
Abstract
Preconditioning of hippocampal CA1 neurons was evaluated in a gerbil model of transient global ischemia using extracellular recording of DC potential shifts characteristic of ischemic depolarization to precisely define the duration of both priming and test insults. Brief ischemia resulting in depolarizations of 2.5 to 3.5 minutes consistently induced maximal tolerance (95% protection) against subsequent challenges 2 days later with an approximate doubling of the insult duration required for complete CA1 neuron loss from 6 to 12 minutes depolarization when evaluated 1 week after the test insult. Significant protection persisted at 2 months survival, although the apparent injury threshold regressed to approximately 8 minutes, indicating delayed progression of injury after longer test insults. In situ hybridization was used to evaluate depolarization thresholds for induction of mRNAs encoding the 70 kDa heat shock/stress protein, hsp72, as well as several immediate-early genes (c-fos, c-jun, junB, and junD). Immediate-early genes were prominently expressed after short insults inducing tolerance, whereas appreciable hsp72 induction only occurred after insults approaching the threshold for neuron injury. These results establish an ischemic preconditioning model with the predictability needed for mechanistic studies and demonstrate that prior transcriptional activation of the postischemic heat shock response is not required for expression of delayed tolerance.
Phenomena of inducible tolerance are evident in the context of diverse insult conditions and cell types. In the brain, such effects have been most extensively studied after cerebral ischemia, as was recently reviewed (Dirnagl et al., 2003; Kirino, 2002). These were initially demonstrated for hippocampal neurons after global ischemia (Kirino et al., 1991; Kitagawa et al., 1990), as well as for vulnerable neurons in other brain regions (Kitagawa et al., 1991), and later for infarct volume after focal insults (Chen et al., 1996; Simon et al., 1993). Most studies have used short duration priming challenges otherwise identical to the subsequent test insults, although forms of cross-tolerance have also been reported. It is unlikely that protection involves identical mechanisms in all such models.
Initial reports proposed induction of the 70 kDa heat shock protein, hsp72, as a potential contributor to tolerance mechanisms in hippocampus (Kirino et al., 1991; Nishi et al., 1993), as well as in cell culture models (Lowenstein et al., 1991; Rordorf et al., 1991). However, considerable evidence associates hsp72 expression more closely with injurious stimuli in brain (Abe and Nowak, 1996a, 2000; Pringle et al., 1999; Simon et al., 1991; Sloviter and Lowenstein, 1992), and examples of in vitro thermotolerance in the absence of hsp72 induction have been reported (Bader et al., 1992; Smith and Yaffe, 1991). Furthermore, brief ischemia leads to widespread changes in mRNA and protein expression (Koistinaho and Hökfelt, 1997; Nowak and Kiessling, 1999). Particularly prominent are the products of immediate-early genes (Kiessling et al., 1993; Nowak et al., 1993), which are typically induced at lower stimulus thresholds than hsp72 (Abe and Nowak, 1996a; Ikeda et al., 1994). An expanding literature documents modulation of diverse signaling pathways in preconditioning models (Dirnagl et al., 2003; Kirino, 2002), suggesting multiple candidate mechanisms by which priming insults could alter responsiveness to subsequent ischemia.
Rigorous mechanistic studies of ischemic tolerance in vivo, particularly preconditioning with prior global ischemia, have been somewhat impeded by the variability of the models used. Whereas statistically significant differences in CA1 neuron survival between naive and primed groups are routinely demonstrated, it has generally not been possible to predict the effectiveness of protection that could be expected in a given hippocampus. Improved protection and reproducibility were evident in the gerbil model with repeated preconditioning intervals of brief carotid artery occlusion and relatively short test insults (Kitagawa et al., 1990; Ohtsuki et al., 1996), but some animals could still show detectable pathology (Corbett and Crooks, 1997; Kitagawa et al., 1990). Nearly complete protection could be achieved in rats after a single preconditioning episode, but this also required that the test challenge not be too severe (Shamloo and Wieloch, 1999). Generation of robust, optimized preconditioning models demands more precise definition of both priming and test insult conditions.
Direct Current (DC) potential shifts concurrent with ischemic depolarization can be monitored as an indicator of insult duration in various brain regions (Abe and Nowak, 1996a; Li et al., 2000; Sorimachi et al., 1999; Xu and Pulsinelli, 1994) and are known to be better predictors of brain injury than failure of electroencephalographic activity (Kaminogo et al., 1998). In the present study, we demonstrate that depolarization monitoring is particularly effective in reducing variability in a gerbil model of ischemic preconditioning. Combined with rigorous control of brain temperature during ischemia and postischemic recirculation, this allows precise insult thresholds for both neuron injury and tolerance induction to be defined. Optimal preconditioning doubles the threshold for CA1 damage when evaluated 1 week after test challenges, although results at 2 months indicate significant delayed injury progression after severe insults. It is demonstrated that ischemic tolerance is closely associated with maximal induction of mRNAs encoding immediate-early genes. Hsp72 transcription occurs at a longer depolarization threshold and is therefore dissociable from tolerance induction. Preliminary reports have been presented (Abe and Nowak, 1996a,b).
MATERIALS AND METHODS
Experimental ischemia and depolarization monitoring
Ischemia was produced according to a protocol approved by the Animal Care and Use Committee, University of Tennessee. Mongolian gerbils (female; 50–70 g) (Charles River/Tumblebrook, West Brookefield, MA, U.S.A.) were anesthetized with 2% halothane in 70% N2, 30% O2. The common carotid arteries were exposed bilaterally and snared with a suture loop, and the animal was fixed in a stereotaxic frame. A rectal temperature probe (Yellow Springs Instruments, Yellow Springs, OH, U.S.A.) was inserted, and temperature was maintained at 37°C throughout subsequent procedures by a feedback-controlled heating lamp. The skull was exposed by a midline scalp incision, burr holes were drilled overlying hippocampus (2 mm caudal to bregma, 3 mm lateral), and glass microelectrodes filled with 2 Mol/L potassium acetate were lowered to a depth of 1.8 mm. These coordinates targeted hippocampus caudal to the region to be evaluated by histology and in situ hybridization (Loskota et al., 1974). Brain surface temperatures were independently monitored and maintained via a bare wire epidural thermistor and temperature control unit (Physitemp Instruments, Clifton, NJ, U.S.A.). DC potential in each hippocampus was monitored relative to a subcutaneous chloridized silver wire ground, and continuously recorded via a digital interface (GW Instruments, Somerville, MA, U.S.A.), after which midpoints of ischemic depolarization and repolarization were subsequently estimated to the nearest 5 seconds. Ischemia was produced by applying tension (approximately 10 g) to the carotid artery suture loop for varied intervals, as indicated for each experiment. The electrodes and epidural probes were then removed, and the scalp incision was closed. Anesthesia and rectal temperature control were maintained for 1 hour after release of occlusion to avoid postischemic hyperthermia characteristic of the gerbil model (Kuroiwa et al., 1990), which is known to modulate transcriptional responses (Abe and Nowak, 2000; Suga and Nowak, 1998). Thereafter, temperature was maintained with a feedback-controlled heat lamp until full recovery from anesthesia. Animals involved in studies of induced ischemic tolerance were reanesthetized at intervals of 6 hours to 10 days and subjected to a second interval of carotid artery occlusion using identical procedures.
Histology and quantitative evaluation of hippocampal neuron injury
One week, or in several cases two months, after the last ischemic insult, animals (n = 130) were anesthetized with ketamine/xylazine (85/15 mg/kg) and perfusion fixed with 10% formalin, 10% acetic acid, and 80% methanol. After overnight postfixation in situ, brains were removed and embedded in paraffin. Coronal sections (8 μm) were cut at the level of medial habenula (approximately 1.7 mm caudal to bregma) and stained with hematoxylin-eosin. Sections were examined under a 50x objective using an eyepiece equipped with a calibrated reticle. CA1 neuron densities (cells/mm) for each hippocampus were obtained by summing the counts obtained in five contiguous 200 μm segments of CA1 pyramidal layer arc dorsal to dentate gyrus, averaged over three sections per animal.
In situ hybridization
Gerbils (n = 58) subjected to varied durations of ischemia were briefly halothane anesthetized and killed by decapitation at 1 hour or 6 hours recirculation. Brains were removed and rapidly frozen in hexane at −40°C. Sections (16 μm) were cut in a cryostat, thaw-mounted on polylysine coated slides, and stored dessicated at −70°C. Hybridizations were carried out essentially as previously described (Nowak, 1991). All tissues in which signals for a given mRNA were to be quantitative compared were processed simultaneously. In brief, sections were fixed for 5 minutes in 4% paraformaldehyde and 100 mmol/L sodium phosphate (pH 7.4), and they were rinsed twice in 70% ethanol, followed by two rinses in 2X SSC (300 mmol/L NaCl, 30 mmol/L Na citrate). Slides were transferred to 100 mmol/L triethanolamine (pH 8) and acetylated by dropwise addition of acetic anhydride to a final concentration of 0.25% with rapid agitation for 10 minutes using a magnetic stirrer. Slides were then incubated for 30 minutes in 200 mmol/L Tris and 100 mmol/L glycine (pH 6.5), rinsed in 2X SSC, dehydrated through graded alcohol, extracted 5 minutes in chloroform, and air dried. To each section, we applied 10 μL of hybridization mix consisting of 20 nmol/L labeled probe (see below) in 2X SSC, 50% formamide, 10% dextran sulfate, 1X Denhardt's solution (0.02% Ficoll, 0.02% polyvinylpyrrolidone, 0.02% bovine serum albumin), 0.5 mg/ml salmon DNA, 0.25 mg/ml yeast tRNA, and 100 mmol/L dithiothreitol. Sections were cover-slipped and hybridized at 37°C for 3 hours, followed by stringency washes in 2X SSC and 50% formamide at temperatures specified for individual probes. After rinses in 2X SSC and dehydration through graded alcohols, slides were dried and placed against Kodak SB-5 film together with 14C standards (Amersham Life Sciences, Arlington Heights, IL, U.S.A.). Hybridization signal was determined by quantitative densitometry of CA1 pyramidal and dentate granule cell layers in calibrated autoradiograms using the program NIH Image.
Hybridization probes
We used previously characterized oligonucleotide sequences for detection of mRNAs encoding immediate-early genes (Wisden et al., 1990) and hsp72 (Nowak et al., 1990). Probes were end-labeled (Roychoudhury and Wu, 1980) with [35S]α-thiodATP (Perkin Elmer Life Sciences, Boston, MA, U.S.A.) using a terminal deoxynucleotide transferase kit (Boehringer Mannheim, Indianapolis, IN, U.S.A.). Probe sequences and temperatures used for stringency washes were as follows: c-fos, 5'-GCAGCGGGAGGATGACGCCTCGTAG-3' (43°C); c-jun, 5'-GCAACTGCTGCGTTAGCATGAGTTGGCACCCACTGTTAACGTGGTTCATGACTTTCTGTT-3' (48°C); junB, 5'-GAAGGCGTGTCCCTTGACCCCTAGCAGCAACTGGCAGCCGTTGCTGACATGGGTCATGAC-3' (56°C); junD 5'-CGCCGGGACCTGGTGCTGGGGCAGCAGCTGGCAGCCGCTGTTGACGTGGCTGAGGACTTT-3'(62°C); hsp72, 5'-CGATCTCCTTCATCTTGGTCAGCACCATGG-3'(37°C).
RESULTS
Characteristics of ischemic depolarization in naive and preconditioned animals
Occlusion of the common carotid arteries usually led to ischemic depolarization in hippocampus within 1 to 2 minutes, with a similar lag in repolarization following reperfusion (Fig. 1). However, occasional hippocampi exhibited delays of 4 minutes or longer in depolarization onset or failed to depolarize during occlusions as long as 6 to 7 minutes. The duration of ischemic depolarization was generally proportional to the duration of successful occlusions, although depolarizations tended to be shorter than occlusion time for insult durations less than 5 minutes and slightly longer with increasing occlusion time. This shift arises primarily from a progressive delay in repolarization with increasing duration of ischemic depolarization (see below). Approximately one in ten gerbils exhibited only unilateral depolarization in this study, yielding an overall failure rate of 5% for individual hippocampi.
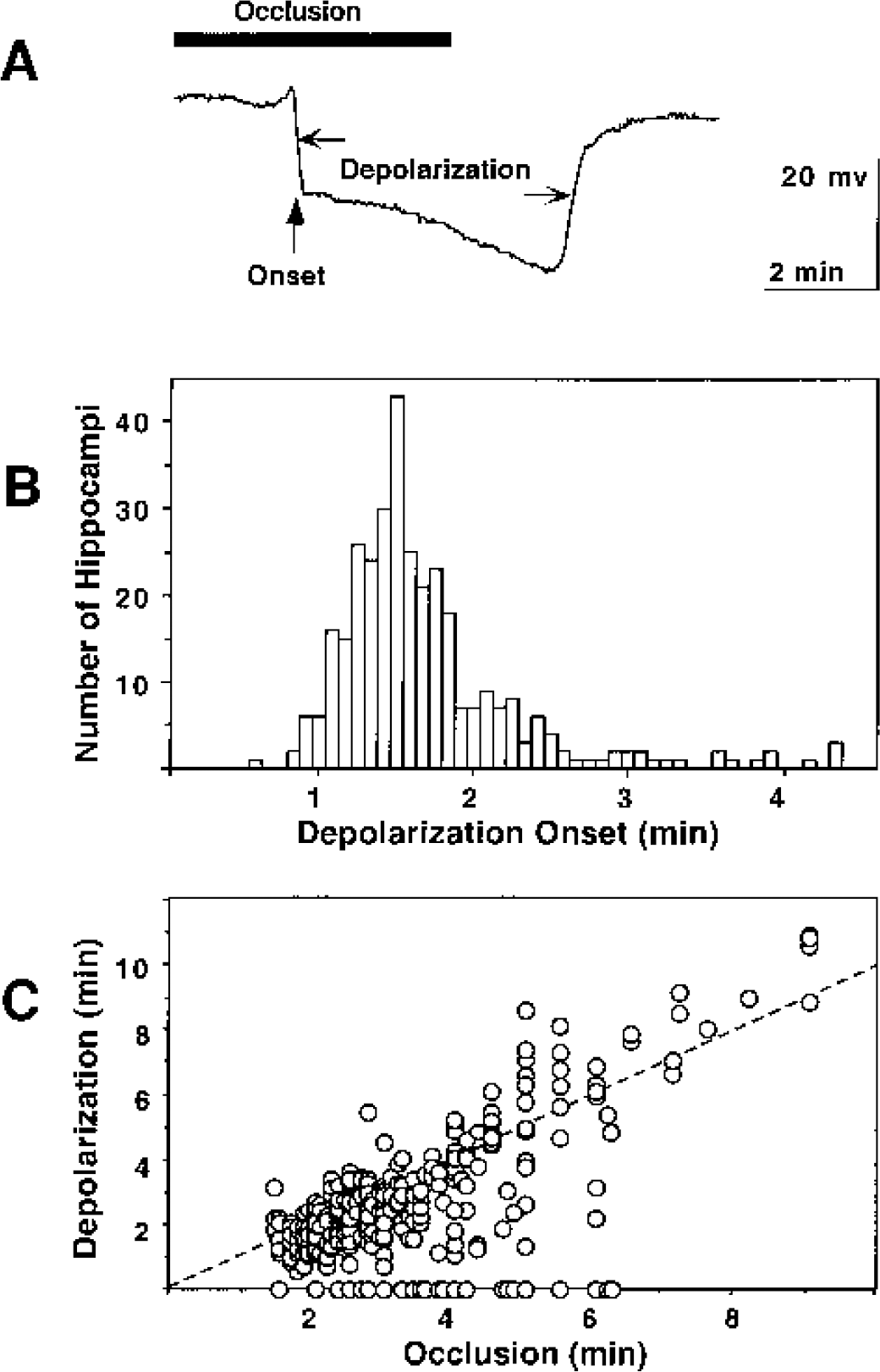
Characteristics of ischemic depolarization after bilateral carotid artery occlusion in naive gerbils. Animals were surgically prepared, and glass microelectrodes were positioned bilaterally in hippocampus, as described in the text. Carotid arteries were then transiently occluded for varied intervals, with simultaneous recording of DC potential changes. (A) Representative tracing identifying features of ischemic depolarization. Onset of depolarization exhibits a characteristic lag following occlusion, and repolarization occurs with a similar lag after recirculation. The depolarization time was determined as the interval between inflection points of depolarizing and repolarizing shifts. (B) Distribution of depolarization onset times. Lag to depolarization is indicated for those hippocampi in which depolarization was successfully observed (n = 334). Most hippocampi depolarized within 1 to 2 minutes, but delays as long as 5 minutes were occasionally observed. (C) Relationship between occlusion time and depolarization interval. The depolarization interval was generally proportional to occlusion duration, although approximately 5% of hippocampi failed to depolarize within the selected occlusion interval, and a several-fold range of depolarization times was observed for a given occlusion duration, particularly for shorter occlusion intervals (n = 375). Dashed line indicates identity.
The characteristics of ischemic depolarization remained quite similar in animals subjected to repeated ischemia (Fig. 2). There was a trend toward later depolarization during test occlusions in preconditioned hippocampi (102 ± 23 seconds) versus that observed during production of the priming insult (94 ± 22 seconds), which was slight but statistically significant (P < 0.0001, n = 144, paired t-test). In comparing priming and test insults, it also became evident that the interval between release of occlusion and repolarization was strikingly dependent upon insult duration. When plotted as a function of total depolarization time, the delay in repolarization ranged from as little as 30 seconds after the shortest priming occlusions to more than 4 minutes after the longest test insults, with an intermediate plateau value of approximately 2 minutes over the broad range of 3 to 7 minutes of depolarization. It is important to note that this relationship did not differ in naive versus preconditioned hippocampi. Although fewer naive animals were subjected to long occlusions during the course of the present studies, their hippocampi exhibited repolarization delays completely overlapping the distribution of responses observed during long second occlusions in preconditioned animals.
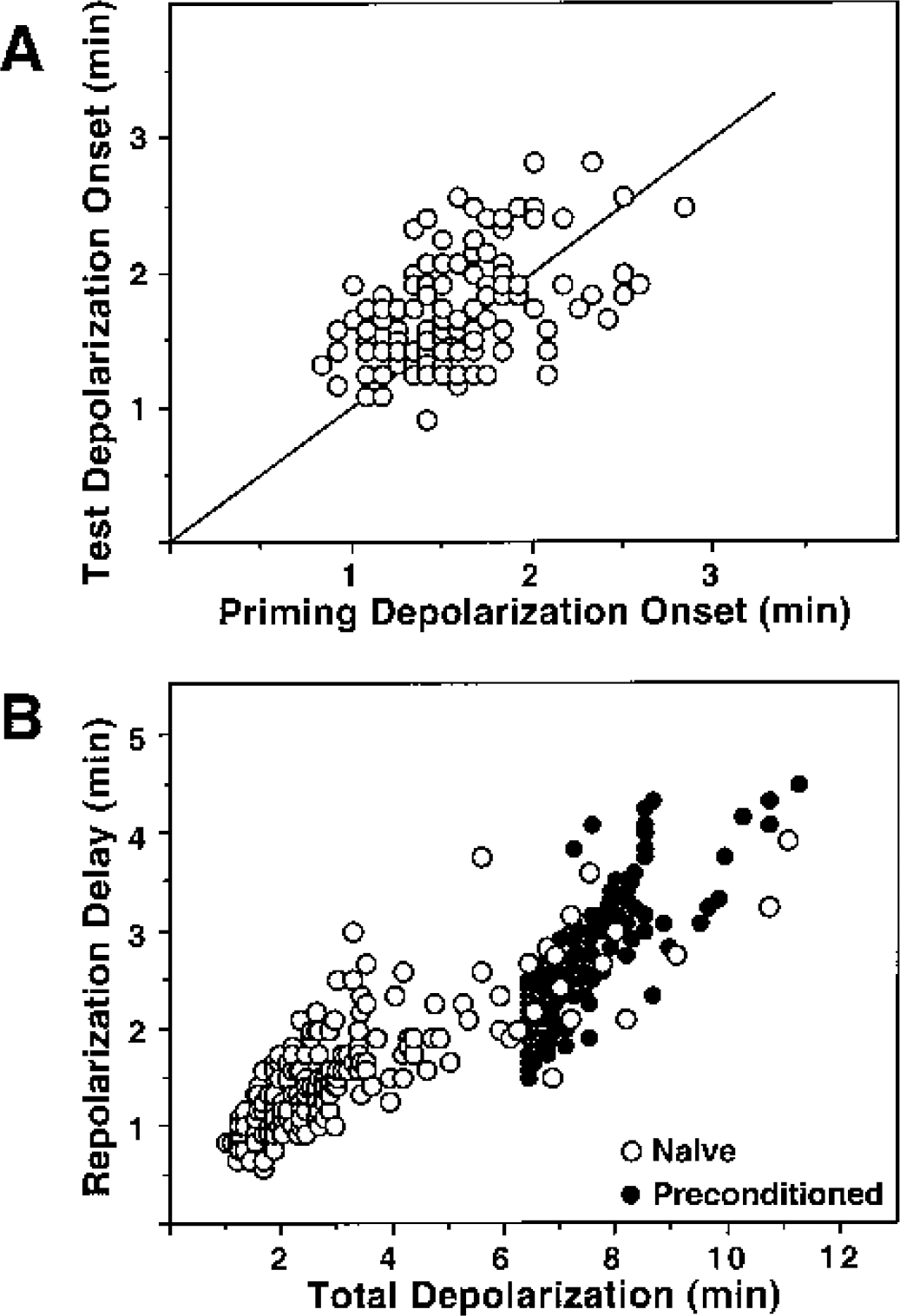
Comparison of depolarization characteristics in naive and previously ischemic animals. Gerbils were subjected to repeated intervals of bilateral carotid artery occlusion with DC potential recording in hippocampus during each ischemic insult. (A) The delay in onset of depolarization after occlusion tended to be slightly longer during test occlusions than during initial priming insults. Line indicates identity. (B) The delay in repolarization after release of occlusion increased with total depolarization time, but this response did not differ in naive and preconditioned hippocampi.
Depolarization thresholds for neuron injury and depolarization windows for tolerance induction
Using depolarization time as an index of insult severity, CA1 neuron damage at 1 week predictably progressed after depolarizations longer than 4 minutes in naive animals, with almost complete losses after insults longer than 6 minutes (Fig. 3). This depolarization threshold for maximal CA1 injury doubled to 11 to 12 minutes in hippocampi subjected to optimal priming insults (2.5–3.5 minutes depolarization at an interval of 2 days, as defined below). It should be noted that the density of CA1 neurons in optimally preconditioned hippocampi (265 ± 7 cells/mm, n = 23) was slightly reduced relative to that of control or subthreshold depolarized hippocampi (278 ± 6 cells/mm, n = 20), and this small difference was statistically highly significant (P < 0.0001, unpaired t-test). Whereas preconditioning led to persistent CA1 neuron preservation at a survival interval of 2 months after the test insult, the injury threshold was detectably reduced in this group relative to preconditioned animals examined at 1 week.
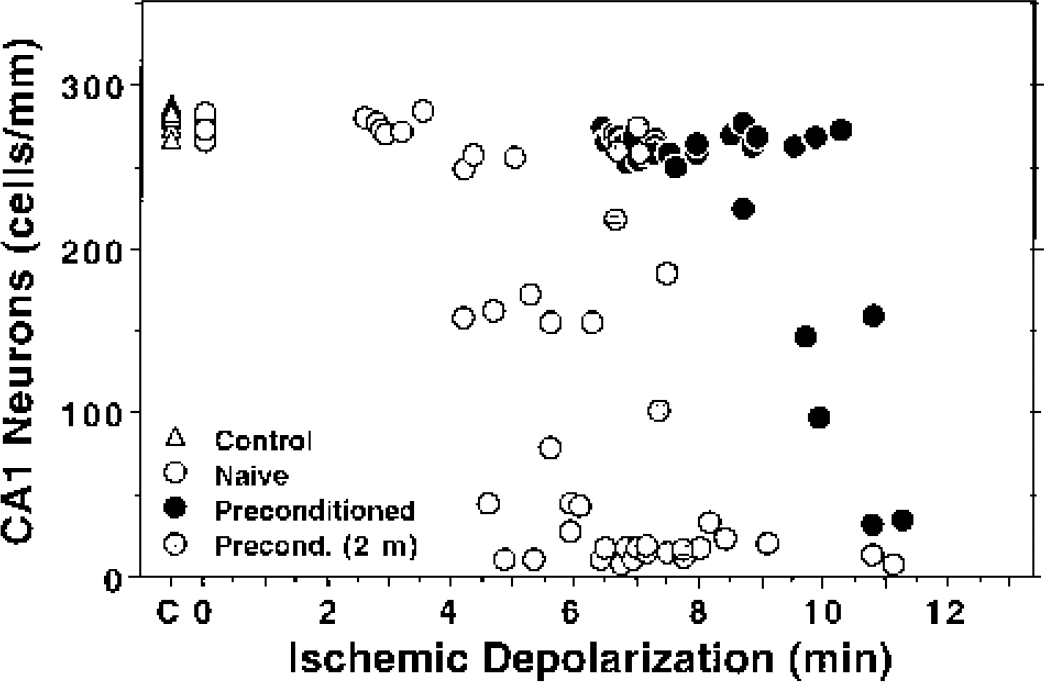
Depolarization threshold for CA1 neuron loss in naive and tolerant hippocampi. Ischemic depolarizations were recorded during test occlusions in naive hippocampi or in those rendered tolerant by an optimized priming insult, as described in the text. CA1 neuron density was evaluated after 1 week, or in a few cases 2 months, in paraffin sections stained with hematoxylin-eosin. Depolarizations less than 4 minutes in duration produced negligible damage in naive hippocampi (open circles), with cell counts indistinguishable from those of control animals (open triangles), whereas depolarizations of 6.5 minutes or longer resulted in essentially complete CA1 neuron loss. The injury threshold was increased to approximately 10 minutes of depolarization in tolerant hippocampi examines at 1 week after the test insult (closed circles). This threshold regressed to an intermediate range in those hippocampi evaluated at 2 months (shaded circles).
To define conditions for optimal tolerance induction in the above studies, gerbils were subjected to priming occlusions resulting in a range of short depolarization times, followed at varied intervals by standard test occlusions yielding 6.5- to 8.5-minute depolarizations, which were shown to produce maximal CA1 loss in naive animals. Histologic analysis at 1 week after the test insult demonstrated partial protection as early as 6 hours after 1- to 2-minute priming depolarizations (Fig. 4). Protection became more complete 24 hours after priming insults, but there remained a tendency toward attenuated protection after depolarizations exceeding 2 minutes. At 2 days after the priming insult, a clear window of optimal protection was evident between 2.5 and 3.5 minutes of depolarization, and hippocampi experiencing preconditioning depolarizations within this range were used to generate the protection data illustrated above in Fig. 3. A few animals allowed 10 days between priming and test insults also exhibited significant protection, but cell counts tended to be lower than seen at the 2-day interval.
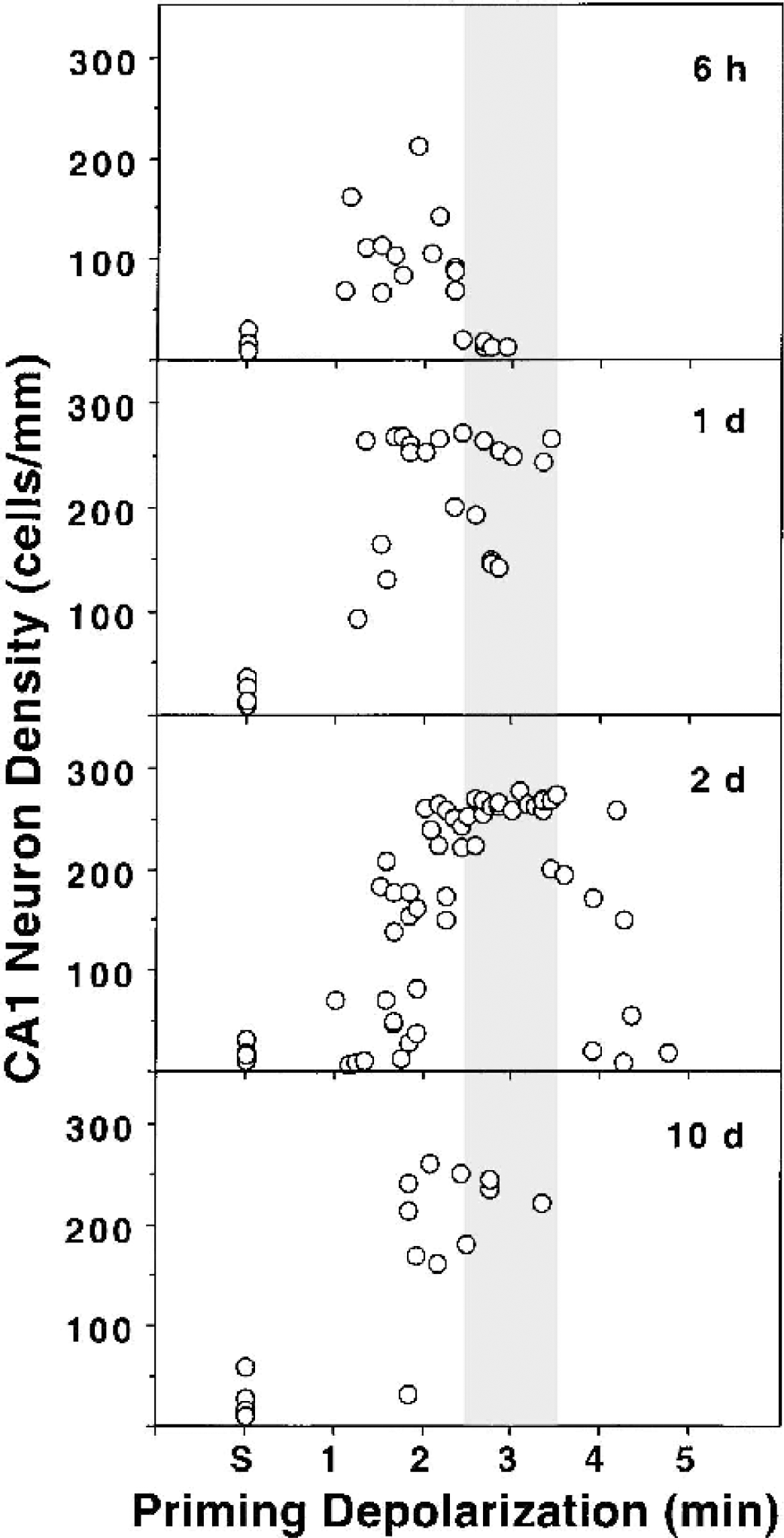
Depolarization thresholds and time course for induction of ischemic tolerance. Gerbils were subjected to priming occlusions resulting in varied durations of depolarization in individual hippocampi, followed at the indicated interval by standard test insults resulting in 6.5- to 8.5-minute depolarization, and CA1 counts were obtained at 1 week after the test insult. Partial protection was observed within 6 hours after short 1.0- to 2.5-minute depolarizations, and more robust protection was evident 24 hours after somewhat longer priming challenges. Optimal tolerance was observed 2 days after 2.5- to 3.5-minute depolarization (shaded bar), and significant protection persisted in animals tested 10 days after such pretreatments.
Depolarization dependence of ischemia-induced changes in gene expression
Relationships between ischemic depolarization and levels of mRNA expression were examined for hsp72 and several immediate-early genes. Representative autoradiograms in Fig. 5 illustrate baseline and ischemia-induced in situ hybridization signals for the several probes in one animal that showed unilateral hippocampal depolarization. As shown in Fig. 6, levels of c-fos, c-jun, and junB hybridization signals were markedly increased in CA1 at 1 hour recirculation after depolarizations that produce optimal tolerance, whereas hsp72 hybridization remained near baseline, and junD showed an intermediate response. Hsp72 hybridization increased with depolarization duration and reached a plateau above 6 minutes, but the other mRNAs showed progressive declines in signal intensity with depolarization times beyond 4 minutes. Hybridization signals were stronger in the densely packed dentate granule cell layer but showed a response pattern generally similar to that in CA1, although signals for immediate-early genes did not decline as markedly with increasing depolarization time. The relationship between depolarization and mRNA expression was also compared at 6 hours recirculation for those mRNAs showing persistent increases (Fig. 7). Hybridization for c-jun was heterogeneous, with some hippocampi at the control level and others showing a stronger signal than seen at 1 hour over a broad range of depolarization durations (2–6 minutes). JunB and junD mRNAs exhibited persistently elevated hybridization, somewhat reduced in magnitude but with a depolarization dependence comparable with that seen at the earlier time point. Hsp72 hybridization was more intense at 6 hours, especially after insults yielding longer depolarizations. However, the induction threshold did not change appreciably, and a number of hippocampi failed to show detectable increases after depolarizations that would have produced marked neuroprotection.
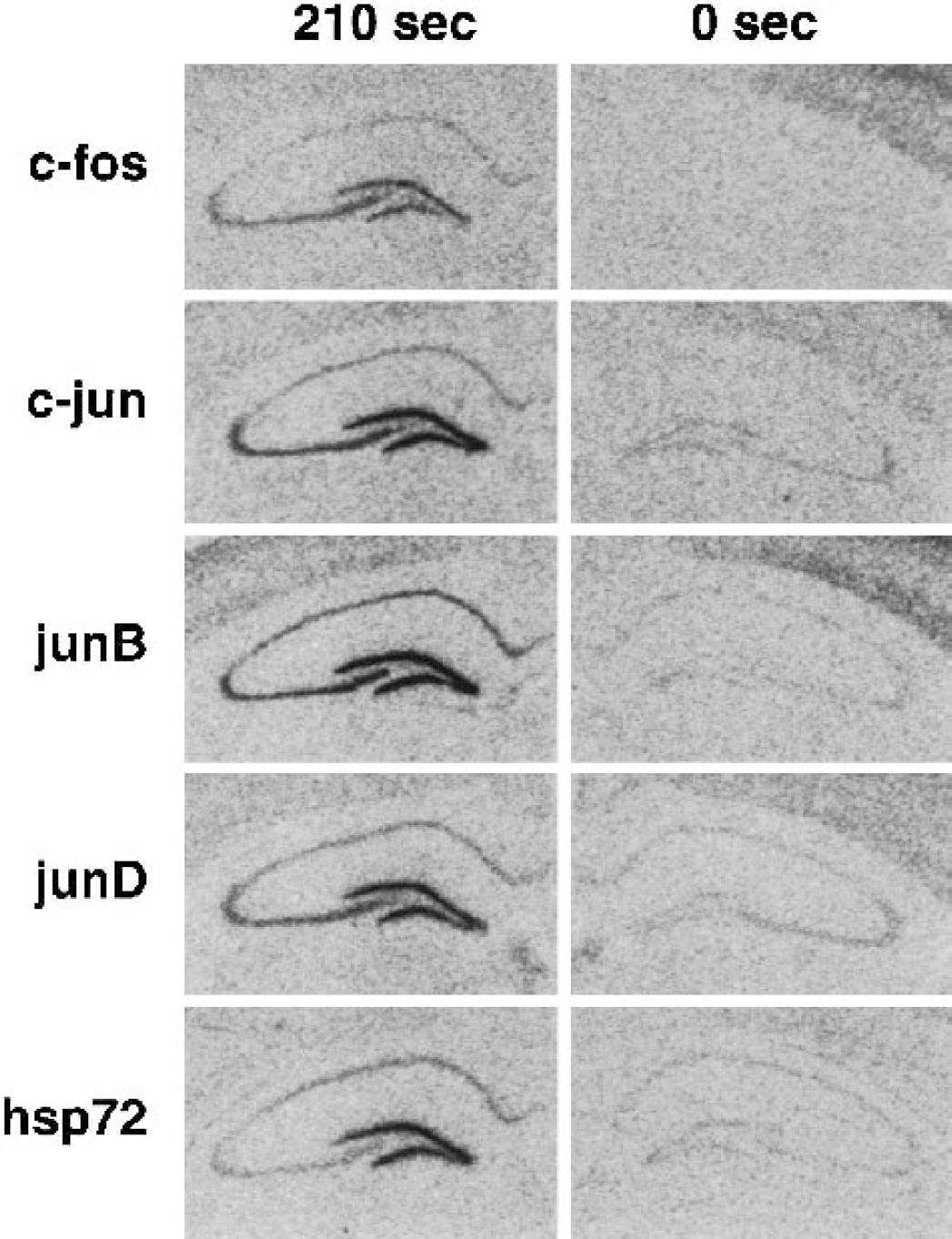
Representative in situ hybridization autoradiograms illustrating hippocampal gene expression and the effect of ischemic depolarization. Sections from one animal that experienced unilateral depolarization of 210 seconds duration, followed by 1 hour recirculation, were prepared and hybridized with the several probes, as described in the text. All mRNAs showed robust expression after depolarization of this duration, whereas a signal identical to that of control animals was evident in the hippocampus that failed to depolarize.
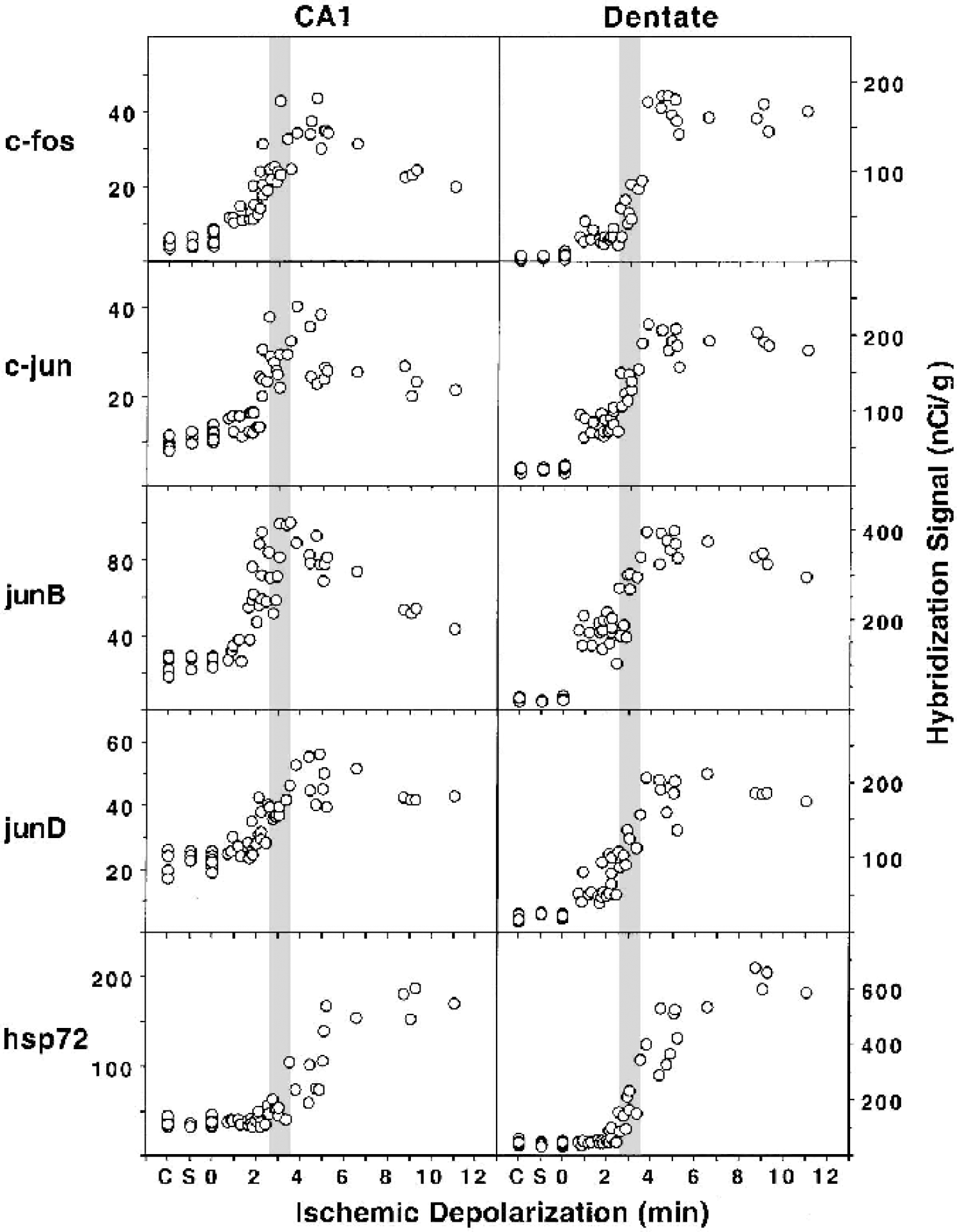
Depolarization threshold for induction of ischemia-inducible mRNAs evaluated at 1-hour recirculation. Sections from control gerbils (C), sham operated animals (S), and gerbils subjected to transient ischemia resulting in varied durations of ischemic depolarization were hybridized with probes specific for the indicated sequences. Hybridization signals of CA1 pyramidal neurons and dentate granule cells were quantitated for each hippocampus as described in the text and plotted as a function of ischemic depolarization. Shaded bars, range of optimal tolerance induction for comparison.
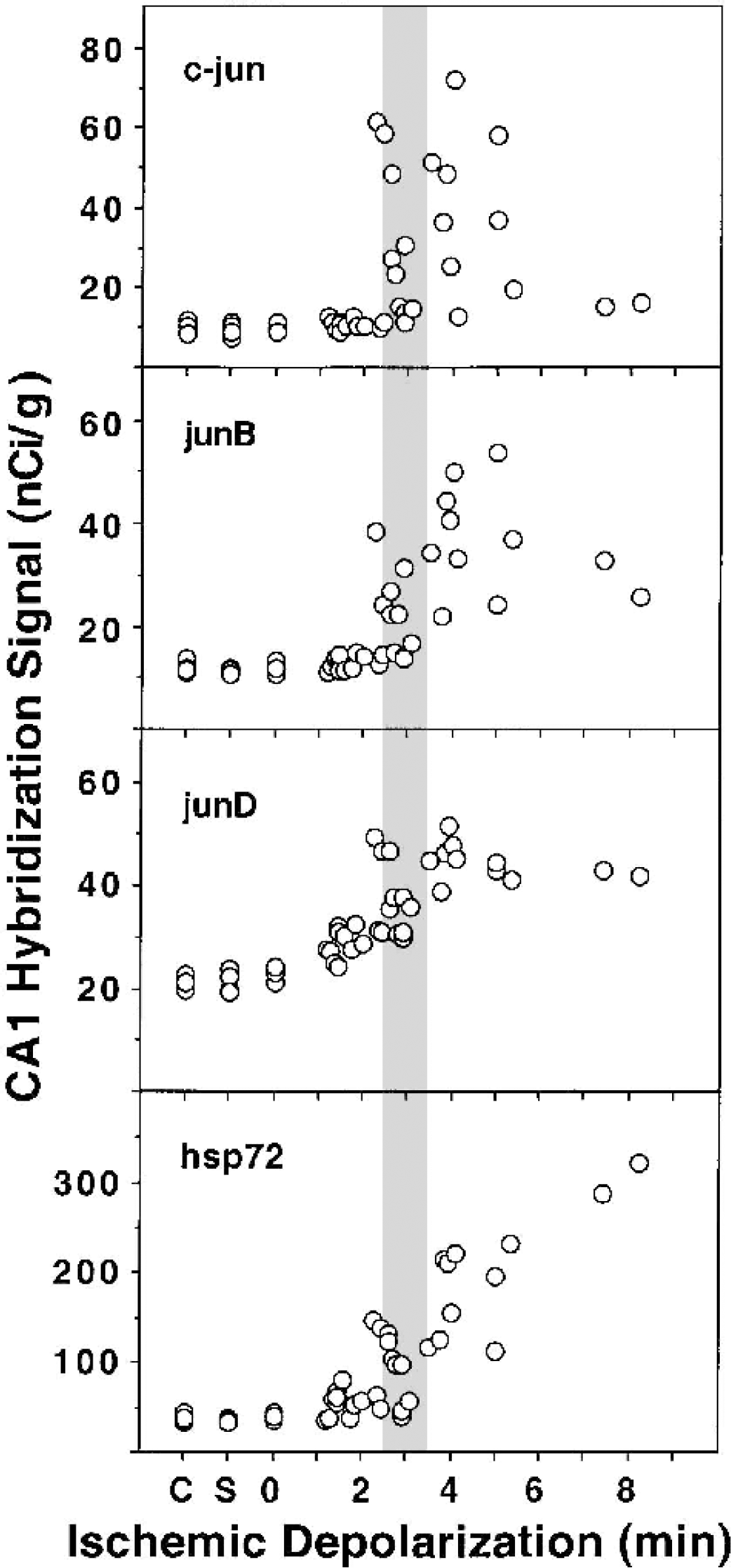
Depolarization thresholds for persistent induction of ischemia-inducible mRNAs in CA1 neurons at 6-hours recirculation. Hybridization signals for mRNAs showing persistent expression in CA1 neurons at this later recirculation interval were analyzed as indicated for Fig. 6. Sections were processed together with different sections from the same control animals (C) used previously and those from additional sham animals (S) killed 6 hours after the surgical procedure. Shaded bar, range of optimal tolerance induction.
DISCUSSION
Depolarization monitoring and control of model variability
DC potential changes have been increasingly applied to monitor the severity of global ischemic insults in hippocampus (Abe and Nowak, 1996a; Bart et al., 1998; Sorimachi et al., 1999, 2002; Xu and Pulsinelli, 1994, 1996), striatum (Xu, 1995), and cortex (Kaminogo et al., 1998; Li et al., 2000). In contrast to the early loss of EEG in the initial seconds of global ischemia (Astrup et al., 1977; Raffin et al., 1991), apparently secondary to rapid hypoxia-induced alterations in synaptic vesicle release (Fleidervish et al., 2001), ischemic depolarization coincides with the ultimate loss of ion homeostasis concurrent with energy failure (Ekholm et al., 1993) and therefore reflects the net impact of CBF reduction, substrate availability, and metabolic activity over time. Differences in the duration of ischemic depolarization for a given occlusion interval clearly account for most of the heterogeneity in the model (Figs. 1–3). The use of DC potential monitoring to define insult duration is particularly important in the gerbil because the severity of forebrain ischemia is entirely dependent upon a vascular anatomy that limits extracarotid blood supply, and this can vary considerably among animals (Levy and Brierley, 1974). For example, a previous report noted a case of unilateral hippocampal depolarization in association with a bilateral response in overlying cortex (Abe and Nowak, 1996a). Whereas unilateral cortical monitoring may generally permit inference of effective hippocampal ischemia in rat brain after carotid occlusion plus hypotension (Pérez-Pinzón et al., 1997), it would clearly not be sufficiently reliable in the gerbil. Ongoing experience with depolarization monitoring in the rat four-vessel occlusion model suggests that a skilled operator can achieve a somewhat better correlation between occlusion and depolarization durations than reported here for the gerbil (Howard, Ueda, and Nowak, unpublished observations, 2000).
It is well recognized that factors such as temperature (Astrup et al., 1981; Bart et al., 1998; Nakashima et al., 1995; Xu, 1995; Xu and Pulsinelli, 1994), blood glucose levels (Hansen, 1978), and anesthetics or other agents that affect energy demand (Astrup et al., 1981; Nakashima et al., 1995; Xie et al., 1995) can dramatically affect the timing of depolarization onset. However, temperature and glucose levels can modulate insult severity independent of effects on depolarization kinetics (Bart et al., 1998; Dempsey et al., 1996), and small variations in such parameters undoubtedly contribute to the remaining heterogeneity in the model. Under consistent experimental conditions, several independent studies have now derived virtually identical depolarization thresholds for CA1 loss (Abe and Nowak, 1996a; Sorimachi et al., 1999, 2002) (Nishino and Nowak, 2003). It should be emphasized that the present experiments used an extended interval of postischemic anesthesia to eliminate spontaneous postischemic temperature increases that can markedly amplify ischemic injury in the gerbil (Abe and Nowak, 2000; Kuroiwa et al., 1990; Suga and Nowak, 1998). Studies in a rat four-vessel occlusion model under similar conditions of temperature control have established a depolarization threshold for CA1 injury identical to that observed here for the gerbil (Yufu, Howard, Ueda, Nowak, manuscripts in preparation), supporting the suggestion that postischemic hyperthermia is primarily responsible for the greater ischemia sensitivity generally recognized for gerbil versus rat hippocampus. A somewhat higher depolarization threshold for CA1 injury was found in a previous rat study (Xu et al., 1999), reflecting in part the use of a global CA1 rating scale rather than the present evaluations at a discrete, relatively medial locus. Given the consistency of depolarization thresholds for CA1 damage in gerbil and rat, it is somewhat surprising that a recent study of cardiac arrest in mice failed to demonstrate significant hippocampal damage after even 7 minutes depolarization (Kawahara et al., 2002). Whether this reflects a true species variation in ischemia sensitivity or is a consequence of unrecognized differences in physiologic control in such a technically demanding model remains to be clarified.
Changes in repolarization kinetics were observed as a function of insult duration, but these were identical in naive and preconditioned animals (Fig. 2B). The repolarization delay upon release of occlusion was more closely correlated with depolarization time than with total occlusion duration (data not shown), suggesting that, like neuron loss, it was primarily dependent upon the effective duration of the resultant metabolic insult. More rapid repolarization after very short insults may reflect earlier metabolic recovery after incomplete depletion of energy stores or faster reequilibration of ion and neurotransmitter pools caused by smaller net shifts during brief ischemia. Depolarizations of 3 to 7 minutes, spanning a range from below to well above the threshold for CA1 injury, resulted in a relatively constant repolarization lag of approximately 2 minutes, which then tended to increase for longer insults. The rate of metabolic recovery per se, therefore, is not of direct pathophysiologic significance for selective neuron vulnerability, although delayed repolarization will clearly result in a longer effective insult for a given occlusion duration. Regarding a possible mechanism for such an effect, grossly impaired reperfusion (‘no-reflow’) is recognized after prolonged ischemia (Ames et al., 1968; Fischer and Hossmann, 1995; Kagstrom et al., 1983), and regionally heterogeneous restoration of CBF has been reported after global ischemic insults of even moderate duration, depending upon reperfusion conditions (Bottiger et al., 1997; Fischer and Hossmann, 1995; Kagstrom et al., 1983). Initially impaired reperfusion is therefore a likely contributor to slow metabolic recovery and delayed repolarization after the longer occlusion intervals examined here, resulting in sporadic worsening of insult severity. This may be expected to vary with details of occlusion methods and among animal models, although ongoing studies indicate a generally similar phenomenon in rat hippocampus after four-vessel occlusion (Howard, Ueda and Nowak, unpublished observations, 2000), and others have reported a tendency toward longer and more variable delays in repolarization with increasing insult duration in rat cortex (Li et al., 2000).
Characteristics of optimized preconditioning
These results demonstrate a robust tolerance phenomenon after priming ischemia in the gerbil, with highly consistent outcome. The factors that impact reproducibility in the model, considered above, acquire added importance in the context of preconditioning because two separate occlusions are produced, and control of insult severity is particularly difficult during short ischemia. Previous studies in rat models have demonstrated striking preconditioning effects after single priming challenges, provided the test insults were not too severe (Liu et al., 1992; Shamloo and Wieloch, 1999). CA1 survival averaged only 50% to 70% under comparable conditions in the gerbil (Abe and Nowak, 2000; Kirino et al., 1991; Kitagawa et al., 1990; Nakano et al., 1998). Whereas repeated priming insults improved protection to as much as 95% after relatively short test occlusions (Kitagawa et al., 1990; Nakano et al., 1998; Ohtsuki et al., 1996), individual animals could nevertheless show slight (Kitagawa et al., 1990) or even substantial CA1 damage (Corbett and Crooks, 1997). Temperature control is a particular consideration in the context of priming insults because spontaneous postischemic hyperthermia characteristic of the gerbil undoubtedly narrowed the insult window for optimal preconditioning in many studies involving this species (Abe and Nowak, 2000). Conversely, intraischemic cooling has been shown to reduce the preconditioning effect of brief ischemia (Wada et al., 1997), probably by delaying depolarization and therefore shortening insult duration as well as slowing metabolic changes during ischemia and reperfusion. The present approach eliminates all of these variables.
A detailed analysis of depolarization characteristics identified a slight (<10%) increase in the average delay in depolarization after test versus priming insults (Fig. 2A). This was quantitatively small and did not occur in all animals, indicating that potential contributing factors such as reduced metabolic rate, improved collateral flow, or increased substrate availability do not in aggregate contribute significantly to the neuroprotection observed. Because test insults in the present study were in any case defined upon the basis of total depolarization time, such delays did not contribute to even small attentuations of insult severity during the second occlusions. These results are in agreement with a number of previous studies indicating no gross impact of preconditioning on CBF or metabolic changes during subsequent insults in vivo (Chen et al., 1996; Matsushima and Hakim, 1995; Nakata et al., 1994; Vannucci et al., 1998) or in slices (Kawai et al., 1998; Paschen and Mies, 1999). However, they contrast with observations of better preserved blood flow and sustained metabolic integrity during forebrain ischemia in rat cortex ipsilateral to a prolonged prior interval of unilateral carotid artery ligation (Bronner et al., 1998), as well as attenuated glutamate release in rats preconditioned by spreading depression (Douen et al., 2000). Such findings reinforce the view that distinct adaptive mechanisms can contribute to the protection observed in different preconditioning models.
There was a noticeable lengthening of the optimal duration of preconditioning depolarization with increasing interval between priming and test insults (Fig. 4). Shorter depolarizations produced rapid but partial preconditioning that became more robust by 24 hours, whereas longer priming insults achieved nearly quantitative protection but required 2 days to fully develop. The latter finding is in general agreement with previous observations of the temporal progression of ischemic tolerance in global ischemia models (Kirino et al., 1991; Kitagawa et al., 1990). It is clear that lengthening the interval between insults primarily impacts outcome after the longer priming depolarizations, suggesting a requirement for more prolonged recovery after more severe priming insults. The present results do not formally distinguish between the contributions of a lag in expression of positive protective mechanisms versus the gradual elimination of negative consequences of the prior insult, and both factors may be involved. For example, it is well established that even brief periods of ischemia can synergize to produce severe damage if repeated at short intervals (Kato and Kogure, 1990; Tomida et al., 1987). Using methods identical to those of the present study, it has been shown that depolarizations longer than 90 seconds reduce the depolarization threshold for subsequent CA1 injury, although intervening recovery periods of only 30 minutes or less were evaluated (Sorimachi et al., 1999). Results in organotypic hippocampal slice cultures indicate that exacerbation of injury can be the predominant consequence of repeated anoxic/aglycemic insults even at recovery intervals of 24 to 48 hours (Pringle et al., 1999).
The modest protective effect seen here within 6 hours after short priming insults may correspond to the very rapid preconditioning reported in rat brain within 30 minutes of a priming insult, which slowed but did not prevent the progression of ischemic injury after subsequent severe ischemia (Pérez-Pinzón et al., 1997). The present results would suggest that the early phase of protection, though partial, may be somewhat more lasting after very mild preconditioning insults (Fig. 4). However, concerns regarding the permanence of protection are well founded, even after optimal preconditioning. A progressive loss of CA1 neurons has been reported in a gerbil model that used repeated priming insults (Corbett and Crooks, 1997), with mean neuron survival of 80% at 10 days declining to 50% by 30 days. Subsequent studies from the same laboratory documented somewhat less dramatic losses, but the frequency of hippocampi showing substantial CA1 loss was increased at survival intervals longer than 10 days (Dowden and Corbett, 1999). In contrast, others have reported nearly complete preservation through 1 or 2 months in gerbil or rat preconditioning models (Ohta et al., 1996; Shamloo and Wieloch, 1999). The limited data collected here at 2 months strongly suggest a basis for such heterogeneity in longterm protection. Delayed attenuation in preconditioning presented as a regression of the injury threshold, such that hippocampi experiencing slightly longer test depolarizations exhibited a more pronounced late decline in surviving cell number (Fig. 3). We have since confirmed this observation with a larger study in a rat model (Ueda and Nowak, 2003) (Ueda and Nowak, manuscript in preparation). These results establish that cell death can continue in preconditioned animals after severe test challenges but also demonstrate a component of lasting protection, with a persistent increase in the depolarization threshold for cell loss relative to naive hippocampi. Insult heterogeneity therefore provides a unifying explanation for the divergent results obtained in earlier studies. A critical inference from these observations is that a 1- to 2-minute difference in depolarization time during a test insult separates “protected” from “slowly dying” neurons, placing considerable demands on the control of experimental models to clearly discriminate between these conditions.
It should also be noted that, although the densities of CA1 neurons in optimally preconditioned hippocampi exceed 95% of control 1 week after the test insult, the small reduction observed is nevertheless statistically significant. This is consistent with the slightly less than 100% protection reported under the best conditions in most previous studies (Kitagawa et al., 1990; Ohta et al., 1996; Shamloo and Wieloch, 1999). Recent results in a rat four-vessel occlusion model have identified occasional eosinophilic CA1 neurons 24 hours after test insults in optimally primed hippocampi (Howard and Nowak, unpublished observations, 2000). The slight decrement in cell number must represent the loss of a particularly vulnerable CA1 subpopulation rather than an initial phase of generalized cell death because the density of surviving cells thereafter remains essentially unchanged even 2 months after the shorter 6- to 7-minute test challenges (Fig. 3). Given the relatively medial position sampled for cell counts, this may reflect in part the known gradient in CA1 vulnerability, with initial injury to subiculum that progresses laterally (Hatakeyama et al., 1988; Iwai et al., 1995). Such results are perhaps also consistent with the failure of ischemic preconditioning to protect highly vulnerable neurons of dentate hilus (Sugimoto et al., 1993). Indeed, preconditioning appears to be unavoidably associated with hilar neuron damage in this model (Matsuyama et al., 1993; Nishino and Nowak, 2003).
Depolarization dependence of postischemic mRNA induction
Depolarization monitoring allows the severity of an ischemic insult to be defined with sufficient reliability for accurate correlational studies because the magnitude of protection can be predicted for individual hippocampi. This permits comparisons between histopathologic outcomes and diverse experimental endpoints across groups of animals evaluated at different recirculation intervals. Transcriptional responses to ischemia occur rapidly relative to pyramidal cell loss, and heretofore there have been risks in suggesting relationships between early changes in gene expression and subsequent injury or protection. The present in situ hybridization results distinguish distinct thresholds for accumulation of hsp72 versus immediate-early gene mRNAs (Fig. 6) and strongly indicate that prior induction of hsp72 mRNA is not required for expression of ischemic tolerance. Optimal preconditioning rather occurs in association with maximal expression of mRNAs for a number of transcription factors encoded by immediate-early genes, as noted in preliminary reports (Abe and Nowak, 1996a,b). This supports data from previous studies associating upregulation of c-Jun immunoreactivity with induction of ischemic tolerance (Nowak et al., 1993; Sommer et al., 1995), although a broader response is clearly identified at the transcriptional level. Many ischemia-induced responses undoubtedly exhibit a comparable threshold, and the full range of mechanisms that may contribute to ischemic preconditioning in such models remains to be established.
Many previous studies noted significant increases in hsp72 protein or mRNA expression in CA1 neurons after brief insults of durations that also induced tolerance (Kirino et al., 1991; Nishi et al., 1993). However, the time course of hsp72 expression did not always correlate well with the interval of tolerance expression (Kirino et al., 1991), and the magnitude of hsp72 expression was not increased by repetition of priming insults that improved the reliability of preconditioning (Kitagawa et al., 1997). In addition, the gerbil work was often carried out under conditions that permitted postischemic hyperthermia, which is known to both worsen damage (Kuroiwa et al., 1990) and amplify hsp72 mRNA expression (Abe and Nowak, 2000; Suga and Nowak, 1998). Normothermic recirculation has been associated with reduced hsp72 mRNA expression but improved tolerance, partially dissociating these responses (Abe and Nowak, 2000). The present results firmly reinforce this conclusion because appreciable increases in hsp72 mRNA in normothermic animals only occurred after depolarizations longer than required for optimal preconditioning (Figs. 6 and 7). CA1 hsp72 hybridization was moderately elevated in some hippocampi after insults in the preconditioning range, but many failed to show any change from the control level, whether examined at 1- or 6-hour recirculation. It therefore appears that prior hsp72 induction is not required for ischemic preconditioning. This still permits a role for subsequent hsp72 expression in the successful response to severe ischemia in preconditioned hippocampi. For example, it has been previously noted that hsp72 mRNA induced after test insults may be better translated in tolerant CA1 neurons, in association with improved recovery of protein synthesis activity (Aoki et al., 1993; Furuta et al., 1993; Kanemitsu et al., 1994; Nakagomi et al., 1993). This would be consistent with protective effects of selective overexpression of hsp70 family proteins described in diverse in vitro and in vivo injury models (Dedeoglu et al., 2002; Hoehn et al., 2001; Kelly et al., 2001; Kelly et al., 2002; Li et al., 1991; Rajdev et al., 2000; Uney et al., 1993; Yenari et al., 1998). However, whereas parallel studies examining hsp72 immunoreactivity in this depolarization-monitored gerbil model do indicate improved hsp72 expression after severe insults to tolerant CA1 neurons, they suggest that the predominant effect of preconditioning is to increase the insult threshold for hsp72 accumulation (Nishino and Nowak, 2003).
It is somewhat surprising that CA1 neurons and dentate granule cells exhibit essentially identical depolarization thresholds for hsp72 mRNA induction (Fig. 6). Previous studies in several models have consistently demonstrated that brief insults result in much more pronounced hsp72 immunoreactivity in vulnerable neurons of CA1 and dentate hilus than observed in resistant cells in the dentate granule layer and CA3 (Kirino et al., 1991; Nishi et al., 1993; Simon et al., 1991). It is well established that the duration of hsp72 mRNA expression after severe insults reflects the relative vulnerability of hippocampal neuron populations (Nowak, 1991; Suga and Nowak, 1998), raising the possibility that more prolonged mRNA expression leads to increased accumulation of the encoded protein after mild insults that allow translational recovery in neurons. In the present study, the levels of hsp72 hybridization were quantitatively similar in CA1 and dentate granule cells at 6 hours (data not shown), reflecting reciprocal changes in the CA1 and dentate signals, which respectively increased and decreased relative to the 1-hour time point (Fig. 6). This supports the suggestion that persistent hsp72 mRNA expression is largely responsible for preferential accumulation of hsp72 immunoreactivity in more vulnerable neurons. It is important to note that whereas hsp72 mRNA expression in CA1 increased between 1 and 6 hours, there was no substantial shift in the depolarization threshold for expression (Figs. 6 and 7). Later time points were not examined in these experiments because previous studies have shown a decline in hsp72 mRNA expression in CA1 neurons between 6 and 24 hours after brief insults in the absence of postischemic hyperthermia (Abe and Nowak, 2000; Suga and Nowak, 1998). In addition, recent results in this model have detected subsequent CA1 hsp72 immunoreactivity in only half of the hippocampi subjected to optimal preconditioning and 48hour recirculation (Nishino and Nowak, 2003). This is entirely consistent with the frequency of above-baseline hybridization in the current study, strongly indicating that there is no shift in the threshold for hsp72 induction at later recirculation times not examined here.
Attenuated mRNA accumulation after longer depolarizations was observed to varying degrees for all of the immediate-early genes, but not for hsp72 (Figs. 6 and 7). Hybridization signals peaked after insult durations just below the threshold for initiation of CA1 damage (approximately 4 minutes of depolarization), and the decline with increasing depolarization time was more prominent in CA1 than in the less vulnerable dentate granule cells, suggesting a correlation with neuron injury. A blunting of c-fos mRNA expression has been noted previously in the context of exacerbated injury following hyperglycemic ischemia (Combs et al., 1992). Because mRNA levels are determined by the relative rates of gene transcription, intranuclear processing, cytoplasmic transport, and degradation, effects at any of these sites could alter the balance of transcript accumulation detectable by in situ hybridization. Stress-induced hsp70 mRNAs do not require splicing, and their transport to the cytoplasm can occur under insult conditions such as heat shock that are associated with impaired processing of other mRNAs (Parsell and Lindquist, 1994; Yost and Lindquist, 1986). Whereas there are known alterations in splicing, editing, or processing of a number of mRNAs after transient ischemia (Daoud et al., 2002; Paschen et al., 1996, 2003; Saito et al., 1995), no generalized deficits in overall mRNA synthesis and processing have been described. Nevertheless, subtle reductions in the rates of such steps could provide one explanation for the observed effect. Considerable evidence indicates that transient DNA damage can be a prominent consequence of ischemia (Charriaut-Marlangue et al., 1999; Cui et al., 2000; Hayashi et al., 1999; Jin et al., 1999), and it has been suggested that this could impair proper transcription of encoded mRNAs (Cui et al., 1999). This would not contribute to differential effects on mRNA levels unless there were mechanisms by which the hsp72 gene was selectively protected from DNA damage. It is interesting that whereas degradation of aberrant mRNAs harboring premature termination signals provides a quality control measure to avoid expression of damaged proteins, recent evidence indicates that such mRNA surveillance is avoided by hsp70 and other intronless genes (Maquat and Li, 2001). Finally, signals controlling immediate-early gene expression might be selectively downregulated with increasing insult severity, or the decline in expression could reflect the impact of competition for active polymerase by hsp72 and other ischemia-induced genes that are preferentially upregulated in CA1 after more severe insults (Chen et al., 1998; Nowak, 1991). It remains to be determined whether the observed decrements in expression of these ischemia-induced transcripts might share common mechanisms with the relative declines in GluR2 expression reported in rat and gerbil ischemia models (Gorter et al., 1997; Pellegrini-Giampietro et al., 1992; Sommer and Kiessling, 2002).
CONCLUSIONS
Variability in gerbil models of ischemia and ischemic tolerance is largely eliminated by the combination of depolarization monitoring and postischemic temperature control. This has allowed the definition of a narrow but consistent depolarization window for optimal ischemic preconditioning. The approach can be readily extended to other global ischemia models and would improve the reliability of tolerance evaluation after any preconditioning protocol. Short depolarizations that induced ischemic tolerance also induced near-maximal levels of mRNAs encoding several immediate-early genes, whereas longer depolarization intervals were required to induce hsp72. Although even precise correlative evaluations cannot establish causality, they provide the capability to discriminate between responses that constitute viable candidate mechanisms and those that do not. Prior hsp72 induction can therefore be excluded as a requirement for ischemic preconditioning in this model. A wide range of potential mechanisms remains to be evaluated.
Footnotes
Acknowledgment
The authors thank William A. Pulsinelli for the suggestion to monitor ischemic depolarization in the model and Yoshimasa Takeda and Zhao-Cheng Xu for sharing their experience.