
Abstract
The mechanisms and role of nerve cell death after traumatic brain injury (TBI) are not fully understood. The authors investigated the effect of pretreatment with the oxygen free radical spin trap α-phenyl-N-tert-butyl-nitrone (PBN) on the number of neurons undergoing apoptosis after TBI in rats. Apoptotic cells were identified by the TUNEL method combined with the nuclear stain, Hoechst 33258, and immunohistochemistry for the active form of caspase-3. Numerous neurons became positive for activated caspase 3 and TUNEL in the cortex at 24 hours after injury, suggesting ongoing biochemical apoptosis. In PBN-treated rats, a significantly greater number of cells were found to be TUNEL positive at 24 hours compared with controls. However, PBN treatment resulted in a reduced cortical lesion volume and improved behavioral outcome two weeks after injury. The authors conclude that a treatment producing an increase in DNA fragmentation in the early phase may be compatible with an overall beneficial effect on outcome after TBI. This should be considered in the screening process for future neuroprotective remedies.
Apoptosis, or programed cell death, is a strictly regulated and evolutionary conserved mechanism through which multicellular organisms dispose of unwanted cells, which are no longer needed or are harmful to the organism (Kerr et al., 1972). Apoptosis is necessary for normal brain development, and gene deletion of the apoptosis effector enzyme caspase-3 is associated with an excessive growth of the central nervous system (Kuida et al., 1996). However, neurons also die in various neurodegenerative conditions, which are characterized by death of specific subsets of neurons. The causes and types of nerve cell death in these diseases and in several neuropathologic conditions, such as brain trauma and ischemia, are not fully understood. Recent data point to the involvement of apoptosis in different, acute and chronic, disease processes afflicting the brain (Thompson, 1995). Both necrosis and apoptosis have been shown to occur after traumatic brain injury (TBI) (Colicos and Dash, 1996; Conti et al., 1998; Rink et al., 1995; Skoglösa et al., 1999). Several pathologic mechanisms are involved in TBI—for example, intracellular calcium overload, amino acid excitotoxicity, and oxygen free radical stress (Siesjö, 1993). Therefore, it is of great interest to know which factors determine if an injured cell enters the necrotic or apoptotic pathway.
In the current study, the authors addressed this issue by pretreating the rats with the drug α-phenyl-N-tert-butyl-nitrone (PBN), which quenches reactive oxygen species (ROS) (Carney and Floyd, 1991) and has neuroprotective potentials (Folbergrova et al., 1995, 1999; Zhao et al., 1994), and the authors analyzed whether PBN could influence posttraumatic apoptosis after controlled cortical contusion injury produced by the weight drop technique in rats (Feeney et al., 1981; Lewén et al., 1996; Nilsson et al., 1990). Apoptosis was studied using terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL) technique (Gavrieli et al., 1992; Skoglösa et al., 1999), together with Hoechst nuclear staining and immunohistochemistry for the active, processed form of caspase-3. Cellular identification of apoptotic cells was made by double staining with TUNEL and the neuronal nuclear marker NeuN (Mullen et al., 1992). Morphologic and behavioral outcome was assessed two weeks after severe contusion injury by measurements of cortical cavity volume and performance of rats in a Morris Water Maze task (Morris, 1984). Results show that PBN pretreatment significantly increased the number of neurons showing apoptotic criteria 24 hours after injury. The same treatment resulted in sparing of cortical tissue and improved behavioral outcome at later time points. The findings suggest that a shift from necrosis to apoptosis produced by free radical scavenger treatment is compatible with an overall favorable outcome in TBI.
MATERIALS AND METHODS
Animals and production of trauma
The current study was approved by the Uppsala ethics committee for animal research. Forty male Sprague-Dawley rats, weighing 310 to 469 g (BK Laboratories, Stockholm, Sweden), had free access to food and water and were divided into 2 groups for sham operation and brain trauma. Rats were killed at 12 hours (trauma, n = 12), 24 hours (sham, n = 2; trauma, n = 12), and 72 hours (sham, n = 2; trauma, n = 12) after injury. In each trauma group, 6 animals were pretreated intravenously with 30.0 mg/kg PBN (Centaur Pharmaceuticals, Sunnyvale, CA, U.S.A.) dissolved in saline, and 6 animals received an equal volume of saline. The treatment was given blindly. Conditions for surgery and monitoring of physiologic parameters have been given previously (Nilsson et al., 1990). Rats were placed in a stereotaxic frame and a craniotomy was made over the right parietal cortex with its center 3.5 mm behind bregma. Trauma was performed by a 21-g free-falling weight that was dropped from a height of 35 cm on a piston (4.5 mm in diameter) resting on the exposed dura. The piston compressed the cortex 2.0 mm at a speed of 2.7 m/sec (Skoglösa et al., 1999). Morphologic and cellular changes occurring in this model of TBI have been described in detail elsewhere (Lewén et al., 1996; Nilsson et al., 1990, 1993). In a separate experiment, morphologic and behavioral outcome was studied after a more severe injury (2.5 mm compression trauma) to facilitate investigation of eventual long-term neuroprotection by PBN. In this series, an additional 18 animals were used—6 sham-operated, 6 saline-treated traumatized, and 6 PBN-pretreated traumatized. Animals were killed 15 days after injury.
Brain preparation
Animals were killed using a mixture of halothane and O2. Brains were rapidly dissected and frozen in isopentane on dry ice. Coronal sections were cut at −3.5 mm from bregma and sectioned at 14 μm on a Leitz cryostat (Leitz Digital 1702, Wetzlar, Germany). Sections were mounted onto poly-L-lysine-coated glass slides (50 μg/mL; Sigma P-1399, St. Louis, MO, U.S.A.) and stored at −80°C.
TUNEL assay
The TUNEL method to detect DNA strand breaks was performed essentially as described earlier (Gavrieli et al., 1992; Skoglösa et al., 1999). Briefly, 4 frozen consecutive brain sections taken −3.5 mm from bregma from each animal were mounted and postfixed using 4% paraformaldehyde in phosphate-buffered saline (PBS). After washing in PBS, the sections were incubated for 1 hour at 37°C in a buffer containing TdT and fluorescein-conjugated 12-dUTP (Roche, Mannheim, Germany). Sections were coated with Vectashield mounting medium (Vector Laboratories, Burlingame, CA, U.S.A.) and analyzed using a Zeiss fluorescence microscope (Carl Zeiss HB, Stockholm, Sweden). In the majority of the TUNEL-positive cells, the labeling was located in condensed nuclei (Fig. 1). In each animal taken 12 to 72 hours after moderate injury, all cortical TUNEL-positive cells in 4 consecutive coronal sections were counted by an observer blinded to treatment and survival times. Sections were taken from the middle of the lesion, that is, 3.5 mm behind bregma. The average number of TUNEL-positive cells from those four sections from each animal was used for statistical analysis. This level of injury did not produce any cortical cavity formation during the 72 hours and thus no tissue loss was in these sections. For double staining with Hoechst 33258 (Sigma), sections treated with the TdT working solution (ApopTag, Intergen, Oxford, U.K.) were further incubated for 30 minutes with anti-digoxygenin-fluorescein and counterstained with Hoechst (0.1 mg/mL) for 2 minutes. Slides were rinsed in PBS three times and mounted with Vectashield.
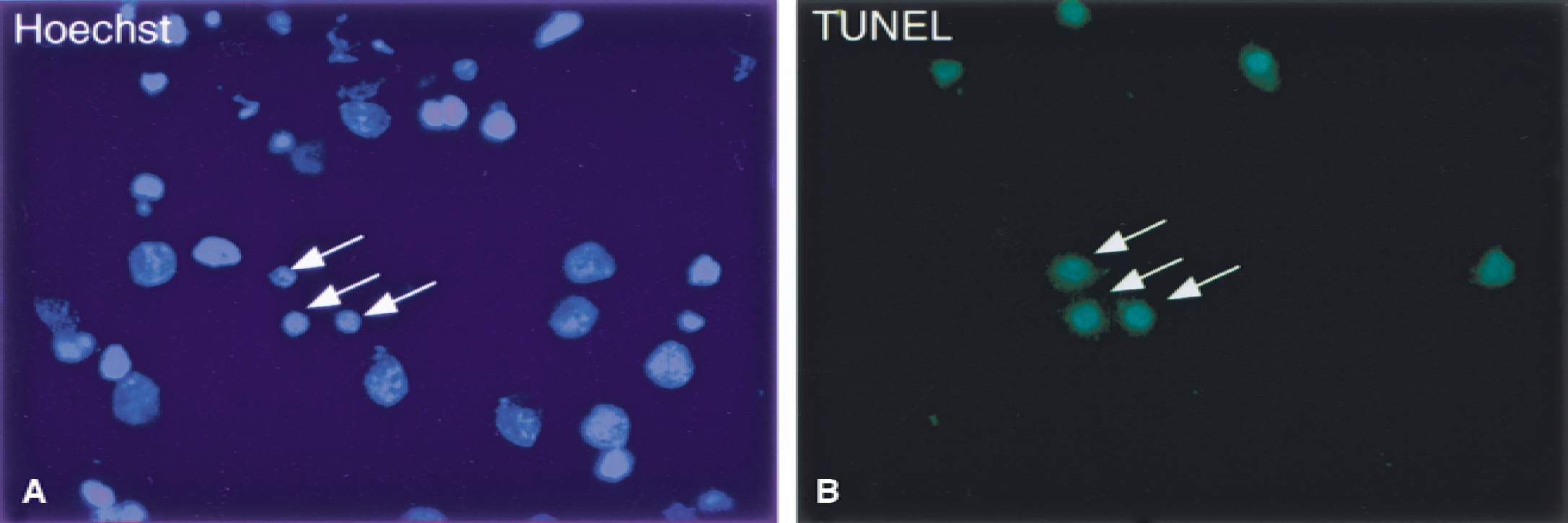
Apoptotic cells in the cortex 24 hours after traumatic brain injury. Arrows indicate cells double labeled with the nuclear staining Hoechst 33258
Immunohistochemistry
To verify the neuronal identity of apoptotic cells, the anti-NeuN antibody (diluted 1:150; Chemicon, Temecula, CA, U.S.A.) was added to fixed brain sections and mounted on SuperFrost Plus slides (Fisher Scientific, Pittsburgh, PA, U.S.A.), which were incubated overnight at room temperature. Slides were washed with PBS and a secondary biotinylated anti-mouse-IgG antibody (diluted 1:200; Vector Laboratories) was added for 30 minutes, washed, and incubated further with streptavidin-Texas Red for 30 minutes. After washing in PBS, sections were incubated for 1 hour at 37°C with the TdT working solution and rinsed in the stop/wash buffer for 10 minutes. Anti-digoxygenin-fluorescein and block buffer were added for 30 minutes, and the slides were washed three times with PBS, mounted with Vectashield, and photographed using the fluorescence microscope. Sections adjacent to those stained for TUNEL/Hoechst and TUNEL/NeuN were used to study the expression of the small, p17 and p12, segments of activated caspase-3. Sections were fixed for 10 minutes in cold acetone (−20°C). After rinsing with PBS, the sections were permeabilized for 2 minutes in 0.1% Triton-X/0.1% sodium citrate and rinsed with PBS. Endogenous peroxidase activity was blocked with 0.3% H2O2. Sections were rinsed with PBS and the primary antibody was applied (Anti-Caspase-3 Polyclonal Rabbit Antibody, Cat. No. 557035, dilution 1:100 in PBS; PharMingen, San Diego, CA, U.S.A.). Sections were incubated overnight at 4°C. After further rinsing with PBS, the secondary antibody was applied (Goat-anti-Rabbit IgG 1:200; Vector Laboratories). Avidin-biotin complex (Vectastain Elite; Vector Laboratories) was applied with 3,3′-di-amino-benzidine (DAB; Sigma) as chromogen. Sections were rinsed with tap water and counterstained with Mayer's hematoxylin (Histolab, Gothenburg, Sweden). Sections were dehydrated and coverslips mounted. Control experiments were performed by omission of the primary antibody, showing no labeling of cells.
Morphologic and behavioral outcome
At 15 days, the animals used for morphologic and behavioral outcome were sedated with pentobarbital (60 mg/kg) and perfused through the heart with PBS followed by a 4% formaldehyde solution. Brains were kept in fixative overnight and dehydrated, and coronal blocks were embedded in paraffin. Six-micrometer-thick sections were cut and stained with hematoxylin and eosin (H&E). After staining, the cortical lesion was identified as a region with pallor, or loss of H&E staining, and traced out. The area of the lesion was calculated by using the Neurozoom software program (Scripps Research, San Diego, CA, U.S.A.) and the volume of the lesion was calculated by adding the lesion area from each section multiplied with the distance between the sections. To analyze behavior in control and PBN-treated rats, a modified Morris Water Maze task was performed at days 11 to 14 after TBI (Morris, 1984). A pool (140 cm in diameter) was filled with 26°C clear water and placed in a room designed for this experiment. Four curtains each containing one visual cue surrounded the pool. A hidden platform was submerged 1 cm under the surface and placed in the northwest quadrant. A computer system (HVS Image, Hampton, U.K.) attached to a video camera was used to track latency for the animals to find the hidden platform and to analyze swim speed. Each rat was allowed to swim four times each day. Animals were randomly placed from four different positions into the water facing the pool wall. If they did not find the platform after 120 seconds, they were placed on the platform by hand and remained there for 30 seconds.
Statistical analysis
Factorial analysis of variance with Fisher's PLSD test was applied to compare different groups of animals at individual time points. For all calculations, a commercial computer program was used (StatView 4.51; Abacus Concepts, Berkeley, CA, U.S.A.). Differences with a P value ≤ 0.05 were considered statistically significant. Values presented are mean ± SD.
RESULTS
Previous studies using this trauma model at a mild injury level showed a scattered loss of nerve cells with occasional eosinophilic neurons, most prominent in the perimeter surrounding the impact of the lesion (Lewén et al., 1996; Nilsson et al., 1993). The hippocampus is relatively spared, displaying occasional loss of nerve cells in the hilus of the dentate gyrus (Lewén et al., 1999). In the current study, moderate contusion injury did not result in any appreciable necrotic cortical cavity during the 72 hours of the study. However, tissue edema and small intraparenchymal hemorrhages were present in the perimeter of the lesion and in the underlying subcortical white matter. After severe contusion injury, a large cavity was formed at the site of the cortical impact, which extended down to the underlying white matter two weeks after injury. There was an enlargement of the ipsilateral ventricle and bilateral cell loss in the hippocampus.
After moderate cerebral contusion injury, several cells in the perimeter of the lesion were TUNEL positive at 12 to 72 hours, suggestive of DNA fragmentation (Fig. 1). The maximal number of TUNEL-positive cells was observed at 24 hours, with the number of cells declining thereafter. The authors previously have shown that the sham operation does not induce cell death, nor were any TUNEL-positive cells present in intact rat brain tissue (Skoglösa et al., 1999). The authors conclude that most of the TUNEL-positive cells were undergoing cell death because of extensive DNA fragmentation. A small minority of cells, however, had a more diffuse cytoplasmic staining pattern, which could result from necrosis. To investigate further whether the TUNEL staining was located in the nuclei or the cytoplasm, double staining with TUNEL and the nuclear dye, Hoechst 33258, was undertaken, showing that the TUNEL-positive cells had intact nucleus and that the TUNEL staining resided in the nucleus (Fig. 1).
Immunohistochemistry using the neuronal marker, NeuN, showed that a major part of TUNEL-positive cells were neurons (Fig. 2). The NeuN staining appeared somewhat rugged and was not strictly located to the nucleus, which may be explained by the use of frozen sections. To substantiate the data, the authors used an antibody against the cleaved, processed form of caspase-3, activated during the course of apoptosis (Thornberry and Lazebnik, 1998). The results obtained showed that a significant number of cells were positive for caspase-3 at 24 hours, demonstrating ongoing biochemical apoptosis after TBI (Fig. 3).
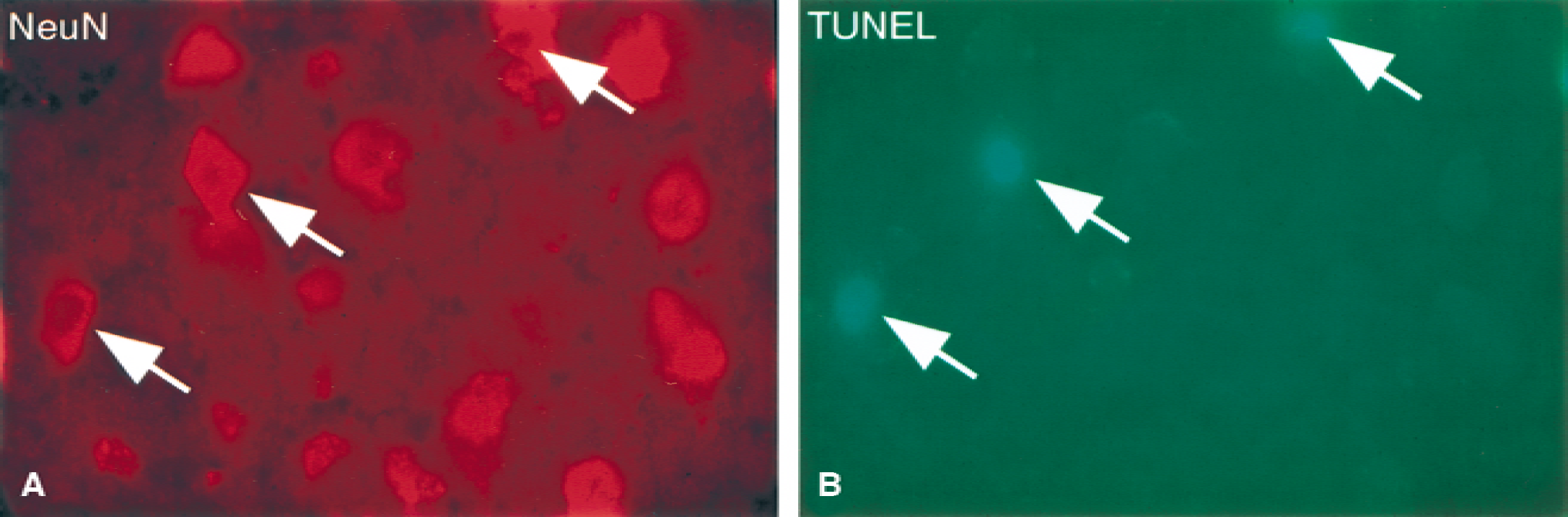
Double-stained sections with NeuN
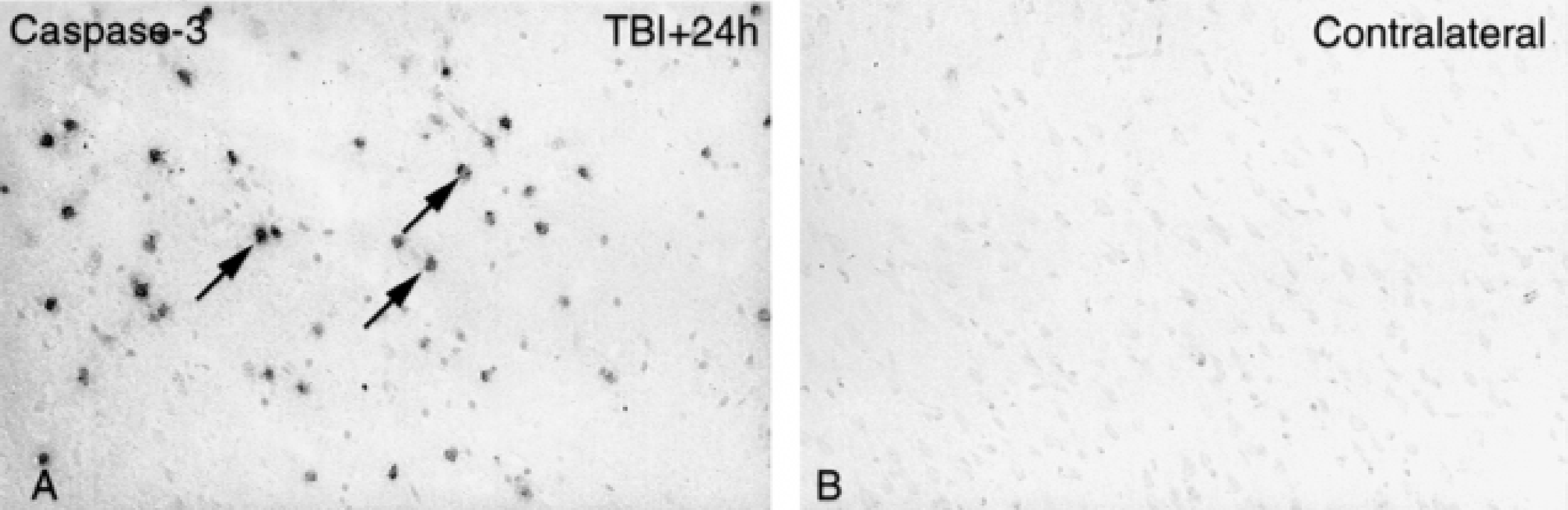
Immunohistochemistry for the activated small fragment of caspase-3.
To study whether the amount of cell death can be altered by pharmacotherapy, the authors used the drug PBN, which has been shown to possess neuroprotective potential. PBN was administrated 30 minutes before the injury, and brains were analyzed for the number of apoptotic cells. Quantification of the number of TUNEL-positive cells from 4 consecutive coronal sections of each animal from the middle of the lesion revealed 80% more TUNEL-positive cells in the PBN-pretreated rats compared with the control group (Fig. 4). Double staining using the NeuN antibody and TUNEL showed that the cells were neurons, with the same proportion of cells double labeled in control and PBN-treated rats (data not shown). Immunohistochemistry for active caspase-3 showed that the amount of immunopositive cells increased after PBN treatment, corroborating the data obtained with the TUNEL staining (Fig. 3).
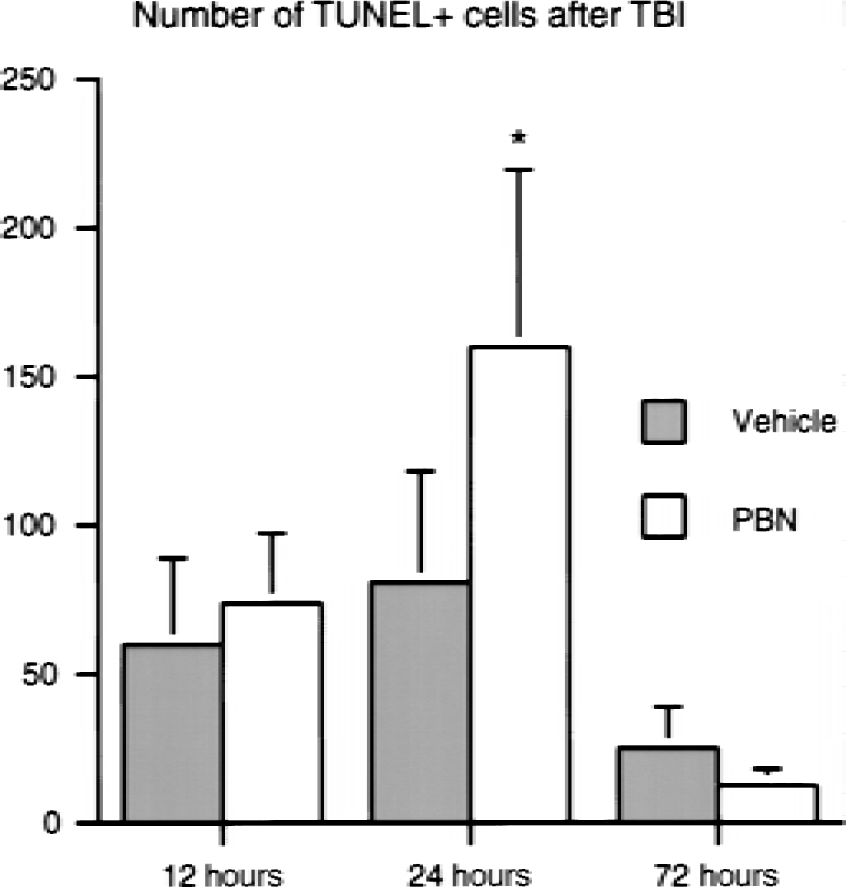
Quantification of number of apoptotic cells in the cortex 12, 24, and 72 hours after traumatic brain injury (TBI). Significantly more apoptotic cells were in the α-phenyl-N-tert-butyl-nitrone (PBN)-pretreated group at 24 hours after trauma. *P < 0.05.
Although PBN increased the amount of cells undergoing apoptosis at 24 hours after TBI, the size of the cortical lesion was significantly smaller in the PBN-treated group (6 ± 3.1 mm3) compared with the vehicle-treated group (15 ± 3.5 mm3) 15 days after severe injury (Fig. 5). Behavioral outcome was studied using a Morris Water Maze task at day 11 to 14 after injury. Mean latency to find a hidden platform was assessed as a measure of recovery of function. After severe injury, the mean latency was 29 ± 4.9 seconds in saline-treated animals, compared with 21 ± 4.1 seconds in the PBN-pretreated group, and 17 ± 2.47 seconds in the sham group (Fig. 5). The value for saline-treated animals was significantly different from both the sham and PBN-treated group (P < 0.05; Fig. 5). However, there was no statistical difference between the sham-operated nontraumatized control group and the PBN-pretreated traumatized group. This indicates that pretreatment with PBN improved functional outcome after severe TBI.
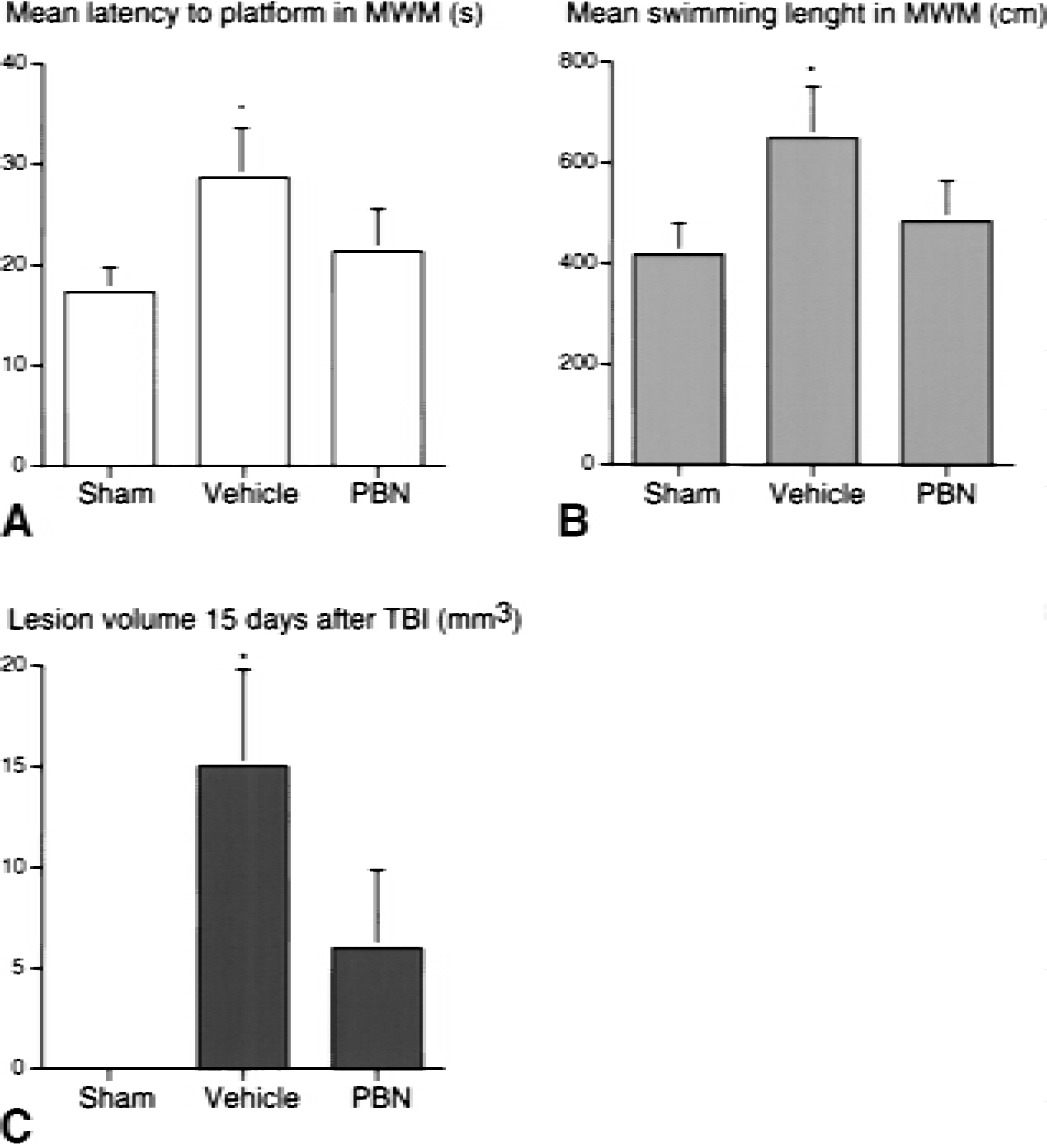
The beneficial effect of α-phenyl-N-tert-butyl-nitrone (PBN) pretreatment on behavioral and morphologic outcome after severe traumatic brain injury (TBI). Memory performance in Morris water maze (MWM) 11 to 14 days after severe TBI.
DISCUSSION
The results in the current study show that neuronal apoptosis occurs extensively in the brain after a moderate cerebral contusion injury in rats, with a peak approximately 24 hours after the injury. Apoptotic cells were identified using the TUNEL method, by double staining for TUNEL and the nuclear dye, Hoechst 33258, and using strict morphologic criteria (Kerr et al., 1972). Immunohistochemistry for the neuronal marker, NeuN, revealed that most of the cells were neurons. In addition, staining with an antibody for the cleavage product of caspase-3, showed that after injury, many neurons exhibited biochemical evidence of an activated apoptotic program. These findings, together with previous studies, showed the presence of TUNEL-positive cells in experimental TBI (Clark et al., 1997; Conti et al., 1998; Fox et al., 1998; Rink et al., 1995; Skoglösa et al., 1999; Yakovlev et al., 1997), which demonstrated that trauma is accompanied by apoptosis of neurons in the brain. Recently, it was shown that traumatic spinal cord injury in the rat also leads to an activation of caspase-3, which occurs within hours followed by activation of various downstream substrates (Springer et al., 1999).
The roles and functions of apoptosis in brain injury and in other neuropathologic processes are not fully understood. It is generally thought that a block or a delay of the apoptotic process may be beneficial with respect to improved brain function. To study this the authors investigated the relation between the number of apoptotic brain cells and the final outcome after TBI, using the free radical scavenger PBN, known to have neuroprotective effects in experimental brain injury, which may involve a preservation of mitochondrial function (Folbergrova et al., 1995, 1999; Zhao et al., 1994). The results showed that a pretreatment with PBN leads to an increase in the number of apoptotic cells at 24 hours, which was transient and not observed at later stages of the injury. However, PBN improved behavior of rats after TBI as shown using the water maze test. In addition, there was a significant sparing of the posttraumatic cortical tissue in PBN-treated rats. These findings suggest that the drug-mediated increase in early apoptosis was compatible with an overall favorable effect on behavioral and morphologic outcome. It is conceivable that the PBN pretreatment resulted not only in a shift from necrosis to apoptosis, but also rescued other injured cells. Admittedly, more work is needed to show if repeated doses, or other doses of PBN, would also attenuate the apoptotic process. Moreover, it remains to be studied if a shift from necrosis to apoptosis per se, for example, due to a more physiologic removal of dead cells, can have a beneficial effect on outcome after brain injury without inducing inflammation.
The mechanism by which PBN influences apoptosis after TBI is not known, but may involve effects on ROS formation. The production of ROS is thought to be involved in the pathogenesis of TBI (Chan, 1988; Lewén et al., 2000; Povlishock and Kontos, 1992), and elevated ROS may impair mitochondrial function (Hillered and Ernster, 1983). Quenching of ROS by application of PBN has been shown to improve mitochondrial function after cerebral ischemia (Folbergrova et al., 1995; Kuroda et al., 1996) and after status epilepticus (Folbergrova et al., 1999), along with a reduced infarct volume, that is, reduced necrosis (Zhao et al., 1994). Adenosine triphosphate (ATP) is involved in several steps controlling the propagation of the apoptotic process and the level of ATP can determine cell death by necrosis or apoptosis (Eguchi et al., 1997; Lelli et al., 1998). Thus, a depletion of ATP can translocate the proapoptotic protein Bax from the cytosol to the mitochondria and activate apoptosis (Saikumar et al., 1998). However, low ATP level also can inhibit some of the caspases in the apoptotic pathway (Eguchi et al., 1999). It has therefore been suggested that ATP in a concentration-dependent manner stimulates cells to undergo either apoptosis (ATP levels 25% to 70% of normal) or necrosis (ATP levels less than 15%) (Lieberthal et al., 1998). The levels of ATP after a brain lesion ultimately are determined by the severity of an insult; however, other downstream events, such as ROS, may also affect posttraumatic ATP levels (Hillered and Ernster, 1983). Recently, ROS has been shown to inhibit caspase activity in cultured cells (Samali et al., 1999). Whether this is also the case in brain in vivo remains to be studied. However, as shown in the current study, pretreatment with PBN, which is a ROS scavenger (Carney and Floyd, 1991), caused a significant increase in apoptosis and caspase-3 activation at 24 hours after injury. The authors recently have shown that PBN pretreatment reduces extracellular lactate accumulation after a mild cortical contusion injury, presumably by improving posttraumatic mitochondrial function (Lewén and Hillered, 1998). It seems reasonable to assume that an entrapment of ROS by PBN leads to better preservation of mitochondrial function and thus higher levels of ATP, thereby allowing an apoptotic pathway instead of ATP-depleted passive necrosis. In a recent study, the glutamate antagonist MK-801 was found to increase apoptosis, whereas treatment with S-PBN, a derivative of PBN that does not cross the blood-brain barrier, reduced apoptosis after brain trauma in the developing rat brain (Pohl et al., 1999). This finding is in contrast with the authors' results regarding PBN and TBI in adult rat brain, which could be because of the age of the animals or the drug treatment itself, or both. Interestingly, the discrepancies in the results reveal a difference in apoptosis and its response to drug treatment in developing compared with mature brain tissue, which has been noted before for immature cortical neurons in culture (McDonald et al., 1997).
To study if PBN could affect morphologic and behavioral outcome after TBI, the authors applied a more severe cerebral contusion injury after pretreatment with PBN. The rationale for increasing the severity was to produce an injury resulting in readily quantifiable functional and morphologic changes. The authors found that the drug treatment resulted in a sparing of cortical tissue and improved behavior of the rats in the water maze task. This is in agreement with the neuroprotective effect ascribed to the drug, but the transient increase in apoptosis observed with PBN was unexpected. This study suggests that a pharmacologic treatment that results in an increase in DNA fragmentation may still have an overall beneficial effect on outcome. Thus, the use of DNA fragmentation as the only outcome measure in the screening process for future neuroprotective remedies in TBI may be disadvantageous.