
Abstract
Earlier studies indicated that sublethal ischemic insults separated by many hours may “precondition” and, thereby, protect tissues from subsequent insults. In Wistar rats, we examined the hypothesis that ischemic preconditioning (IPC) can improve histopathological outcome even if the “conditioning” and “test” ischemic insults are separated by only 30 min. Normothermic (36.5–37°C) global cerebral ischemia was produced by bilateral carotid artery ligation after lowering mean systemic blood pressure. The conditioning ischemic insult lasted 2 min and was associated with a time sufficient to provoke “anoxic depolarization” (AD) (i.e., the abrupt maximal increase in extracellular potassium ion activity). After 30 min of reperfusion, 10-min test ischemia was produced, and histopathology was assessed 3 and 7 days later. After 3 days of reperfusion, neuroprotection was most robust in the left lateral, middle and medial subsections of the hippocampal CA1 subfield and in the cortex, where protection was 91, 76, 70 and 86%, respectively. IPC also protected the right lateral, middle and medial subsections of the hippocampal CA1 region. These data demonstrate that neuroprotection against acute neuronal injury can be achieved by conditioning insults followed by only short (30 min) periods of reperfusion. However, neuroprotection almost disappeared when reperfusion was continued for 7 days. When test ischemia was decreased to 7 min, a clear trend of neuroprotection by IPC was observed. These data suggest that subsequent rescue of neuronal populations could be achieved with better understanding of the neuroprotective mechanisms involved in this rapid IPC model.
In heart and brain, ischemic preconditioning (IPC) has been defined as the protection produced when mild ischemic episodes ameliorate damage from subsequent ischemic insults (Murry et al., 1986; Kitagawa et al., 1990). In heart for example, IPC protected from subsequent sustained coronary occlusion both the region in which IPC was elicited and remote myocardium (Przyklenk et al., 1993). In brain, histopathological damage was less when a single “sublethal” insult preceded a “lethal” ischemic insult by 1 or 2 days (Kitagawa et al., 1990).
In both heart and brain, IPC protection depends upon the intensity of ischemia and the latency between insults. In cardiac muscle for example, the maximum latency between insults that is necessary for IPC protection is 1 h. In brain, however, IPC paradigms are less well defined. In general, IPC is protective against global cerebral ischemia in vivo, but only if many hours of reperfusion separates “conditioning” and “test” insults (Kato et al., 1991). In brain slices, however, preconditioning-induced neuronal protection occurs with shorter latencies between conditioning and test insults. Such latencies range from 30 min to 2 h (Schurr et al., 1986; Schurr and Rigor, 1987; Pérez-Pinzón et al., 1996).
The fact that shorter latencies between conditioning and test insults are sufficient for preconditioning in brain slices suggests that intact brains may have a similar intrinsic neuroprotective potential that was not expressed in earlier studies (e.g., Kato et al., 1991). This is important since IPC may offer unique insights into basic mechanisms of ischemic injury and into potential therapies that may be especially applicable in the early period after ischemic insults. Therefore, the major goal of the present study was to determine whether neuroprotection against neuronal damage could be induced by IPC, as occurs in the heart, when the latency between conditioning and test insults was limited to only 30 min.
MATERIALS AND METHODS
Animal model
All animal procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and were approved by the animal care committee of the University of Miami. Male Wistar rats weighing 300–350 g were fasted overnight and then anesthetized with 3% halothane and 70% nitrous oxide (balance oxygen). The femoral arteries were cannulated for blood pressure measurements and for arterial sampling of blood gases. Arterial blood gases (178 pH/blood gas analyzer, ABL 30, Acid–Base Analyzer, Copenhagen, Denmark), plasma glucose levels (One Touch glucose monitor, Precision Q.I.D.; Medisense, Inc., Waltham, MA, U.S.A.) and hematocrit were measured intermittently throughout the experiment. Goals were to maintain blood gases within normal ranges (arterial P
Rats underwent endotracheal intubation and were artificially ventilated with 0.5% halothane and 70% nitrous oxide (balance oxygen). Rats were immobilized with pancuronium bromide (0.75 mg/kg, i.v.). Both common carotid arteries were exposed by a midline ventral incision and gently dissected free of surrounding nerve fibers. Ligatures of polyethylene (PE-10) tubing, contained within a double-lumen silastic tubing, were passed around each carotid artery.
Brain temperature was monitored with a 33-gauge thermocouple implanted on the temporalis muscle (Dietrich et al., 1993). Temperature was maintained at 36.5–37°C throughout the experiment using a small warming lamp placed above the animal's head. An extracellular potassium electrode was placed in a burr hole opened left of the midline.
Ion selective microelectrodes (ISM)
Extracellular potassium ion activity [K+]o was measured with double-barrel, micropipette electrodes (Pérez-Pinzón et al., 1995) placed in a burr hole opened in the left cortical region of the rat brain. The tip of one barrel was silanized and filled with ion exchanger for potassium (Corning 477317). The remainder of the micropipette was filled with 100 mM KCl. The other barrel was filled with 150 mM NaCl and was used as a reference to monitor local direct current (DC) potentials. These DC potentials were electronically subtracted from the signal obtained from the K+-sensitive micropipette to yield an electrical potential that varied with [K+]o. This difference signal (Ek+) was displayed on a chart recorder and was used to calculate [K+]o by reference to a standard calibration curve obtained with solutions containing varying concentrations of KCl substituted for NaCl to maintain constant ionic strength of 150 mEq/L. Potassium sensitive microelectrodes (K+-ISM) were calibrated before and after each experiment. Calibration data were fitted to the Nikolsky equation to determine electrode slope and interference (Pérez-Pinzón et al., 1995).
Production of cerebral ischemia
Before each ischemic insult, blood was gradually withdrawn from the femoral artery into a heparinized syringe to reduce systemic blood pressure to 40–50 mm Hg. Cerebral ischemia was then produced by tightening the carotid ligatures bilaterally. To allow postischemic reperfusion, the carotid ligatures were removed and the shed blood reinjected into the femoral artery. This infusion usually restored mean arterial blood pressure to 100–120 mm Hg. Vessels were inspected to verify that perfusion was reestablished.
The threshold for ischemia was established as the loss of ion homeostasis, as shown by the abrupt increase in extracellular potassium to 50–70 mM, characterized as “anoxic depolarization” (AD) (Hansen, 1985). Figure 1 illustrates the effect of ischemia in the two-vessel occlusion model with hypotension (<50 mm Hg). The main criterion for complete induction of ischemia was the onset of AD, and the duration of ischemia was determined by AD. If AD did not occur, no further studies were conducted and the animal was discarded.
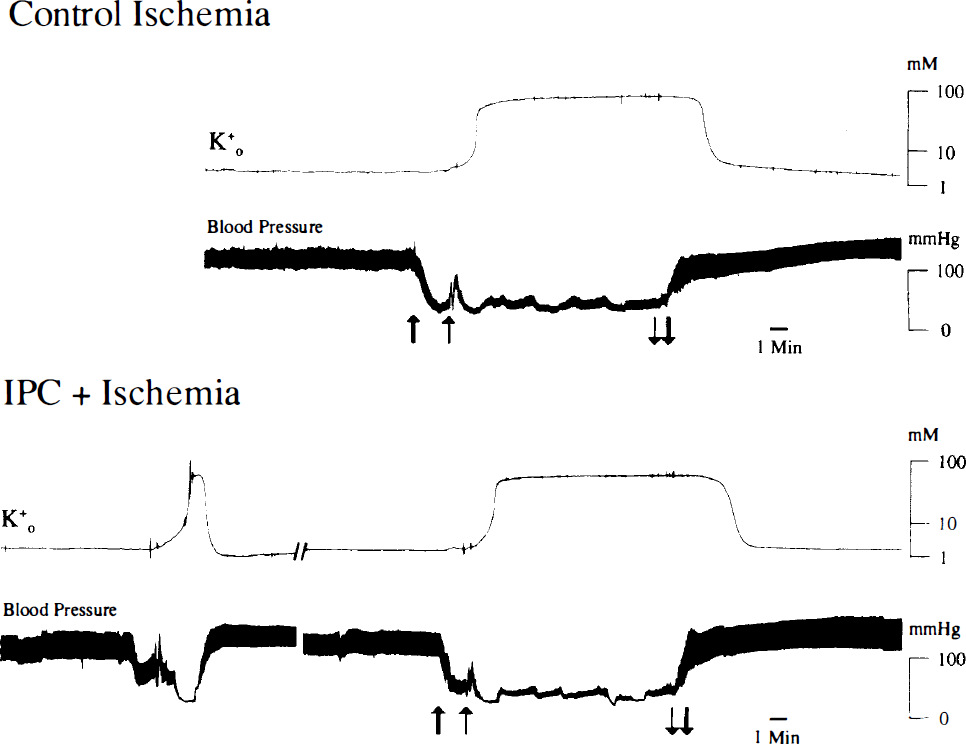
Typical responses to carotid ligation. Top trace shows a typical extracellular potassium ion activity recorded prior to, during, and after carotid ligation and hypotension (blood pressure, second trace) in the control group. First, blood was withdrawn (thick arrow), followed by carotid ligation (thin arrow). Bottom traces show a typical ischemic preconditioning paradigm in which the duration of conditioning ischemia was 2 min (short AD), followed by 30 min of reperfusion, and 10 min of test ischemia, as shown by the arrows (blood withdrawal, thick arrow; carotid ligation, thin arrow).
Histopathology
At 3 or 7 days after test ischemic insults were provoked, rats were again anesthetized with halothane, perfused for 1 min with physiologic saline, and then perfused with a mixture of 40% formaldehyde, glacial acetic acid, and methanol, (FAM) 1:1:8 (vol/vol/vol) for 19 min. The perfusate solution was delivered into the root of the ascending aorta at a constant pressure of 110–120 mm Hg, as described previously (Dietrich et al., 1993). The head was removed and immersed in FAM at 4°C for 1 day. The brain was then removed from the skull, and coronal brain blocks were embedded in paraffin. Coronal sections of 10 μm thickness were stained with hematoxylin and eosin.
Sections containing frontoparietal neocortex and hippocampus were examined by an observer (W.D.D.), blinded from experimental conditions. Ischemic neurons were counted within different high-power fields (400 ×) of cortex and hippocampus. Ischemic damage was defined as the condition in which neurons showed moderate-to-severe shrinkage, eosinophilic cytoplasm, increased nuclear basophilia, and a pyknotic nucleus, as in previous studies (Dietrich et al., 1993).
Study groups and experimental paradigms
Animals were assigned to one of three groups: In control group 1, animals underwent a single ischemic insult of 10 or 7 min, 40 min after onset of the experiments. Animals were killed 3 or 7 days following ischemia and the brains fixed for histological evaluation. In group 2 rats, IPC was induced by a short (2 min) ischemic insult (conditioning ischemia). After 30 min of reperfusion, a second ischemic insult lasting 10 or 7 min (test ischemia) was induced. After 3 or 7 days, animals were killed and their brains fixed for histological analysis. In group 3 rats, only the preconditioning ischemia was induced (2 min) to assess degree of damage. After 3 days, animals were killed and their brains fixed for histological analysis.
Statistical analysis
Statistical significance among the three groups was established with analysis of variance (ANOVA) followed by a Bonferroni's post-hoc test. Significance was accepted with p < 0.05. All data were expressed as means ± SD.
RESULTS
Systemic parameters
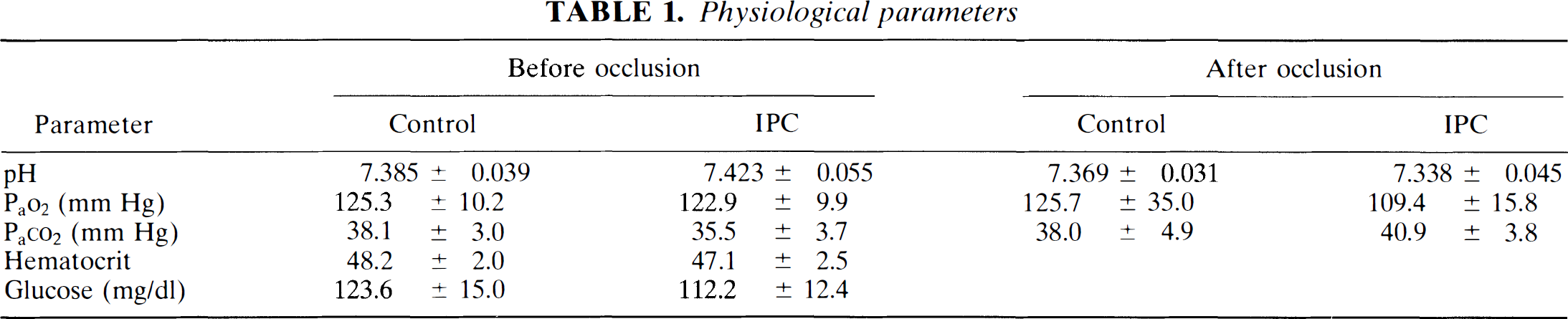
Parameters expressing the physiological condition of the animals prior to, during, and after ischemia are shown in Table 1. These parameters showed that there were no significant differences between groups either prior to or after ischemia, and ischemia itself had no effect on these parameters (Table 1).
Physiological parameters
Histopathology
Figure 1 shows typical responses to carotid ligation. As shown in the top two traces, AD usually occurred during carotid ligation, but only when MABP was decreased below 50 mm Hg. The bottom two traces show a typical preconditioning paradigm. The duration of ischemia during the preconditioning insult was 2 min; 30 min after the conditioning insult, the test ischemic insult was induced. Responses to this latter insult were similar to that described in the top two traces.
In control rats, the test ischemic insult (10 min) produced a high incidence of ischemic neurons within the CA1 hippocampus (Figs. 2A and B ). Ischemic neurons were also commonly observed in layers III, IV, and V of the cerebral cortex immediately dorsal to the entorhinal fissure (Fig. 2C). In contrast, rats that underwent the preconditioning ischemic insult 30 min prior to the test insult had fewer ischemic cells within the hippocampus (Figs. 2D and E ) and cerebral cortex (Fig. 2F).
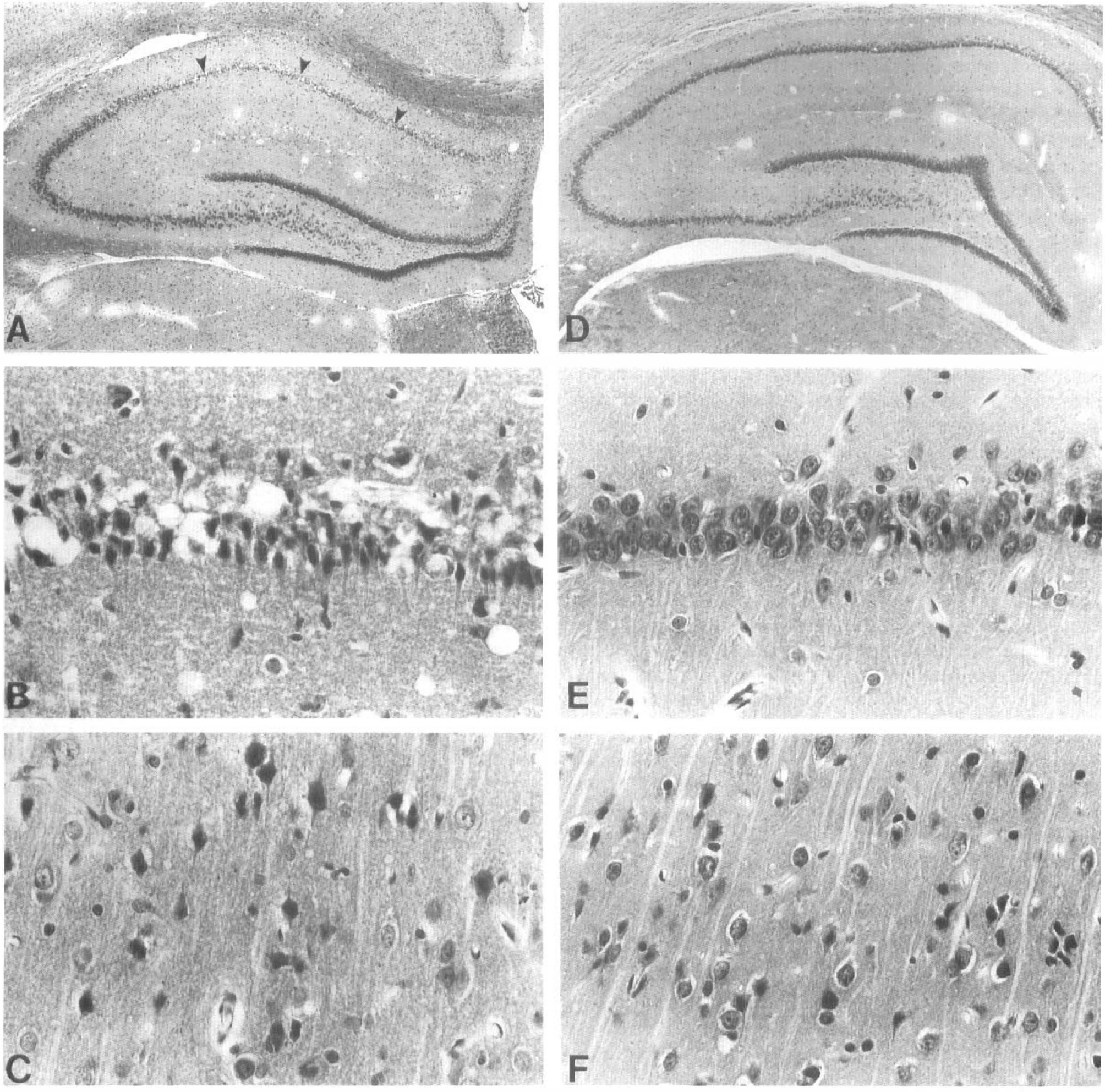
Paraffin sections stained with hematoxylin and eosin from ischemia (
In rats in which only the preconditioning insult was produced (2 min), almost no ischemic neurons were observed. In the left lateral hippocampus, the ischemic cell count was 2.0 ± 3.0, whereas in the right side it was 0.2 ± 0.4 (n = 5). In the left middle hippocampus, the ischemic cell count was 7.0 ± 10.0, whereas in the right side it was 1.0 ± 2.2 (n = 5). In the left medial hippocampus, the ischemic cell count was 14.0 ± 15.0, whereas in the right side it was 3.0 ± 2.0 (n = 5). In the left cortex, the ischemic cell count was 0.6 ± 0.9, whereas in the right side it was 0.2 ± 0.4 (n = 5). These values were all significantly different from those of the control (10 min ischemia) group (p < 0.05), but similar to those of the IPC-treated group.
Ischemic cell counts for rats grouped as controls (n = 11) and IPC-treated (n = 11) rats are shown in Fig. 3. IPC significantly protected by 90, 77, and 69% the left lateral, middle, and medial subsections of the hippocampal CA1, respectively, when compared with control values (Fig. 3). In the lateral hippocampus, the control ischemic cell count was 29 ± 26 versus IPC, which was 3 ± 6 (p < 0.01). In the middle hippocampus, the control ischemic cell count was 26 ± 19 and that of IPC was 6 ± 11 (p < 0.01). In the medial subsection of the hippocampus, the control ischemic cell count was 29 ± 18 and that of IPC was 9 ± 9 (p < 0.01).
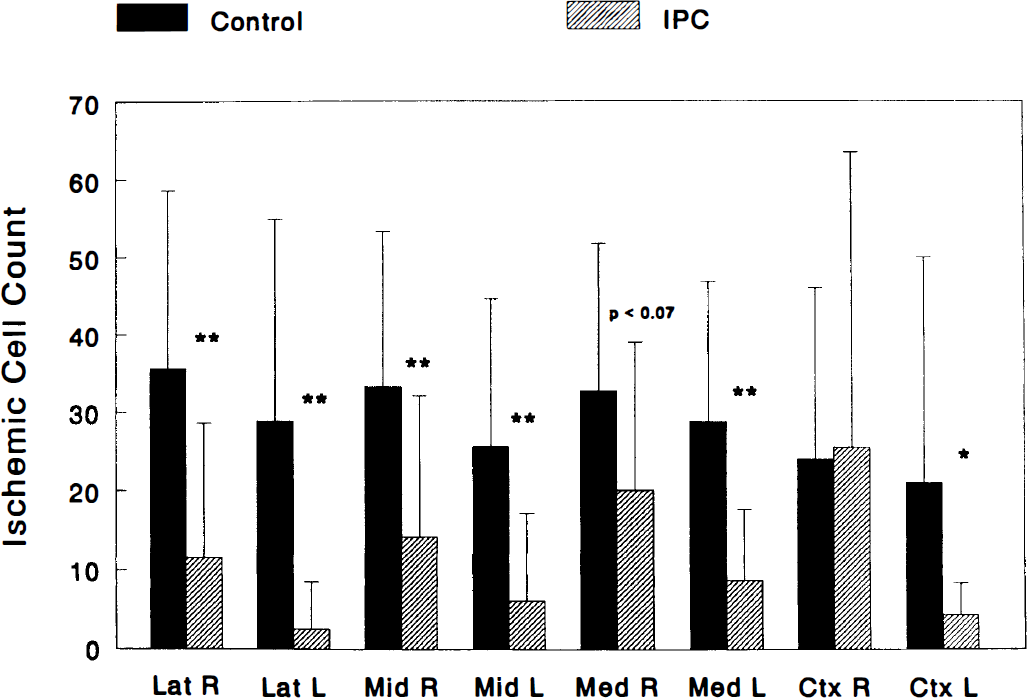
Ischemic preconditioning was neuroprotective 30 min prior to a 10-min test ischemic insult. Ischemic cell count was significantly reduced in all regions studied in IPC-treated rats (n = 11) as compared with controls (n = 11). L, left hemisphere; R, right hemisphere; Lat, lateral subsection of the CA1 subfield of the hippocampus; Mid, middle subsection of the CA1 subfield of the hippocampus; Med, medial subsection of the CA1 subfield of the hippocampus; Ctx, lateral cortex. *,p < 0.05; **, p < 0.01.
IPC also protected by 67, 58, and 39% the right lateral, middle, and medial subsections of the hippocampal CA1 region, respectively, when compared with control values. Significant differences between control and IPC groups were only found in the right lateral and middle subsection of the hippocampal CA1 region. In the right lateral hippocampus, the control ischemic cell count was 36 ± 23 (mean ± SD) versus IPC, which was 12 ± 17 (p < 0.01). In the middle subsection of the hippocampus, the control ischemic cell count was 33 ± 20 versus IPC, which was 14 ± 18 (p < 0.01). Although not significantly different, the medial region of the hippocampus showed a trend towards neuroprotection. The control ischemic cell count was 33 ± 19, whereas IPC was 20 ± 19 (p < 0.07).
Significant IPC protection was also found in the lateral cortex, when compared with controls (Fig. 3). The control ischemic cell count was 29 ± 37 and that of IPC was 4 ± 4 (p < 0.05). In the right neocortex, however, there was no significant effect of IPC. The ischemic cell count in control rats was 38 ± 52 and 21 ± 21 in rats of the IPC group.
In order to determine whether IPC neuroprotection was permanent or whether IPC was just protective against acute neuronal death, four additional groups were examined. In groups 1 and 2 (described above), histopathology was determined at 7 instead of 3 days following test ischemia. Histopathology for the other two groups was determined 7 days following test ischemia, but test ischemia lasted 7 instead of 10 min as in previous groups. Figures 4A and B summarize those results.
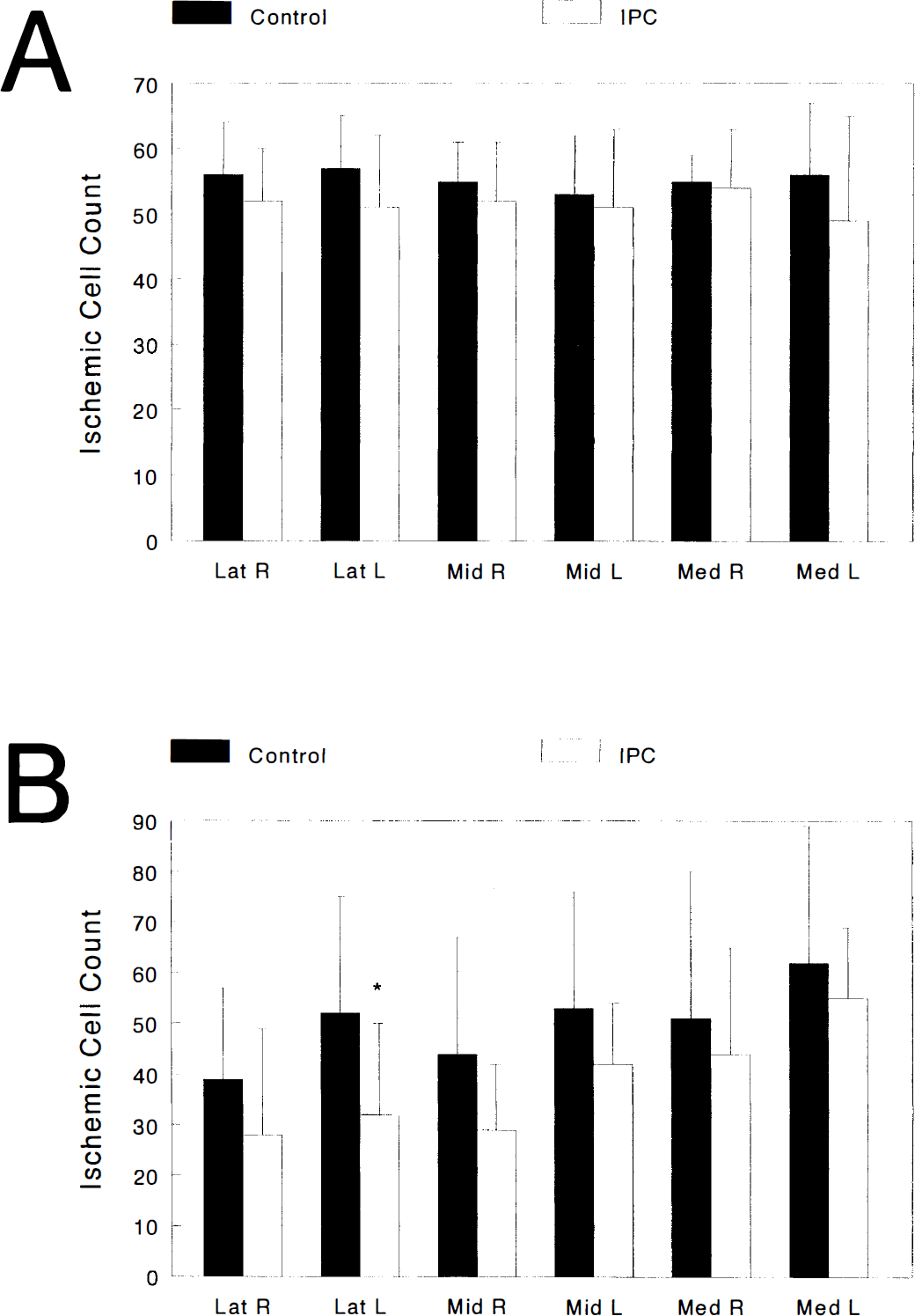
In Fig. 4A, results showed that the acute IPC neuroprotection observed in Fig. 3 almost disappeared when reperfusion was prolonged to 7 days. Although a trend towards neuroprotection is observed, this type of rapid IPC cannot protect against delayed neuronal damage. However, this trend toward neuroprotection was more pronounced when test ischemia was shortened to only 7 min (Fig. 4B). In the latter group, significant neuroprotection was observed only in the lateral hippocampal CA1 subsection.
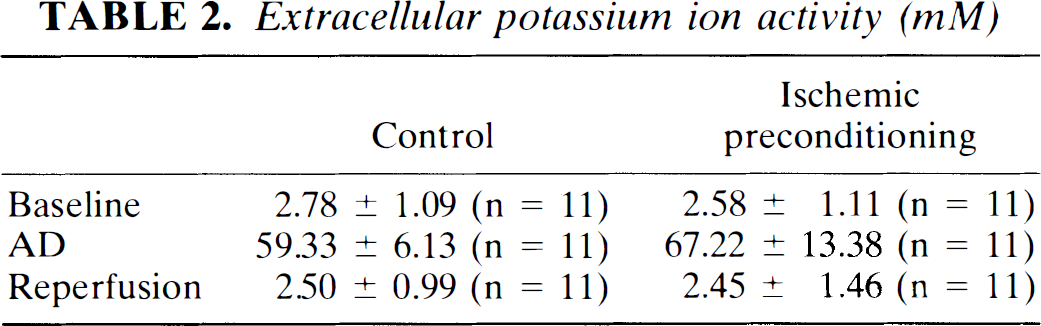
Extracellular potassium ion activity [K+]o
No significant differences were found in the levels of [K+]o prior to, during, and after test ischemia in controls versus the IPC group (Table 2). [K+]o increased from 2.78 ± 1.08 to 59.33 ± 6.13 mM during AD in controls animals (n = 11). Shortly after the onset of reperfusion, [K+]o decreased to 2.50 ± 0.99 mM. In the IPC group, [K+]o increased from 2.58 ± 1.11 to 67.22 ± 13.38 mM during AD (n = 11). Following reperfusion, [K+]o decreased to 2.45 ± 1.46 mM.
Extracellular potassium ion activity (mM)
DISCUSSION
In intact brain, ischemic preconditioning was shown to provide significant protection from subsequent ischemic insults. However, the protective effect of IPC was evident only when the latency between preconditioning and test insults was very prolonged (days) (Kitagawa et al., 1990; Kato et al., 1991; Liu et al., 1992). To our knowledge, the present study is the first to demonstrate that only a short (30 min) reperfusion period between conditioning and test ischemic insults can provide significant protection against acute ischemic neuronal damage in the CA1 hippocampal subfield and neocortex. The rapid induction of IPC shown to be possible in intact brain was seen earlier in brain slices (Schurr et al., 1986; Schurr and Rigor, 1987; Pérez-Pinzón et al., 1996) when latencies between conditioning and test insults ranged from 30 min to 2 h.
In contrast to the neuroprotection achieved by this rapid preconditioning paradigm, multiple ischemic insults separated by short periods worsened ischemic injury (Lin et al., 1992; Lin et al., 1993). However, both paradigms differed in that, in the multiple insult ischemic paradigm, insults were repeated more than twice and each insult was of the same duration. Thus, those studies were not aimed at determining ischemic preconditioning, but at determining the effect of multiple ischemic insults. The paradigm used in the present study was designed to determine a type of metabolic regulation in which the brain adapts, via a sublethal insult to resist a more robust ischemic insult.
Confirmation of the rapid IPC capability in brain leads to the suggestion that there are two phases of IPC. The first phase may arise quickly from events associated with preconditioning insults, e.g., enhanced glycolysis, suppressed electrical activity, loss of ion homeostasis, and depressed ATP consumption. This phase of neuroprotection may disappear rapidly, as in heart (Murry et al., 1986). A second phase of preconditioning may occur later on after the preconditioning insult and may involve expression of specific genes that may limit delayed neuronal damage (Liu et al., 1993a, b ; Kato et al., 1994, 1995).
The protective mechanisms of the first phase of IPC are better understood in cardiac muscle (Liu et al., 1991; Alkhulaifi et al., 1993). One of the primary candidates is adenosine. Liu et al. (1991) suggested that endogenous adenosine mediates IPC in cardiac muscle (see also Yao and Gross, 1994), since IPC was blocked by adenosine A1 receptor antagonists. Also, IPC protection was prolonged by the adenosine regulating agent acadesine, 5-amino-4-imidazolecarboxamide riboside (AICAR), which increases local levels of adenosine (Tsuchida et al., 1993; Gruver et al., 1994; Tsuchida et al., 1994).
Evidence for the involvement of adenosine in brain in this rapid phase of preconditioning is supported by in vitro studies (Pérez-Pinzón et al., 1996). In these studies, evoked potentials (EPs) recovered better in hippocampal slices after a 2-min preconditioning anoxia (similar to the in vivo paradigm in the present study). Transient superfusion with adenosine or 2-chloroadenosine (an adenosine A1 receptor agonist) 30 min prior to test anoxia (as in preconditioning) also improved the recovery of EPs (Pérez-Pinzón et al., 1996). In addition, EP recovery was lessened by 8-cyclopentyl-1,3-dipropylxanthine (an adenosine A1 receptor antagonist) when it was superfused during conditioning anoxia.
Additional support for the role of adenosine during rapid IPC in brain are findings in adult rats that transient infusion of 1 mg/kg cyclopentyl adenosine (an adenosine A1 receptor agonist) only 15 min prior to global cerebral ischemia produced 70% protection of CA1 cells after 3 days of reperfusion (Heurteaux et al., 1995).
Although protection against acute ischemia was very robust in the present study, we observed that protection was asymmetric. The left side of the brain was better protected than the right side. One explanation for the asymmetric ischemic cell count found here could be that spreading depression (SD) from the opening of the burr hole or other manipulations was provoked. This is supported by Kobayashi et al. (1995), who showed that SD protected brain from ischemia. Perhaps the opening of the burr hole in the left hemisphere provoked a wave of SD. When the potassium electrode was placed on the brain, however, very low potassium values were found. Thus, if there was a wave of SD, it was of short duration. Therefore, SD is an unlikely candidate since, in the study of Kobayashi et al., SD was induced for 2 h and ischemia produced 24 h later. Also, that study alluded to the induction of certain genes by SD as a putative mechanism for neuroprotection. In our study, ischemia was induced shortly after the end of the surgical procedures, and gene expression takes many hours.
Another phase in which an SD-like event took place in these experiments was during ischemic insults. However, the rise of extracellular potassium during ischemia was global, i.e., expected to occur all over the brain in this model of global ischemia. Therefore, no asymmetry is expected from AD per se.
It is possible, therefore, that other factors, such as an increase in blood flow in the left hemisphere due to mild trauma (during surgical manipulations), may have partially protected this area. However, since no significant differences were found in control experiments between the left and right hemispheres, it is not expected that these differences significantly affected the results shown in these experiments.
Yet unknown, however, is whether the protection produced by rapid IPC is permanent. Results from the present study suggest that at least with this experimental paradigm (i.e., 30 min between insults), protection is not permanent. This is of concern since recent data indicate that neuronal protection can be temporary with some neuroprotective therapies (Dietrich et al., 1993). Data in the present study showed that IPC neuroprotection disappeared when the reperfusion period was extended to 7 days, indicating that this particular IPC paradigm only protects against acute ischemic neuronal death. Other postischemic events have been linked with this delayed neuronal damage and are independent of the initial insult (Dietrich et al., 1995). It is possible, therefore, that this rapid IPC only protects against the initial ischemic insult. However, it is important to define whether other IPC paradigms (e.g., different times of reperfusion between conditioning and test insults) protect against permanent neuronal damage.
The intensity of the test insult also plays a role. We found an interesting correlation between in vivo (present study) and in vitro (Pérez-Pinzón et al., 1996) studies of preconditioning when comparing insult intensity. For example, in hippocampal slices, preconditioning only protected slices when test anoxia lasted 1 min of AD, but not when the insult was extended to 2 min. Similarly, in the present study, when test ischemia was reduced to 7 min, better protection was observed. Interestingly, when preconditioning was induced with pharmacological agents in hippocampal slices, protection occurred in the 2-min anoxic insults (Pérez-Pinzón, et al. 1996). This suggests that anoxia/ischemia, per se, is associated with some unwanted side effects, which can be bypassed with these pharmacological agents.
In conclusion, the finding that IPC neuroprotection can be induced within 30 min is important because it suggests that strategies may be available for protection of the brain against acute neuronal damage. The extension of the therapeutic window for subsequent rescue of neuronal populations could be achieved with better understanding of the neuroprotective mechanisms involved in this rapid IPC model.
Footnotes
Acknowledgment:
These studies were supported by PHS grants NS 14325, NS 34773, NS 32167, and NS 05820, and a Grant-in-Aid from the American Heart Association. We thank Dr. Baowin Lin, Isabel Garcia, and Susan Kraydieh for technical support.