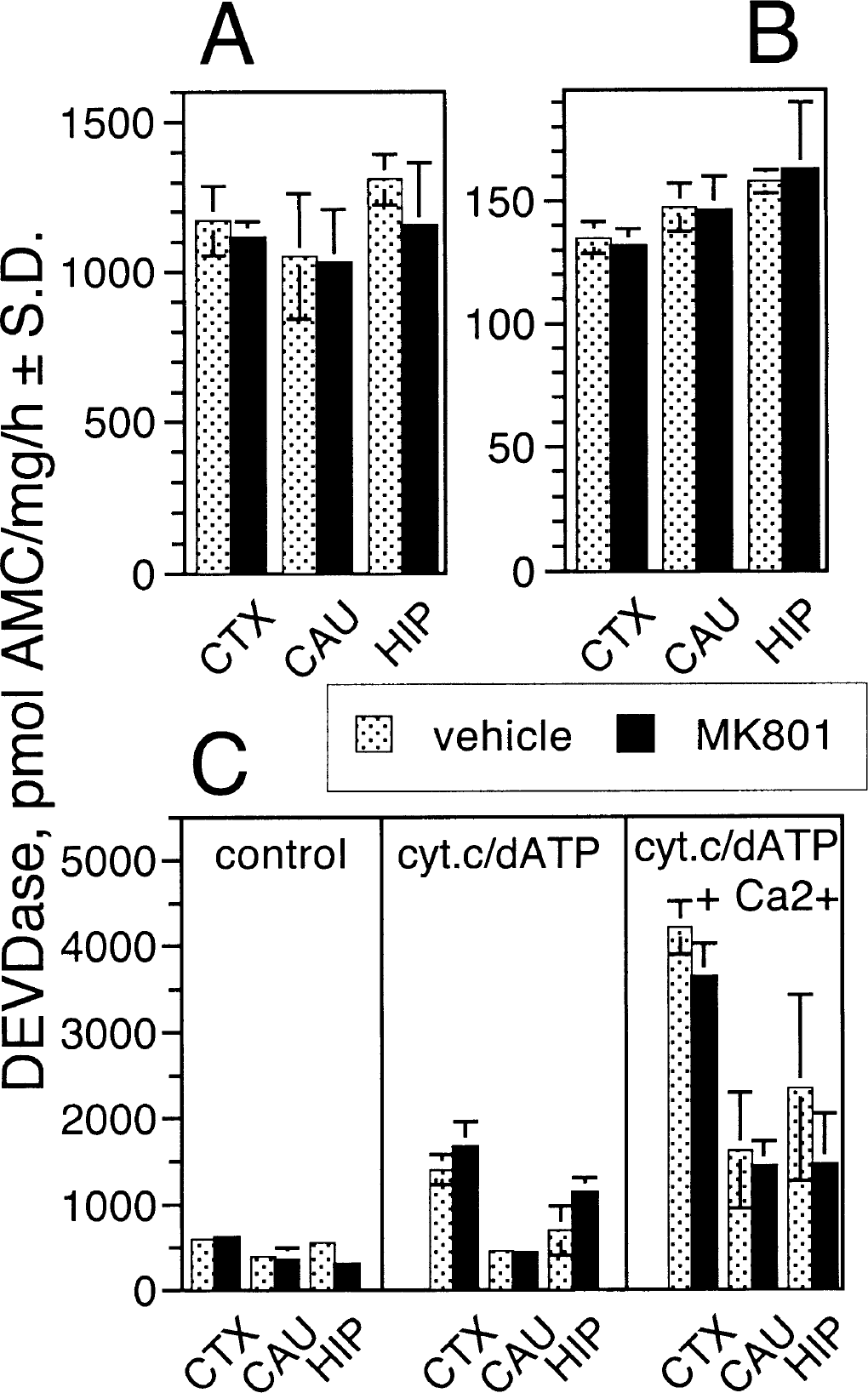
DEVDase activity in various brain regions after administration of MK-801 to neonatal rats. MK-801 (0.5 mg/kg) was given intraperitoneally to postnatal-day-7 rats at 0, 8, and 20 hours. Twenty-four hours after the first dose was administered, the brains were removed and the cortex (CTX), caudate (CAU), and hippocampus (HIP) were dissected. Control animals received vehicle at the same times. Cytosolic
DISCUSSION
The present study has been unable to show induction of DEVDase activity, which would be indicative of procaspase-3 activation, after focal or global ischemia in adult rat or mouse brain. Expression of procaspase-3 in the brain has been reported to be developmentally regulated at the transcriptional level (Krajewska et al., 1997; Ni et al., 1997) with only low levels of messenger RNA detectable in adult brain. Immunoblot analyses has also shown downregulation of procaspase-3 at the protein level at the final stages of brain development (Hu et al., 2000; Yakovlev et al., 2001). We also showed a dramatic downregulation of procaspase-3 in adult rats compared with young neonatal animals, and developmental downregulation in mice. Increases in procaspase-3 messenger RNA have been reported to occur 16 and 24 hours after permanent focal ischemia in rats (Schulz et al., 1999; Ni et al., 1997, 1998). After ischemia, a variety of genes are induced at the transcriptional level; however, under these conditions the translational machinery is severely affected and protein synthesis rapidly decreases (Hossmann, 1993). Therefore, it is important to confirm the presence of any upregulated gene at the protein level, and in the present studies we could not detect induction of procaspase-3 expression in the adult brain after an ischemic insult as analyzed by immunoblot experiments. A similar lack of caspase-3 detection by Western and enzymatic analysis of rat brain samples after permanent MCA occlusion has also been recently presented (Velier et al., 1999). An earlier report by Hara et al., (1997) showed a peak of increased DEVDase activity at 30 minutes of reperfusion after 2 hours of MCA occlusion in a mouse model. We failed to replicate these findings in our assay system because no change in DEVDase activity was noted using the same model and time of ischemia and reperfusion.
We were also unable to induce DEVDase activity in the cytosolic fraction of adult rat or mouse brain tissue by addition of exogenous cytochrome c/dATP and Ca2+, whereas a similar stimulation of neonatal cytosol samples resulted in an increase of DEVDase activity. However, the cytosolic fractions from the adult animals responded to granzyme B treatment with DEVDase activation, thus confirming correct assay design capable of monitoring proteolytic cleavage of the DEVD-AMC substrate. The identity of the granzyme B–induced DEVDase activity in the adult brain extracts is currently unknown. It is unlikely to be caspase-3 because it is downregulated shortly after birth, but potential candidates are caspase-7 or −10 (Talanian et al., 1997; Yang et al., 1998), or other proteases that may or may not be related to the caspase protease family. Granzyme B–induced DEVDase activity in neonatal brain extracts, however, can clearly be attributed to activated procaspase-3, as demonstrated by immunoblotting experiments (Fig. 1D). In summary, the previous morphologic evidence along with our current results suggest that apoptosis and procaspase-3 activation are not involved in the neuronal degeneration seen after permanent or transient focal ischemia in adult animals, which is considered to be a model for human stroke.
We did not see a significant neuroprotection with the rather unselective caspase inhibitor, zVAD-fmk, administered after permanent MCA occlusion; thus, these findings are at odds with those of Loddick et al. (1996). In this earlier study by Loddick et al. (1996), 50% protection was obtained in the striatum. However, in the permanent MCA occlusion model first described by Tamura et al., (1981) and as used in the present study, neuroprotection in the striatum was never obtained (Park et al., 1988; Gill et al., 1992, 1995). This lack of striatal protection is not surprising because the occluded lenticulostriate artery is an end artery, and it is difficult to reconcile how the tissue could survive the ischemic insult in the absence of any blood flow. More recently, additional experiments reported neuroprotective effects of zVAD-fmk in a model of transient focal ischemia in rats and mice with 2 hours of ischemia and 22 hours of reperfusion (Hara et al., 1997). However, in these studies inhibitor treatment was initiated before the ischemic insult.
The data obtained with the neonatal animals are intriguing, particularly in view of a recent report from Ikonomidou et al. (1999) that suggested that repeated administration of MK-801 to rats at postnatal day 7 induced an apoptotic phenotype. Using identical conditions, however, we were able to detect only sporadic TUNEL staining in both control and MK-801–treated animals and did not see any induction of a DEVDase activity, though we could activate the DEVDase in vitro by the addition of exogenous cytochrome c/dATP and Ca2+. These findings further show that, unlike the adult brain, procaspase-3 is present in neonatal brain and can be activated in vitro under appropriate conditions. In contrast, in the hypoxic ischemia model we clearly see an induction of DEVDase activity 24 hours after the ischemic insult that is in agreement with findings of a previous report by Cheng et al. (1998). Furthermore, this report showed reduction of the ischemic damage with BAF (boc-aspartyl(Ome)-fluoromethylketone), a pan-caspase inhibitor. Therefore, the cumulative evidence from these studies suggest that in the neonatal brain hypoxia–ischemia can trigger apoptotic pathways that may contribute to cell death.
The transient forebrain ischemia model results in delayed neuronal degeneration of hippocampal CA1 neurons and it more closely reflects the clinical situation of cardiac arrest. The presence of procaspase-3 and generation of its active fragment after transient forebrain ischemia have been reported (Chen et al., 1998; Gillardon et al., 1997, 1999). Chen et al. (1998) also observed an increase in DEVDase activity in the hippocampus and caudate putamen 24 hours after transient forebrain ischemia produced by four-vessel occlusion for 15 minutes. Here, we used a slightly different model of transient forebrain ischemia, but the time course for delayed degeneration of hippocampal CA1 neurons and for changes in the caudate putamen is similar in the two models (Smith et al., 1984; Pulsinelli et al., 1982). We were unable to show activation of procaspase-3 at various times after transient forebrain ischemia using the enzymatic assay for DEVD-AMC cleavage. The reason for this apparent discrepancy is not known at the present time. However, the findings of Chen et al. (1998) are inconsistent with the very low expression of caspase-3 in adult brain and the inability to activate it in vitro with cytochrome c during the present experiments. The same authors also showed a colocalization of TUNEL and caspase-3 immunoreactivity peaking at 72 hours, at which time cell death is complete in this model, and it is possible that the increased staining is due to increased antigenicity of damaged neurons. Petito et al. (1997) showed that DNA fragmentation was only detected after the development of delayed neuronal death in CA1 neurons subjected to 10 minutes of transient forebrain ischemia.
It has been postulated that in response to an apoptotic stimulus mitochondria release cytochrome c into the cytoplasm, where it interacts with Apaf1 in the apoptosome complex, thereby initiating a cascade of events that finally lead to the activation of procaspase-3 (Liu et al., 1996; Zou et al., 1997; Saleh et al., 1999). We did not obtain any evidence for a functional significance of this pathway in focal ischemia in rats induced by permanent MCA occlusion. Although we occasionally detected cytosolic cytochrome c in the brain samples taken at various times after ischemia, there was no clear correlation of mitochondrial cytochrome c release and the extent of ischemic damage. In addition, this released cytochrome c represented only a minor fraction of cytochrome c still retained in mitochondria. In the mouse model of transient focal ischemia we observed a small amount of cytochrome c in the cytosolic fraction of the ipsilateral hemisphere, but again the majority of the cytochrome c remained in the mitochondria. Whether the difference in cytosolic cytochrome c levels between ipsilateral and contralateral sides reflected a specific mitochondrial cytochrome c release or whether the ischemic conditions rendered mitochondria more vulnerable to subsequent tissue fractionation with a minor loss of cytochrome c remains to be seen. Alternatively, we cannot exclude the possibility that an ischemic insult affects the efficiency of the mitochondrial protein import machinery, and as a consequence mitochondrially targeted, newly synthesized proteins may accumulate in the cytosol. In conclusion, necrotic damage may trigger mitochondrial swelling followed by release of cytochrome c without apoptotic significance, because in the adult brain we were unable to activate DVEDase activity in cytosolic extracts.
There is a lot of controversy in the ischemia field about the relative roles of apoptosis and necrosis in the ongoing neuronal degeneration. All the morphologic evidence, which is the gold standard for defining apoptosis, suggests that in the adult brain there are no signs of apoptosis (Van Lookeren-Campagne and Gill, 1996; Petito et al., 1997; Colbourne et al., 1999). The current studies extend these observations to the lack of a role of mitochondrially derived cytochrome c and procaspase-3 activation in ischemia-induced cell death in adult brain. However, the findings in the neonatal brain, where procaspase-3 is indeed activated after perinatal hypoxia, are important in the context of the present study because it shows the existence of a functional proapoptotic pathway involving procaspase-3 activation in infant but not adult animals. Whether the absence of apoptotic processes in the adult brain is solely due to the downregulation of procaspase-3 expression shortly after birth or whether other essential components of the apoptotic machinery are also developmentally regulated awaits further study.
Footnotes
Acknowledgments:
The authors thank Simone Schleeger and Hanspeter Kurt for expert technical assistance.