
Abstract
Mild hypothermia protects the brain from ischemia, but the underlying mechanisms of this effect are not well known. The authors previously found that hypothermia reduces the density of apoptotic cells, but it is not certain whether temperature alters associated biochemical events. Mitochondrial release of cytochrome c has recently been shown to be a key trigger in caspase activation and apoptosis via the intrinsic pathway. Using a model of transient focal cerebral ischemia, the authors determined whether mild hypothermia altered expression of Bcl-2 family proteins, mitochondrial release of cytochrome c, and caspase activation. Mild hypothermia significantly decreased the amount of cytochrome c release 5 hours after the onset of ischemia, but mitochondrial translocation of Bax was not observed until 24 hours. Mild hypothermia did not alter Bcl-2 and Bax expression, and caspase activation was not observed. The present study provides the first evidence that intraischemic mild hypothermia attenuates the release of cytochrome c in the brain, but does not appear to affect other biochemical aspects of the intrinsic apoptotic pathway. They conclude that necrotic processes may have been interrupted to prevent cytochrome c release, and that the ameliorative effect of mild hypothermia may be a result of maintaining mitochondrial integrity. Furthermore, the authors show it is unlikely that mild hypothermia alters the intrinsic apoptotic pathway.
It is well established that hypothermia is neuroprotective in animal models of global and focal ischemia. There has been a resurgence of interest in hypothermia as a result of observations that even small decreases in brain or body temperature result in marked attenuation of ischemic injury (Ginsberg et al., 1992; Barone et al., 1997). At the clinical level, moderate and mild hypothermia has been applied in the treatment of acute traumatic brain injury (Marion et al., 1997; Clifton et al., 2001) and stroke (Schwab et al., 1997). Although there are numerous reports of the cerebroprotective effect of hypothermia, the mechanisms of this effect remain unclear and controversial. The neuroprotective effects of mild hypothermia have been attributed to alterations in brain metabolism (Chopp et al., 1989), neurotransmitter release (Busto et al., 1989b; Baker et al., 1995; Nakashima and Todd, 1996; Huang et al., 1998), cerebral blood flow (Lo and Steinberg, 1992), preservation of the blood-brain barrier (Dietrich et al., 1996), and suppression of free radicals (Kil et al., 1996). However, it is becoming increasingly clear that a variety of events are involved in ischemia, many of which may be affected by temperature.
Recently, several reports have shown that apoptosis participates in the deterioration of brain injury after cerebral ischemia (Linnik et al., 1993; Li et al., 1995a, 1995b; Asahi et al., 1997; Namura et al., 1998). Furthermore, inhibition of the proteins involved in apoptotic death, such as caspase-1 (Hara et al., 1997, Schielke et al., 1998, Liu et al., 1999) and −3 (Endres et al., 1998), or overexpression of antiapoptotic proteins, such as Bcl-2 (Martinou et al., 1994; Linnik et al., 1995; Lawrence et al., 1996; Lawrence et al., 1997), ameliorate cerebral ischemic injury. Conversely, suppression of Bcl-2 is known to worsen ischemic injury (White et al., 1997; Chen, et al., 2000). We previously reported that mild hypothermia decreased the number of apoptotic cells as detected by transferase TUNEL (terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick-end labeling) staining in the brains of rats subjected to transient focal cerebral ischemia (Maier et al., 1998). Edwards and colleagues (1995) similarly showed that mild hypothermia reduced numbers of apoptotic cells in a piglet model of transient hypoxia plus ischemia, but did not alter numbers of necrotic cells. It is now recognized that apoptosis can occur through one of two main pathways. The extrinsic pathway is triggered via a surface receptor by such ligands as tumor necrosis factor-α or fas ligand (Ashkenazi and Dixit, 1998), whereas the intrinsic pathway is triggered internally via mitochondrial release of cytochrome c (Green and Reed, 1998). Here, we investigate whether elements of the intrinsic apoptotic pathway, such as alterations in Bcl-2 family protein expression, release of cytochrome c, and caspase activation or activity, are altered by mild hypothermia in a rat model of experimental stroke.
MATERIALS AND METHODS
Animal protocols were approved by the Institutional Administrative Panel on Laboratory Animal Care, and procedures were followed according to their guidelines.
Focal cerebral ischemia model
Male Sprague-Dawley rats (Charles River, Wilmington, DE, U.S.A.) weighing 250 to 300 g were anesthetized with 1% halothane in 150 mL/minute oxygen and 850 mL/minute air using a face mask. A rectal probe was placed to monitor body temperature, and the animal was placed on a heating pad to maintain body temperature of 36.5 to 37.5°C (corresponding to brain temperatures of 37.5–38.5°C) (Yenari et al., 2000). The left femoral artery was catheterized to monitor blood pressure and to collect blood samples. Arterial blood gases, serum glucoses, and hematocrits were monitored before, during, and shortly after reperfusion. A cervical midline incision was made and the left carotid artery and branches were isolated. The common carotid, external carotid, and pterygopalatine arteries were ligated with a 6–0 silk suture. Ischemia was induced by inserting a 3–0 nylon monofilament suture (Ethicon, Somerville, NJ, U.S.A.) with a flamed tip into the internal carotid artery approximately 22 mm from the bifurcation of the internal and external carotid artery, creating a lesion within the territory of the middle cerebral artery. The suture was kept in place for 2 hours. In the mild-hypothermia group, systemic body cooling was achieved immediately after inserting the suture by spraying ethanol onto the rat and cooling it with a fan to body temperatures of 30 to 31°C (corresponding to brain temperatures of 33–34°C) (Yenari et al., 2000) for 2 hours. At the end of the ischemic period, the suture was removed and the internal carotid artery was ligated. Animals were rewarmed using a heating pad and a lamp positioned over the rat. Normothermic animals were maintained at a body temperature of 37°C throughout the experiment. At various times (2–72 hours after the onset of ischemia), animals were euthanized by a halothane overdose and brains were prepared for various assays.
Immunohistochemistry
After 2.5, 5 to 6, 24, and 72 hours after the insult, animals were perfused transcardially with heparinized saline (10 U/mL). For Bcl-2 and Bax stains, animals were perfused with 3% paraformaldehyde, and brains were postfixed in 3% paraformaldehyde plus 20% sucrose. For cytochrome c stains, animals were perfused and postfixed in 4% formaldehyde. Brains were stored at 4°C for 1 to 2 days before sectioning.
For Bax and Bcl-2 immunostaining, 25-μm thick sections were cut on a cryostat in the coronal plane, mounted on Superfrost-Plus glass slides (Fisher Scientific, Tustin, CA, U.S.A.), air dried, and stored at −70°C until future use. Cryosections were fixed in 75% acetone and 25% ethanol for 10 minutes and treated with 10 μg/mL proteinase K (Dako, Carpenteria, CA) for 15 minutes, followed by 0.5% triton-X 100/0.03% hydrogen peroxide/0.1% bovine serum albumin for 20 minutes. Sections were incubated in 1.5% normal goat serum for 1 hour and incubated overnight at 4°C in polyclonal rabbit antirat/mouse Bcl-2 (#13456E, 1:500 dilution, rabbit polyclonal, Pharmingen, San Diego, CA, U.S.A.) or Bax (#13686E, 1:1000 dilution, rabbit polyclonal, Pharmingen) antibody (Krajewski et al., 1994). Slides were then incubated for 30 minutes at room temperature in biotinylated antirabbit immunoglobulin G (Vector Laboratories, Burlingame, CA) preabsorbed to normal rat serum (1:200 dilution). After washing in phosphate-buffered saline, the sections were incubated for 1 hour at room temperature in 2% avidin-biotin complex (ABC solution; ABC Elite, Vector Laboratories), followed by 0.02% 3,3′-diaminobenzidine tetrahydrochloride (DAB; Sigma, St. Louis, MO, U.S.A.) in phosphate-buffered saline containing 0.06% hydrogen peroxide (Sigma) as the chromogen. Negative control sections were incubated without the primary antibodies, and rat thymus served as a positive control. Sections were counterstained with hematoxylin and eosin.
Cytochrome c immunostaining was performed as described by Fujimura et al. (1998). After postfixing, brains were cut into 50-μm thick sections using a vibratome. Free-floating sections were blocked in normal sera and reacted with a murine monoclonal antibody against rat cytochrome c (7H8.2C12, 1:500, Pharmingen), followed by rat-adsorbed biotinylated horse antimouse immunoglobulin G (Vector Laboratories) at a dilution of 1:500. The sections were incubated in ABC solution, followed by DAB. Nuclei were counterstained with methyl green.
Western blots
A separate set of animal brains subjected to similar experimental conditions was harvested 2.5, 5 to 6, 24, and 72 hours after the onset of ischemia. Three or four samples were studied per timepoint, and experiments were repeated a minimum of three times. A sham control was prepared from an animal that received no ischemia or surgery. Animals were perfused transcardially with normal saline and the brain was quickly removed. A 2- to 3-mm thick slice corresponding to the region of maximal injury (bregma) was rapidly dissected into regions corresponding to the ischemic striatum and infarct border. Corresponding samples were prepared from the contralateral, nonischemic side (Fig. 1). Brain tissue from neonatal or embryonic animals (developmental stage during which apoptosis is known to occur) were used as positive controls. Whole-brain samples were rapidly frozen on dry ice and stored at −70°C until further use. Thawed samples were homogenized in Laemmli lysis buffer containing protease inhibitors (protease inhibitor cocktail tablets, #1697498; Boehringer Mannheim, Germany). Protein concentration was determined using a bicinchoninic acid protein assay (Pierce Labs, Rockford, IL, U.S.A.) according to the manufacturers' instructions using bicinchoninic acid as a standard. Aliquots containing 50 μg protein in lysis buffer with 2-mercaptoethanol and 5% bromophenol blue were subjected to 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Ten percent sodium dodecyl sulfate-polyacrylamide gel electrophoresis was used for Western blots of poly(ADP-ribose) polymerase (PARP).
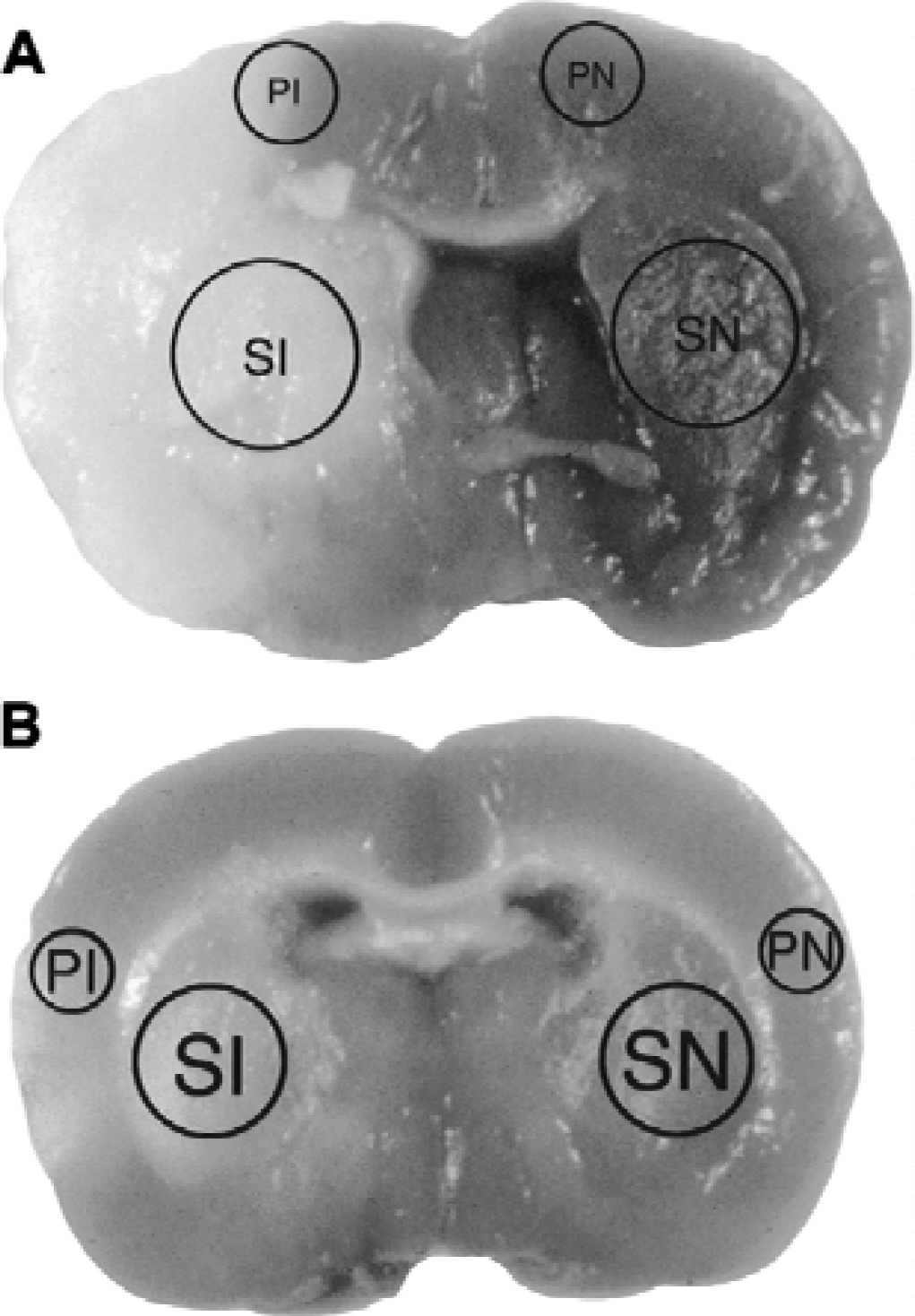
Triphenyl tetrazolium chloride-stained brains 72 hours after the onset of middle cerebral artery occlusion of a representative animal normothermic maintained
For subcellular fractionation, samples were collected as described previously, but were dissected in ice-cold suspension buffer (20mmol/L HEPES-potassium hydroxide (pH 7.5), 250 mmol/L sucrose, 10 mmol/L potassium chloride, 1.5 mmol/L magnesium chloride, 1 mmol/L edetic acid, 1 mmol/L ethyleneglycoltetracetic acid, 1 mmol/L dithiothreitol, 0.1 mmol/L phenylmethylsulfonyl fluoride, 2 μg/mL aprotinin, 10 μg/mL leupeptin, 5 μg/mL pepstatin, and 12.5 μg/mL N-acetyl- l eu- l eu-norleucinal (ALLN)), and gently homogenized using a glass tissue grinder (Wheaton, Millville, NJ, U.S.A.). The homogenates were then fractionated at 4°C as previously described (Liu et al., 1996; Fujimura et al., 1998). Brain samples were first centrifuged at 750 g for 10 minutes. The supernatants were collected and further centrifuged at 8000 g for 20 minutes, and the second pellet containing the mitochondrial fraction was saved at −70°C. The second supernatant was collected and centrifuged at 100,000 g for 1 hour. The final supernatant was collected as the cytosolic fraction. Protein concentration was determined using the Bio-Rad protein assay dye reagent (#500–0006; Bio-Rad, Hercules, CA, U.S.A.). Both cytosolic and mitochondrial fractions (10 μg protein/lane) plus purified cytochrome c from rat heart (positive control, Sigma) were subjected to 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Protein bands were transferred from the gel to polyvinylidinene fluoride (Millipore, Bedford, MA, U.S.A.) membranes overnight. To confirm equal loading of the lanes, membranes were stained with 0.1% Ponceau Red (Sigma) in 5% acetic acid for 5 minutes, and the total amount of protein loaded on each lane was measured using an optical densitometer (GS-700 imaging densitometer, Bio-Rad). Membranes were then blocked with 5% milk in phosphate-buffered saline and probed for Bcl-2 (N-19, 1:500, rabbit polyclonal, Santa Cruz Biotechnologies, Santa Cruz, CA, U.S.A.), Bax (N-20, 1:1000, rabbit polyclonal, Santa Cruz Biotechnologies), caspase-3 (1:4000, rabbit polyclonal, a gift of Idun Pharmaceuticals, La Jolla, CA, U.S.A.), p18 caspase-3 subunit (CM1, 1:4000, gift of Idun Pharmaceuticals), PARP (#9542, 1:1000, rabbit polyclonal, Cell Signaling technology, Beverly, MA, U.S.A.) or cytochrome c (1:1000) by incubating in the primary antibody for 1 hour followed by a horseradish peroxidase-conjugated secondary antibody (1:2000, Santa Cruz Biotechnologies). Membranes were washed between steps with 0.1% tween-20 in 0.01 mol/L phosphate-buffered saline. To determine the specificity of the bcl-2 and Bax antibodies, peptide fragments (Santa Cruz Biotechnologies) were preabsorbed to the antibodies for 24 hours and applied to the membranes as usual. To verify accurate separation of the cellular subfractions, membranes were stripped in 100 mmol/L 2-mercaptoethanol, 2% sodium dodecyl sulfate, 62.5 mmol/L Tris-hydrochloride (pH 6.7) at 50°C for 30 minutes and probed for cytochrome c oxidase (1 μg/mL; 20E8-C12, Molecular Probes, Eugene, OR, U.S.A.). Protein bands were detecting using an enhanced chemiluminescence system (Amersham, Arlington Heights, IL, U.S.A.) and exposed to roentgen film. Films were scanned by an optical densitometer and analyzed using Multi-Analyst software (Bio-Rad).
Caspase assays
Caspase activity was quantified using a fluorometric assay based on cleavage of caspase substrates (Namura et al., 1998). Fresh-frozen tissue samples 2.5, 6, and 24 hours after the onset of ischemia were harvested in the manner described for Western blots. Samples were sonicated in 10 mL lysis buffer (pH 7.5, 100 mol/L HEPES, 1%Triton-X, 1 mmol/L edetic acid, 10%sucrose, and protease-inhibitor cocktail) per gram of tissue. Positive controls were similarly prepared from whole-brain lysates of neonatal rat and mouse brains. Samples were spun for 5 to 10 minutes at 10,000 rpm, and the supernatants were collected. Fifty microliters of each sample and eighty micromoles per liter standards (free 7-amino-4-trifluoromethyl coumarin; #T-7, Enzyme Systems Products, Livermore, CA) were loaded into 96-well, black polystyrene plates with clear flat bottoms (#07200656, Fisher). Two microliters and of ten micromoles per liter caspase-3 (acetyl- a sp- g lut-val- a sp-7-amido-4-trifluoromethyl coumarin; #AFC-138, Enzyme Systems Products) or caspase-9 (acetyl- l eu- g lut- h is- a sp-7-amido-4-trifluoromethyl coumarin; #AFC-154, Enzyme Systems Products) substrates were added into the wells. Baseline readings were obtained using a fluorometric plate reader (F-max, Molecular Devices; excitation = 355 nm, emission = 538 nm), and were taken again after 60 (caspase-3 assay) or 90 minutes (caspase-9 assay) incubation at 37°C.
Statistical analysis
Standard statistical tests were used with the software program StatView 4.5 (Abacus Concepts, Berkeley, CA, U.S.A.). Data were analyzed by Student's t-test for continuous, normally distributed data, whereas the Mann-Whitney test was carried out for data that were not normally distributed. A level of P < 0.05 was considered significant; data are mean ± SD.
RESULTS
A total of 140 animals were studied. Of these, 62 were rendered hypothermic, 70 were maintained normothermic, and 8 were used as sham controls. Fifty-six animals were used for immunohistochemistry, 70 were used for Western blot analysis, and 14 were used for caspase assays. Physiologic parameters (blood gases, serum glucose, hematocrit, mean arterial blood pressure, and heart rate) were no different between groups, except intraischemic temperature. Infarcts among the normothermic group involved most of the middle cerebral artery as shown in Fig. 1A. In contrast, infarcts among the hypothermic group were contained mostly within the striatum, and involved only parts of the overlying cortex (Fig. 1B). We have previously shown that mild hypothermia significantly decreases infarct size measured as early as 2 hours (Yenari et al., 2000) to as late as 2 months after the onset of ischemia in this particular model and paradigm (Maier et al., 1998; Maier et al., 2001).
Bcl-2 family proteins and effect of ischemia and mild hypothermia
Immunostaining for both Bcl-2 and Bax showed widespread expression predominantly within ipsilateral cortical and striatal neurons as early as 2 hours after the onset of ischemia, and was still present at 3 days; however, temperature had no effect on expression. Representative staining patterns for Bax 6 hours after the onset of ischemia are shown in Figs. 2A and B. This finding was confirmed on Western blots of whole-brain homogenates (Fig. 3A). Because Bcl-2 and Bax are both membrane-bound proteins, Western blot analysis was also performed on cytosolic and mitochondrial fractions. Interestingly, Bax was confined to the cytosol 5 hours after the insult, but by 24 hours mitochondrial release was observed (Fig. 4A). Bcl-2, however, was observed mainly in the cytosolic fraction (Fig. 4B) with only minimal redistribution to the mitochondria at 24 hours. However, these phenomena were not altered by mild hypothermia.
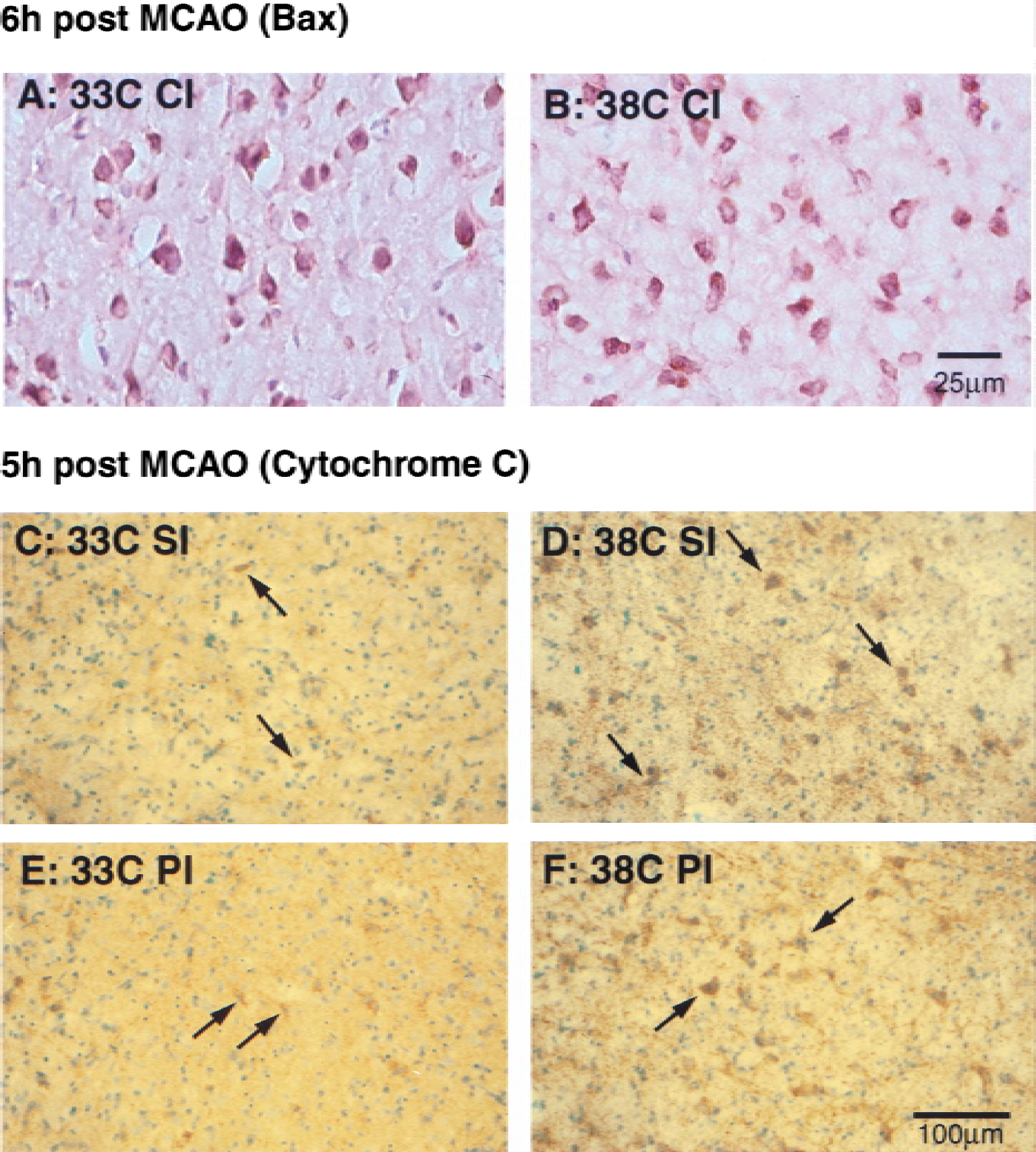
Immunostains of Bax
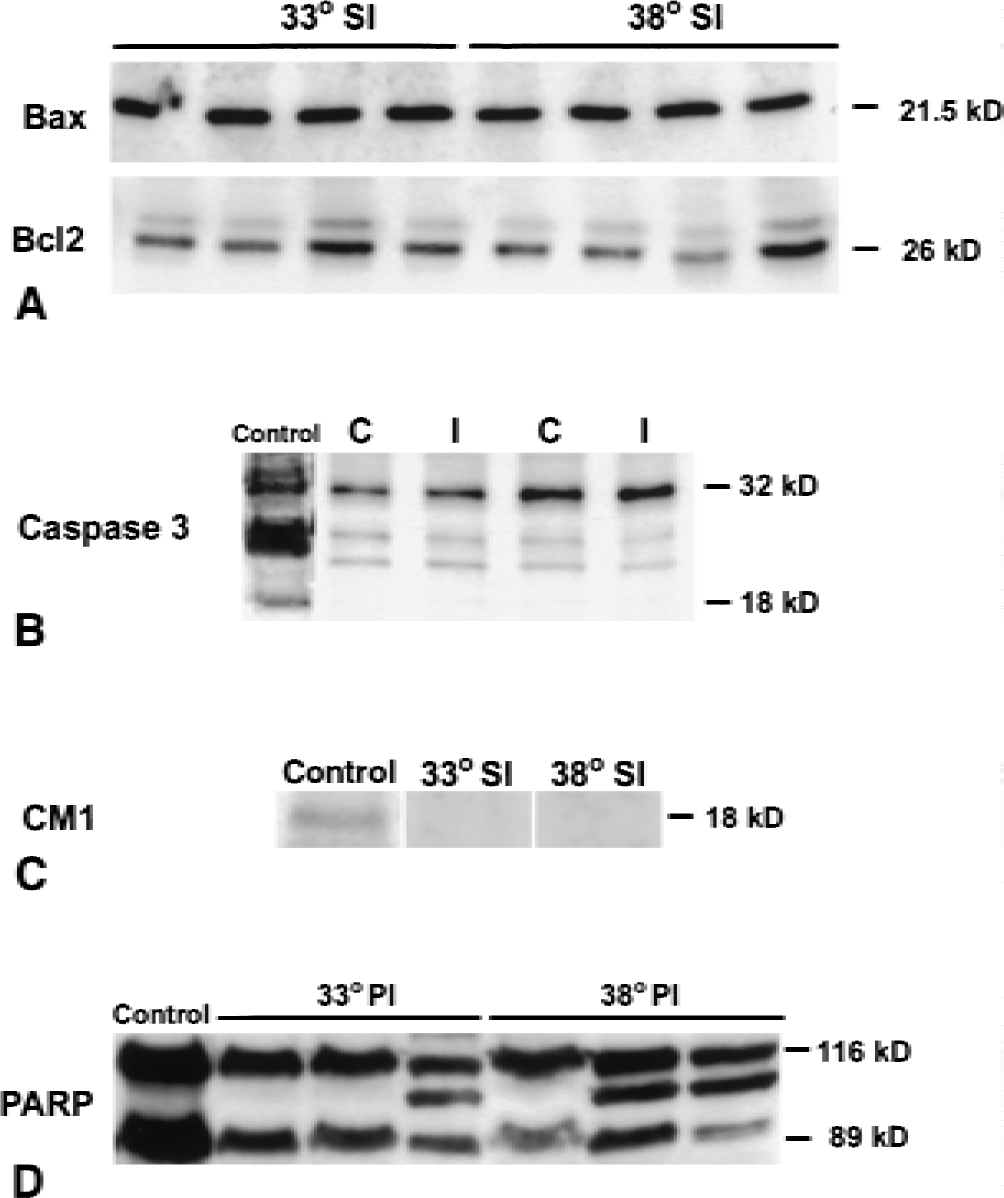
Mild hypothermia does not alter Bcl-2 family protein expression and caspase activity.
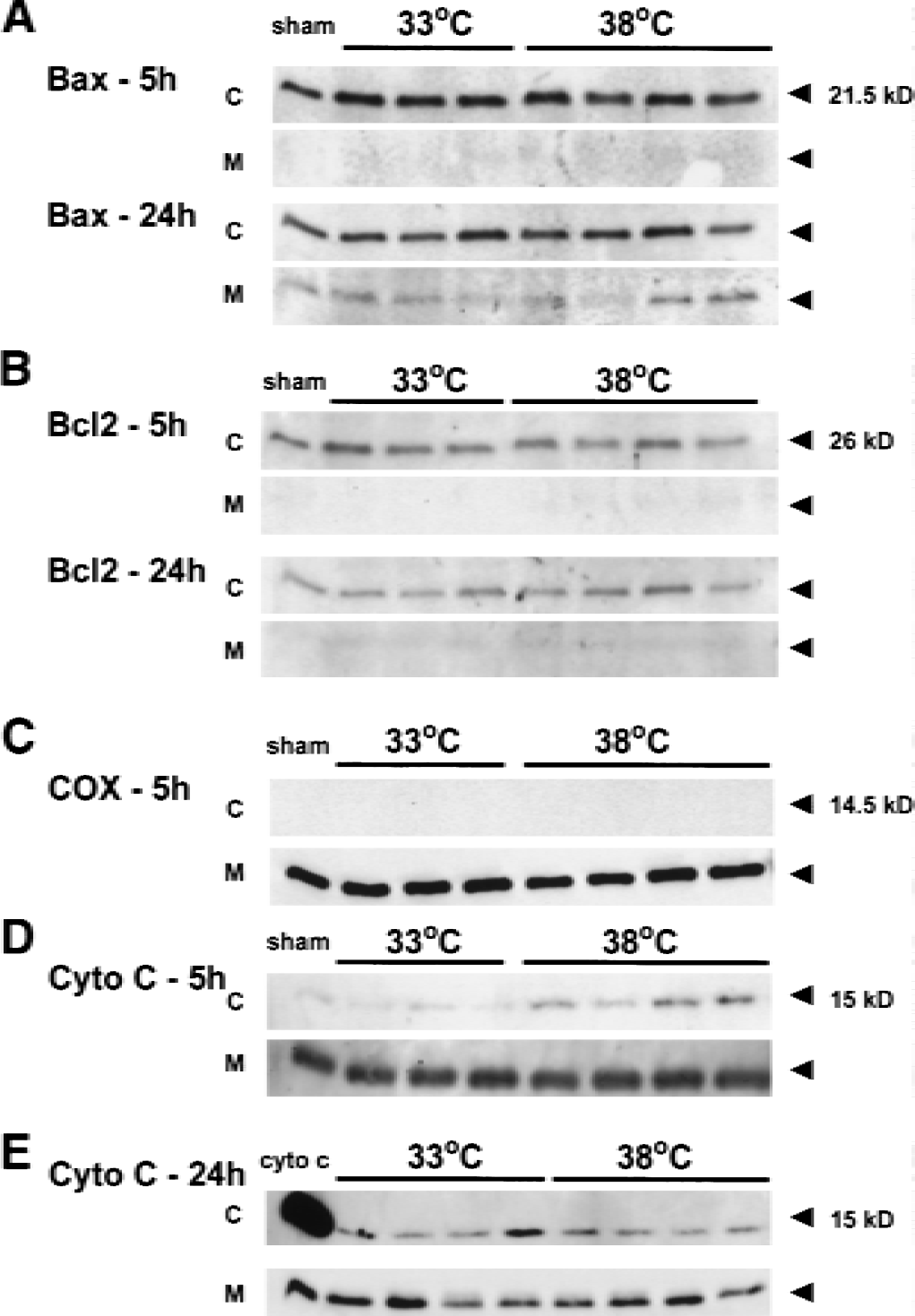
Western blots of subcellular fractions isolated from ischemic striata. Five hours after the insult, both Bax
After stripping, membranes from cytosolic and mitochondrial fractions were probed for cytochrome c oxidase, an enzyme found only within mitochondria. Western blots of cytochrome c oxidase showed positive bands only within the mitochondrial fractions, indicating that subcellular fractionation was complete (Fig. 4C).
Mild hypothermia inhibits cytochrome c release
Immunohistochemical staining was performed 2.5, 5, and 24 hours after the start of occlusion. Cytosolic immunoreactivity in neuronal cells was observed as early as 2 hours after occlusion in the ischemic core of the striatum and preoptic area both in hypothermic and normothermic brains (data not shown). By 5 hours, there was a remarkable difference in the staining patterns. Within ischemic striata, the number and intensity of positively stained neurons was markedly suppressed in the mild-hypothermic group (Figs. 2C and E) compared with the normothermic group (Figs. 2D and F). However, 24 hours after ischemic onset, there was no obvious difference in staining within the ischemic core (striatum) between the two groups (data not shown). No immunoreactivity was observed in the nonischemic contralateral hemispheres. The absence of immunoreactivity in the nonischemic hemisphere is consistent with our previous findings (Fujimura, et al., 1998), and is possibly due to the intensity of the tissue fixation, allowing the antibody to react only with cytosolic but not mitochondria-bound protein (Fujimura et al., 1999).
Western blots from subcellular fractions were consistent with the observations from the immunostains. Significantly less cytosolic cytochrome c was observed in the hypothermic group 5 hours after occlusion (Fig. 4D), but not at 24 hours (Fig. 4E). Cytochrome c was detected as a 15-kD band in the striatum in the ischemic hemisphere and in some ischemic cortical specimens, but was not detected in the contralateral hemispheres. Mitochondrial cytochrome c was also observed, but levels were not affected by mild hypothermia. Optical density measurements from cytosolic cytochrome c bands of samples taken from ischemic striata were significantly lower than that of the normothermic group at 5 hours but not at 24 hours (P < 0.05, Fig. 5C).
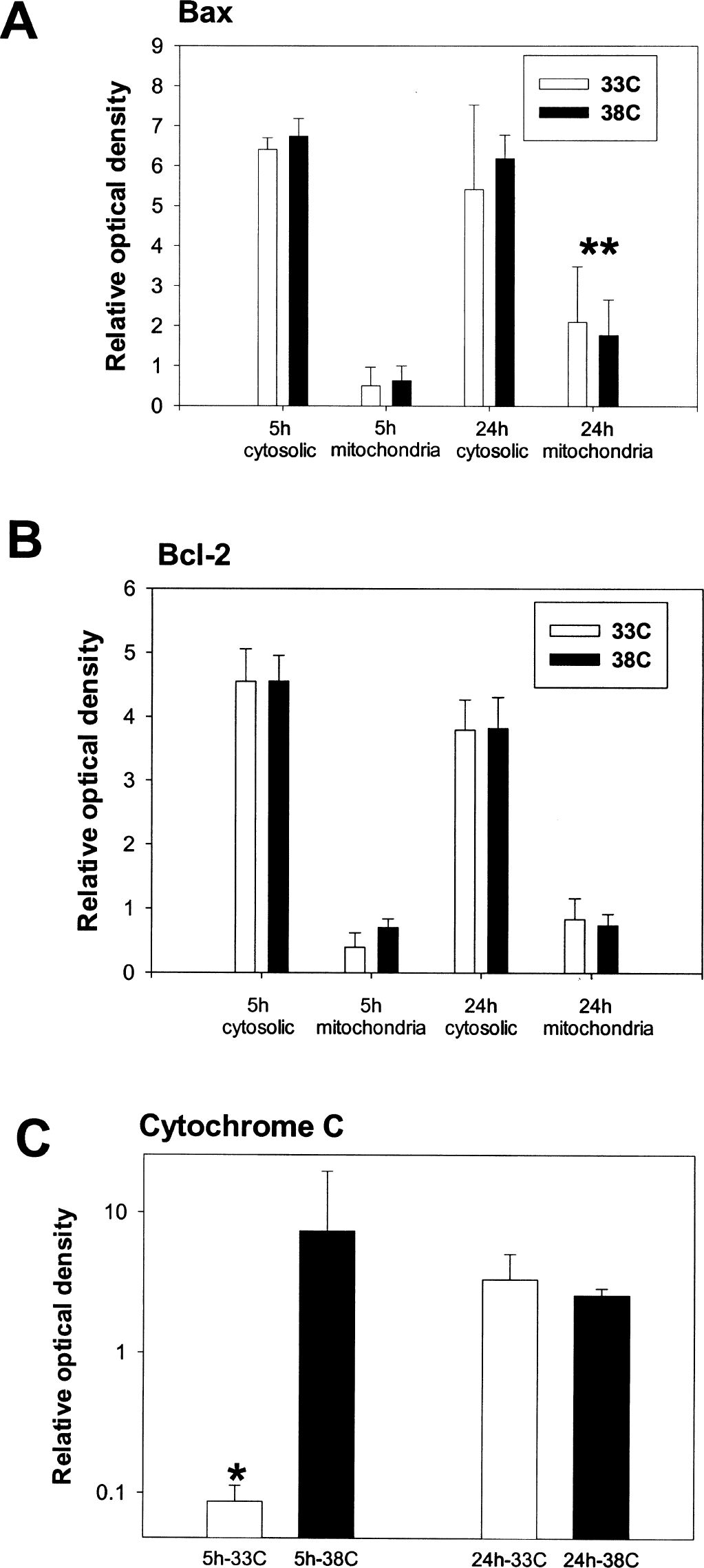
Mild hypothermia inhibits mitochondrial cytochrome c release, but does not alter Bcl-2 family proteins. Optical densities of Western blots show that mild hypothermia does not alter the cytosolic or mitochondrial content of Bax
Caspase activation and effect of mild hypothermia
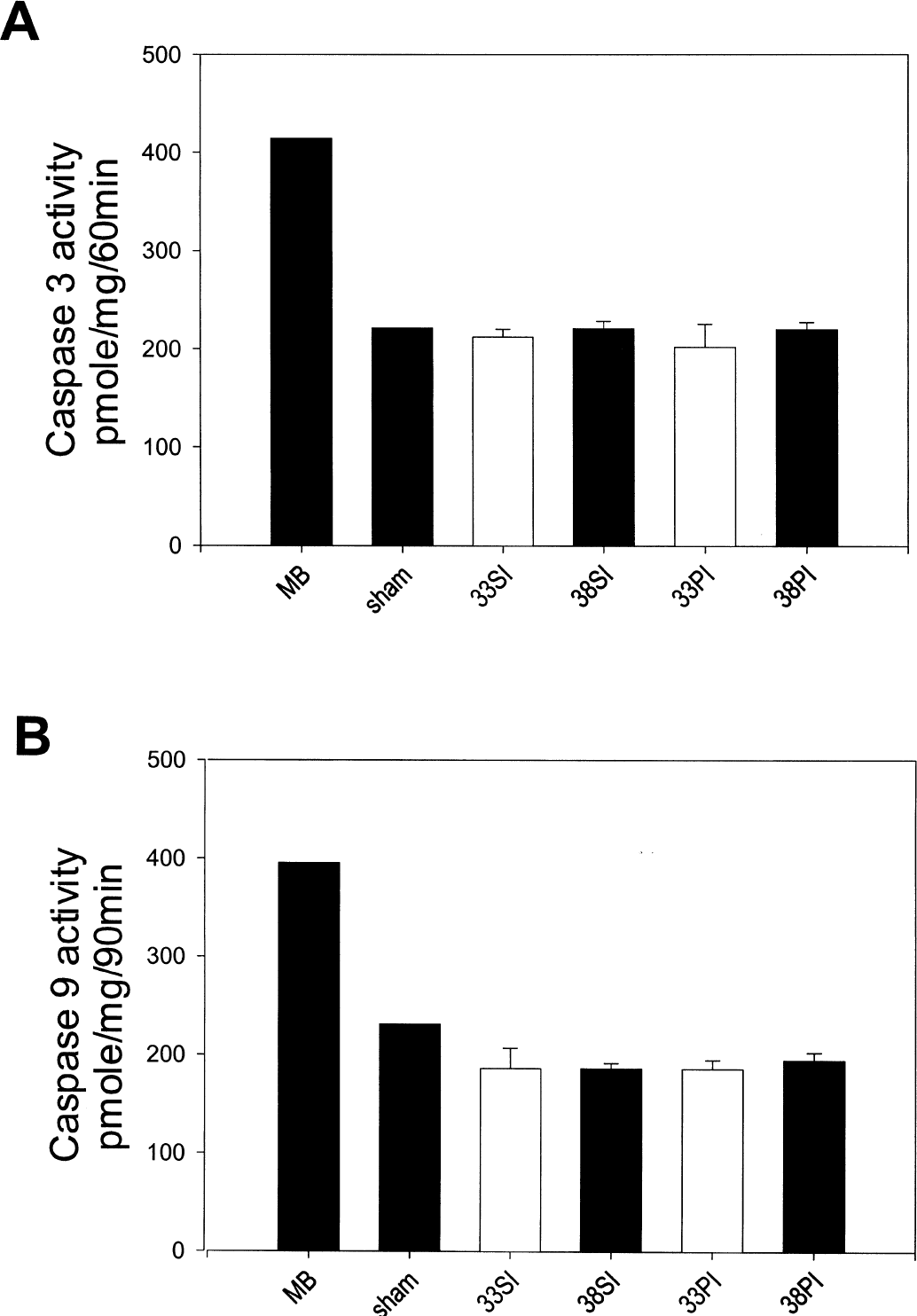
In this model of transient focal cerebral ischemia, no direct evidence of caspase activity could be detected. Figures 3B to 3D show representative Western blots of caspase-3 and PARP 6 hours after the insult. Western blots of caspase-3 showed that both uninjured and injured tissue contained a 32-kD band corresponding to unprocessed caspase-3. However, no 18- or 12-kD bands corresponding to processed caspase-3 were seen within ischemic and nonischemic brain (Fig. 3B). Using an antibody specific for the p18 cleavage product CM-1 (Srinivasan et al., 1998)), Western blots revealed a positive band in neonatal rat brain (positive control, Fig. 3C), but no such band was seen in ischemic tissue regardless of the experimental condition. Interestingly, Western blots of PARP showed that cleavage products of this caspase-3 target protein were present in the ischemic brain (Fig. 3D), and that two of three samples showed somewhat less of the 89-kD cleavage product at 38°C compared with all three 3 samples in the 33°C group. However, caspase-3 and −9 activity was not observed 2.5 to 24 hours after middle cerebral artery occlusion under normothermic and hypothermic conditions (Fig. 6).
Caspase-3
DISCUSSION
Although the protective effect of hypothermia against ischemic brain injury has been recognized for more than 30 years, its mechanisms remain unclear and are still controversial. Mild hypothermia is known to preserve brain energy metabolism and reduce cerebral intracellular acidosis (Chopp et al., 1989), but its cerebroprotective effect may not be due only to the alteration of brain metabolism. Busto et al. (1987) showed that small, intraischemic variations in brain temperature had no significant influence on energy metabolite levels in rat brains subjected to transient global ischemia. Nevertheless, neuronal damage was markedly suppressed by mild hypothermia. Several groups have reported that mild hypothermia attenuates increases in extracellular concentrations of excitatory amino acids (especially glutamate) after global (Busto et al., 1989a; Nakashima and Todd, 1996) and focal cerebral ischemia (Baker et al., 1995; Huang et al., 1998). However, this finding does not explain why hypothermia is protective even when cooling is delayed well after glutamate is released (Colbourne and Corbett, 1994; Maier et al., 2001).
Recently, suppression of apoptotic neuronal cell death has been suggested as a possible mechanism of the protective effect of mild hypothermia. The intrinsic model of apoptosis proposes that Bax, a proapoptotic protein, translocates to the mitochondria and forms a pore within the inner membrane (Green and Reed, 1998; Murphy et al., 2000; Ghatan et al., 2000). Once a mitochondrial pore is formed, cytochrome c is released into the cytosol and activates complexes of procaspase-9 and Apaf-1 (Liu, et al., 1996). This activation leads to cleavage of caspase-9, which in turn activates caspase-3. However, the release of cytochrome c can be inhibited by the antiapoptotic protein, Bcl-2 (Yang et al., 1997; Kluck et al., 1997; Rosse et al., 1998), and may in part explain the protective effect of Bcl-2. Caspase-3 and other effector caspases then act in a final common pathway leading to DNA cleavage and, ultimately, cell death. Evidence arguing in favor of apoptosis occurring during experimental stroke is supported by observations that mitochondria release cytochrome c (Fujimura et al., 1999) and caspase activity is detected in the ischemic brain (Namura et al., 1998; Krajewski et al., 1999). Furthermore, inhibition of caspases (Schielke et al., 1998, Namura et al., 1998) or overexpression of antiapoptotic proteins attenuate infarct size (Linnik et al., 1995; Lawrence et al., 1996, 1997).
The limited literature addressing the effect of temperature on apoptotic cell death is conflicting, and could be dependent on the nature and severity of the insult. Using a model of mild focal cerebral ischemia (1-hour middle cerebral artery occlusion), one group (Prakasa Babu et al., 2000) noted qualitative differences in Bcl-2, Bax, and cytochrome c immunostaining in animals exposed to 1 hour of mild hypothermia. Their data suggest that mild hypothermia decreases the density and intensity of staining for Bax and cytochrome c, but increases Bcl-2 staining. This finding is somewhat in contrast to our observations, but could be because their insult was less severe (1-hour occlusion) than ours (2-hour occlusion). However, using an in vitro injury model, Bossenmeyer-Pourie et al (2000) showed that Bcl-2 expression in cultured neurons exposed to hypoxia was no different under hypothermic conditions, though cell survival was improved. Similarly, mild hypothermia (33°C) did not alter Bcl-2 or Bax expression in cultured neurons exposed to serum deprivation, an apoptosis-specific insult, but did prevent cytochrome c release and caspase activity (Xu et al., 2001). However, Chinese hamster ovary cells exposed to hydrogen peroxide (Slikker W III et al., 2000) and hippocampal CA1 neurons exposed to 10-minute forebrain ischemia (Zhang et al., 2001) showed increased Bcl-2 expression at lower temperatures.
We observed the major difference in cytochrome c release between the mild hypothermic and normothermic groups 5 hours after the start of occlusion (3 hours after hypothermic treatment was terminated), a time when reactive oxygen species and intracellular calcium are known to be increased in the brain after stroke (Chan, 1996; Iadecola, 1997). Cytochrome c release in this model could have been a result of other factors known to cause damage to the mitochondria, such as free radicals and calcium ions (Kristian and Siesjo 1998; Fiskum, 2000), and hypothermia is known to decrease free radical generation (Globus et al., 1995; Kil et al., 1996; Lei et al., 1997; Maier et al., 2000). We show that although mild hypothermia transiently inhibits cytochrome c release, its protective effect does not appear to be due to alterations in other apoptosis-related proteins. Hypothermia does not appear to affect expression and redistribution of Bcl-2 family proteins. Although cytochrome c release was suppressed 5 hours after ischemia onset, Bax translocation to the mitochondria was not observed until 24 hours. Because Bax translocation is thought to precede cytochrome c release, it appears unlikely that the observed inhibition of cytochrome c release influenced Bax-mediated apoptosis. Furthermore, we did not detect evidence of caspase-9 or −3 activation.
Interestingly, immunoblots of PARP, a caspase-3 substrate, showed that PARP inactivation occurred. However, PARP could have been cleaved by other proteases not involved in apoptosis. Furthermore, the 89-kD cleavage product was actually increased by mild hypothermia. Because PARP has been shown to potentiate ischemic injury because of high-energy substrate needs (Eliasson et al., 1997), its increased breakdown would be consistent with a protective effect by hypothermia. Although beyond the scope of this study, this observation warrants further study. Nevertheless, our results indicate that neuroprotection by mild hypothermia in this particular experimental paradigm may be related to an interruption of a necrotic process such mitochondrial swelling, or may interfere with other apoptotic pathways. The extrinsic pathway involves cell surface-receptor activation by ligands, such as tumor necrosis factor-α or fas ligand, with subsequent activation of caspase-8 and Bid, another proapoptotic Bcl-2–related protein (Ashkenazi and Dixit, 1998). Recent studies have shown that fas and activated caspase-8 are also present after ischemic insults to the central nervous system (Velier et al., 1999; Matsushita et al., 2000). Thus, it is possible that mild hypothermia might influence this pathway. However, both the intrinsic and extrinsic pathways ultimately lead to activation of caspase-3, which was not observed in this study. Others have also reported that caspase-3 activation and apoptosis do not occur after focal cerebral ischemia (van Lookeren Campagne and Gill, 1996; Colbourne et al., 1999; Loetscher et al., 2001). The discrepancies in the literature are not easily rectified; however, it should be noted that the intensity of the ischemic insult in our model may have been more severe than that used by others who detected caspase-3 activity (Endres et al., 1998; Velier et al., 1999). Less severe insults may have lead to better preservation of energy stores needed for the execution of apoptosis. Other groups who observed caspase-3 activity studied murine models (Endres et al., 1998; Namura et al., 1998), whereas the current study used rats; therefore, there may be unforeseen species differences.
We previously reported that 2 hours of intraischemic mild hypothermia (33°C) decreased numbers of TUNEL-positive cells in a similar model of rat transient focal cerebral ischemia (Maier et al., 1998). Using a model of global hypoxic-ischemic injury in newborn piglets, Edwards et al. (1995) showed that postinsult moderate hypothermia (34.9°C) significantly reduced the number of apoptotic cells compared with a normothermic group (38.5°C), though the number of necrotic cells did not differ between the two groups. Although the presence of TUNEL-positive cells does not necessarily establish that apoptotic death is the predominant form of injury in experimental stroke (van Lookeren Campagne and Gill, 1996), our previous study suggested that mild hypothermia was also associated with decreased DNA laddering (Maier et al., 1998). However, apoptosis may also occur without caspase activation. Others have recently shown that Bax and other factors can induce mitochondrial damage and apoptosis even if caspases are blocked (Friedlander et al., 1996; Hansen and Braithwaite, 1996). In this scenario, apoptosis-inducing factor is released from the mitochondria and translocated to the nucleus, resulting in chromatin condensation and DNA fragmentation (Daugas et al., 2000; Loeffler et al., 2001). Whether this occurs during cerebral ischemia is not yet known (Graham and Chen, 2001), but is a potential explanation for the observations here, especially given the remarkable lack of caspase activity.
The present study is the first to quantitatively show that intraischemic mild hypothermia reduces mitochondrial release of cytochrome c after experimental stroke. This reduction is associated with the ameliorative effect of the mild hypothermia, but was not associated with other biochemical markers of apoptosis, such as alterations in Bcl-2 family members or caspase-3 and −9 activation. We suggest that under more severe focal cerebral ischemic conditions (2-hour middle cerebral artery occlusion versus shorter durations or permanent ischemia), mild hypothermia is capable of ameliorating this injury by inhibiting mitochondrial damage, but does not appear to interfere with components of the intrinsic apoptotic pathway.
Footnotes
Acknowledgments:
The authors thank Lijun Xu, David Kunis, and David Onley for expert technical assistance, and Beth Houle for figure preparation.