
Abstract
Preconditioning with sublethal ischemia induces tolerance to subsequent lethal ischemia in neurons. We investigated electrophysiologic aspects of the ischemic tolerance phenomenon in the gerbil hippocampus. Gerbils were subjected to 2 minutes of forebrain ischemia (preconditioning ischemia). Some of them were subjected to a subsequent 5 minutes of forebrain ischemia 2 to 3 days after the preconditioning ischemia (double ischemia). Hippocampal slices were prepared from these gerbils subjected to the preconditioning or double ischemia, and field excitatory postsynaptic potentials were recorded from CA1 pyramidal neurons. Capacity for long-term potentiation triggered by tetanic stimulation (tetanic LTP) was transiently inhibited 1 to 2 days after the double ischemia but then recovered. Latency of anoxic depolarization was not significantly different between slices from preconditioned gerbils and those from sham-operated gerbils when these slices were subjected to in vitro anoxia. Postanoxic potentiation of N-methyl-D-aspartate (NMDA) receptor-mediated transmission (anoxic LTP) was inhibited in slices from gerbils 2 to 3 days after the preconditioning ischemia, whereas it was observed in slices from sham-operated gerbils and gerbils 9 days after the preconditioning ischemia. These results suggest that protection by induced tolerance is (1) not only morphologic but also functional, and (2) expressed in inhibiting postischemic overactivation of NMDA receptor-mediated synaptic responses.
Keywords
Brief ischemia, which is not lethal in itself, induces tolerance to subsequent lethal ischemia in neurons (Kitagawa et al., 1990; Kirino et al., 1991). This phenomenon was first found in pyramidal neurons of the hippocampal CA1 sector using a forebrain ischemia model in gerbils. Subsequently, ischemic tolerance phenomenon was found in various other regions of the brain using the same gerbil model and also was found in rat models of global and focal cerebral ischemia (Kitagawa et al., 1991; Liu et al., 1992; Simon et al., 1993; Glazier et al., 1994). In the previous study by Kirino et al. (1991), a significant decrease in morphologic damage of CA1 neurons was demonstrated in gerbil hippocampus 7 days after 5 minutes of forebrain ischemia when gerbils had been preconditioned with 2 minutes of ischemia 2 to 4 days beforehand. Protein synthesis and binding of various neurotransmitters were preserved even after lethal ischemia when gerbils had been preconditioned (Kato et al., 1992a, Nakagomi et al., 1993; Furuta et al., 1993). Although these results suggested the possibility that preconditioning ischemia rescued CA1 neurons from lethal ischemia, it remains undetermined whether surviving CA1 neurons properly function. An activity-dependent change in synaptic efficacy in neuronal circuits of the brain has been considered as an electrophysiologic background of memory and learning. It is well-known that brief tetanic stimulation to the input fibers induces a long-term potentiation (LTP) of excitatory postsynaptic response in CA1 neurons in the hippocampal slice preparation (Bliss and Collingridge, 1993). We previously reported loss of LTP in CA1 neurons a few days after 5 minutes of ischemia in gerbil hippocampus (Kirino et al., 1992; Tsubokawa et al., 1992). In the present study we examined capacity for LTP in hippocampal slices prepared from gerbils that were subjected to double ischemia. The objective of the experiment was to evaluate the functional recovery of hippocampal CA1 neurons protected by the preconditioning ischemia.
The mechanisms by which the preconditioning ischemia induces tolerance and the induced tolerance protects neurons are not fully understood. An important role of stress proteins such as heat shock proteins (HSP) and ubiquitin has been suggested (Kirino, et al., 1991; Aoki et al., 1993a, 1993b; Kato et al., 1993; Liu et al., 1993; Nishi et al., 1993). Expression of HSP70 seems essential in ischemic tolerance since tolerance disappeared by the procedures which counteracted HSP70 (Nakata et al., 1993). However, other studies have demonstrated that the involvement of HSP, at least of HSP72, was less likely (Kato et al., 1992c; Abe et al., 1995; Kitagawa et al., 1995), whereas the role of other HSP and ubiquitin in ischemic tolerance remains elusive. Other genetic, biochemical, and pharmacologic studies suggested the possible involvement of a variety of substances, such as superoxide dismutase (Ohtsuki et al., 1992; Kato et al., 1995), adenosine and ATP-sensitive K+ channels (Heurteaux et al., 1995), the oncogene bcl-2 (Shimazaki et al., 1994), and interleukin-1 (Ohtsuki et al., 1996). However, little work has been done on the electrophysiologic properties in neurons that have acquired tolerance. It is known that when oxygen is deprived from hippocampal slices, anoxic depolarization occurs in the CA1 area. Latency from oxygen deprivation to anoxic depolarization generally correlates reversely with intensity of neuronal damage (Somjen et al., 1990, Haddad and Jiang, 1993). If preconditioning ischemia protects neurons from damage by affecting energy metabolism or membrane properties in the early phase of anoxic or ischemia injury, the latency of anoxic depolarization could change in hippocampal slices from preconditioned gerbils. Therefore, we compared latencies of anoxic depolarization during oxygen deprivation in hippocampal slices that were prepared from preconditioned and sham-operated gerbils. Another electrophysiologic property associated with neuronal vulnerability is anoxic LTP, which was found in in vitro anoxia using the hippocampal slice preparation. Deprivation of oxygen from hippocampal slices resulted in a selective long-lasting potentiation of N-methyl-
MATERIALS AND METHODS
Animal treatments
Forebrain ischemia was produced in gerbils similarly to the previous morphologic studies from our laboratory (Kirino, et al., 1991; Nakagomi, et al., 1993). Briefly, male Mongolian gerbils (Meriones unguiculatus, 60 to 80 g) were subjected to preconditioning ischemia (2 minutes) in an awake condition using Tone's method (Tone et al., 1987). A nylon thread, which had been looped around the carotid arteries under halothane anesthesia on the previous day, was gently pulled and the bilateral carotid arteries were occluded. The thread was only removed in the sham-operated group. In the double ischemia group, gerbils were anesthetized with halothane 2 or 3 days after the preconditioning ischemia and the carotid arteries were exposed and occluded for 5 minutes with Sugita aneurysm clips. During the operation, the cranial temperature was regulated at 37.5° ± 0.3°C using a heating pad and lamp.
Hippocampal slice preparation and electrophysiologic recording
Transverse sections (300 to 400 μm) were prepared by a previously described method (Kirino et al., 1992; Tsubokawa et al., 1992). The slices were maintained at 34° to 35°C, and were perfused (0.5 to 1 mL/min) with an artificial cerebrospinal fluid (ACSF) containing (in mmol/L) NaCl, 124; NaHCO3, 26; KCl, 5; KH2PO4, 1.24; CaCl2, 2.4; MgSO4, 1.3; and glucose, 10; The ACSF was equilibrated with 95% O2 and 5% CO2. Slices were kept in an interface chamber for at least 2 hours before experiments. Field excitatory postsynaptic potentials (fEPSP) were recorded extracellularly from the stratum radiatum of the CA1 region with a glass electrode filled with the ACSF (1 to 2 MΩ). Direct current (DC) potentials were recorded simultaneously using the same electrode. Schaffer collateral and commissural afferents were stimulated (0.05 to 0.08 Hz) with bipolar platinum-iridium electrodes (tip diameter of 10 μm) placed in the stratum radiatum. In experiments of tetanic LTP and anoxic LTP, fEPSP were continuously monitored before and after tetanic stimulation and in vitro anoxia, respectively. Records were digitized at 10 kHz and analyzed by an on-line computer to measure the maximal rate of the initial rise of fEPSP. The computed slope values were displayed as a constantly updated time series, permitting the time course of synaptic potentiation to be monitored accurately during experiments. In experiments to examine anoxic depolarization and anoxic LTP, slices were subjected to in vitro anoxia. The perfusate was transiently changed to the anoxic-aglycemic medium containing 10 mmol/L sucrose instead of 10 mmol/L glucose, equilibrated with 95% N2 and 5% CO2. For examination of tetanic LTP and anoxic LTP, experiments were planned so that gerbils from a single group were not prepared consecutively, i.e, gerbils from different groups were analyzed in random order to avoid interexperimental differences.
Tetanic long-term potentiation
For the experiment evaluating changes in tetanic LTP after ischemia, hippocampal slice preparations were made from four normal control gerbils (2 to 5 slices from each gerbil for a total of 17 slices), five gerbils 2 to 3 days after the preconditioning ischemia (4 to 5 slices from each gerbil, total 21 slices), five gerbils 1 to 2 days after the double ischemia (3 to 5 slices from each gerbil, total 20 slices), and eight gerbils 7 to 8 days after the double ischemia (2 to 9 slices from each gerbil, total 34 slices). After obtaining stable recording of fEPSP for at least 10 minutes, tetanic stimulation, which consisted of two trains of 100 pulses at 100 Hz with an interval of 20 seconds, was given to Schaffer collateral and commissural afferents using the same bipolar electrodes for the baseline stimulation. Monitoring of fEPSP was continued for at least 30 minutes after tetanic stimulation.
Latency of anoxic depolarization
Hippocampal slice preparations were made from five gerbils 2 days after the sham operation and five gerbils 2 days after the preconditioning ischemia. Two slices from each group were placed in a single chamber, and DC potentials were continuously recorded from both slices (30 pairs). After obtaining stable recording of DC potentials, the perfusate was changed from the ACSF to the anoxic-aglycemic medium. The latencies from the start of in vitro anoxia to anoxic depolarization were recorded in both slices. The perfusate was changed back to the normal ACSF a few minutes after the onset of anoxic depolarization in both slices or otherwise at 10 minutes after the initiation of in vitro anoxia.
Anoxic long-term potentiation
Hippocampal slice preparations were made from 13 normal control gerbils, 8 gerbils 2 to 3 days after the sham operation, 11 gerbils 2 to 3 days after the preconditioning ischemia, and 5 gerbils 9 days after the preconditioning ischemia (1 to 4 slices from each gerbil). Slices were first perfused with the ACSF for 2 hours and then the CA1 region was isolated from the CA3 by a knife-cut to prevent spread of bursting activity from the latter regions. Slices were then perfused for at least 30 minutes with a modified ACSF containing 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 15 μmol/L), bicuculline (10 μmol/L), and a low concentration of MgSO4 (0.1 mmol/L) to completely block
Statistical analysis
Data are presented as means ± SD. Comparisons of percentages of slices exhibiting tetanic or anoxic LTP (LTP-positive slices) in each animal in four groups were made using one-way analysis of variance followed by Scheffé's F test. Percentages of LTP-positive slices to all slices in each group were also compared using χ2 test for independence. Comparison of latencies of anoxic depolarization was made using the unpaired Student's t test.
RESULTS
Capacity for long-term potentiation is inhibited 1 to 2 days after double ischemia but later recovers
Typical synaptic responses before and after tetanic stimulation in hippocampal slices from gerbils in each group are shown in Fig. 1. In slices from normal control gerbils, tetanic stimulation of Schaffer collateral or commissural fibers induced a potentiation of fEPSP in the dendritic field of the CA1 region. Long-lasting synaptic potentiation was manifested as an increase in maximum rate of the initial rise of fEPSP, which was more than 150% and lasted for at least 30 minutes (Fig. 1A). This type of robust LTP was observed in 76.5% of the examined slices prepared from normal control gerbils. Similar potentiation of fEPSP after tetanic stimulation (Fig. 1B) was observed in 81.0% of the examined slices prepared from gerbils 2 to 3 days after the preconditioning ischemia. In contrast, in the slices prepared from gerbils 1 to 2 days after the double ischemia, tetanic stimulation failed to induce robust potentiation of fEPSP. The increase in the initial slope of fEPSP in these slices, if any, was less than 150% or did not last as long as that observed in slices prepared from normal control gerbils (Fig. 1C). Robust LTP was detected in only 15.8% of the examined slices prepared from gerbils in this group. However, 71.9% of the slices from gerbils 7 to 8 days after double ischemia again manifested robust potentiation of fEPSP after tetanic stimulation (Fig. 1D). The difference in the percentages of slices that exhibited robust LTP was statistically significant between normal control gerbils and gerbils 1 to 2 days after double ischemia. It was also significant between gerbils 1 to 2 days after the double ischemia and gerbils 7 to 8 days after the double ischemia, but was not significant between normal control gerbils and gerbils 7 to 8 days after the double ischemia (Fig. 2).
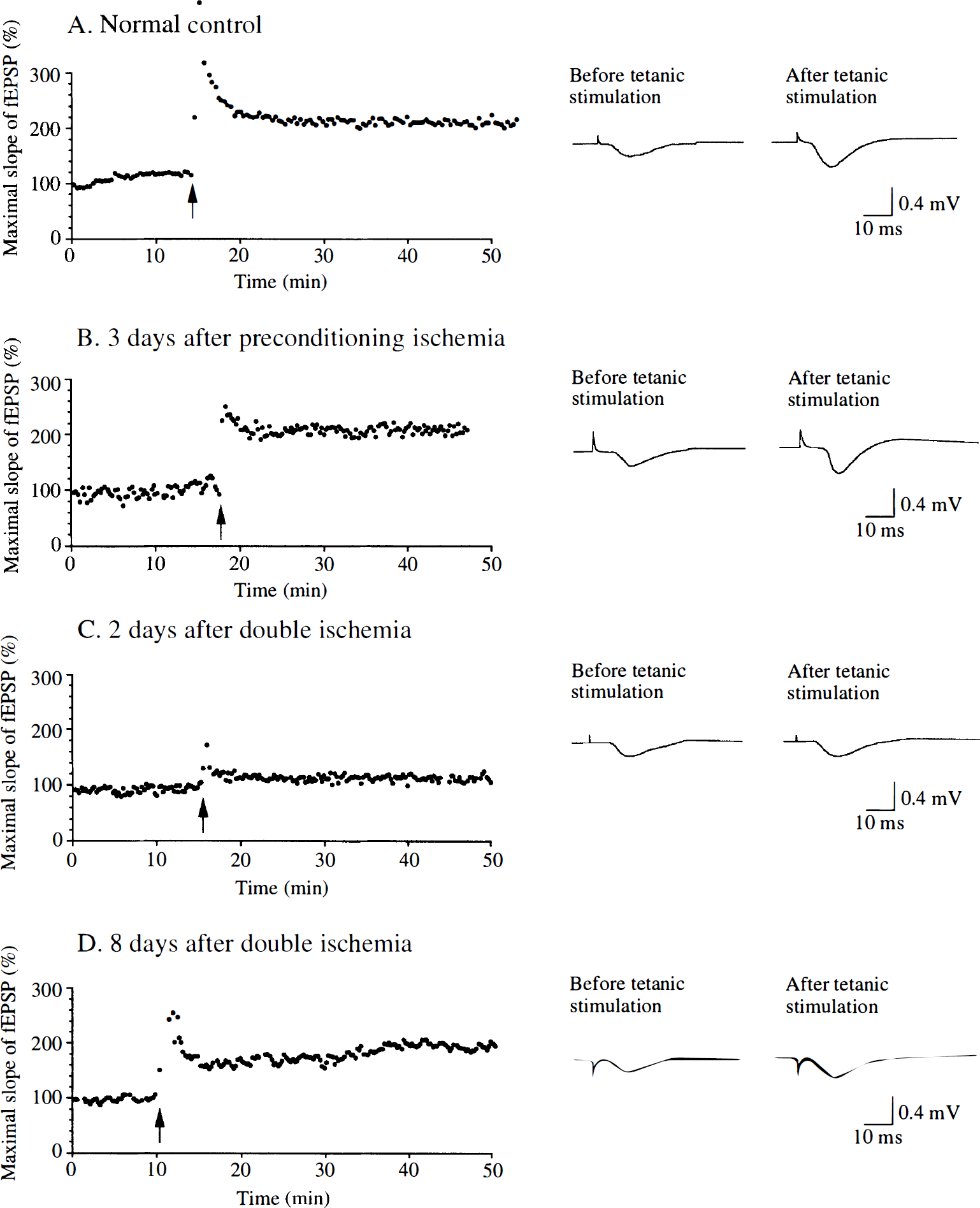
Representative recordings of continuous monitoring of the initial slope of field excitatory postsynaptic potentials (fEPSP) extracellularly recorded from the stratum radiatum of the CA1 region in hippocampal slices. Schaffer collateral and commissural afferents were stimulated (0.05 to 0.08 Hz) and fEPSP were continuously monitored before and after brief tetanic stimulation (arrows). Insets are specimen records of fEPSP taken before and 30 minutes after tetanic stimulation. Long-term potentiation (LTP) of fEPSP was observed in hippocampal slices prepared from normal control gerbils (
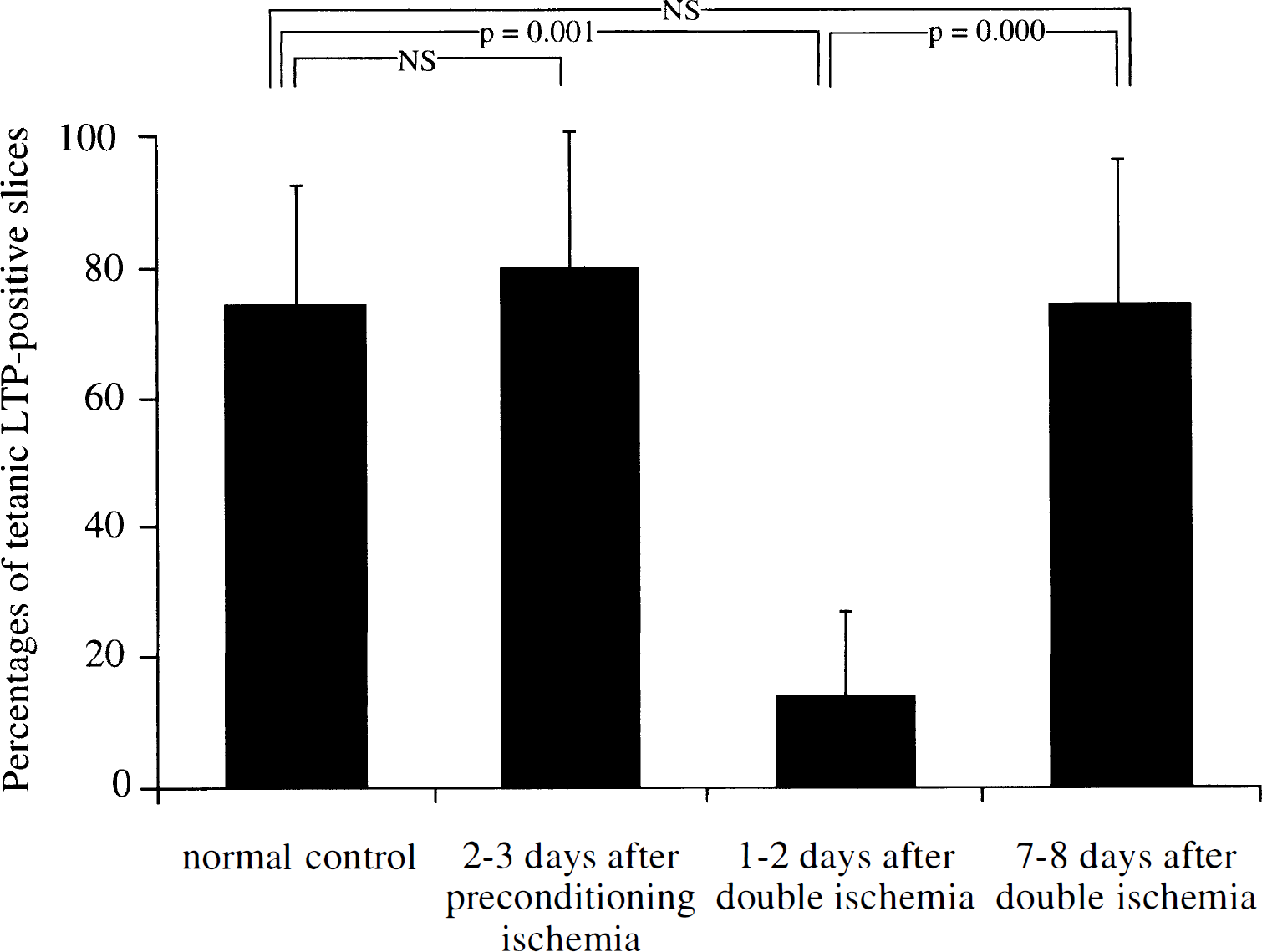
Bar graphs showing the percentages of hippocampal slices that exhibited robust LTP triggered by tetanic stimulation. Hippocampal slices were prepared from four normal control gerbils (14 slices), five gerbils 2 to 3 days after the preconditioning ischemia (21 slices), five gerbils 1 to 2 days after the double ischemia (20 slices), and eight gerbils 7 to 8 days after the double ischemia (34 slices). The percentage of slices that exhibited tetanic LTP in gerbils 1 to 2 days after the double ischemia was significantly smaller than those in normal control gerbils and in sham-operated gerbils.
Preconditioning ischemia does not change latency of anoxic depolarization
In most hippocampal slices, DC potential suddenly decreased by 5 to 10 mV within 5 minutes after changing the perfusate to the anoxic-aglycemic medium (Fig. 3A). In a small number of slices, anoxic depolarization was not observed during in vitro anoxia for as long as 10 minutes. Thus, in 22 of 30 pairs of slices from sham-operated gerbils and preconditioned gerbils, anoxic depolarization was observed in both slices of the pair. In the 22 pairs, anoxic depolarization occurred earlier in a slice from a sham-operated gerbil in 14 pairs and earlier in a slice from a preconditioned gerbil in 8 pairs of slices. When the mean latencies of anoxic depolarization were calculated in slices that exhibited anoxic depolarization within 10 minutes, they were 2.88 ± 1.90 minutes (28 slices) in the sham-operated group and 3.00 ± 1.85 minutes (24 slices) in the preconditioned group, which were not significantly different (Fig. 3B). The amplitudes of the anoxic depolarization in the two groups were 19 ± 7 and 20 ± 7 mV, which were also not significantly different.
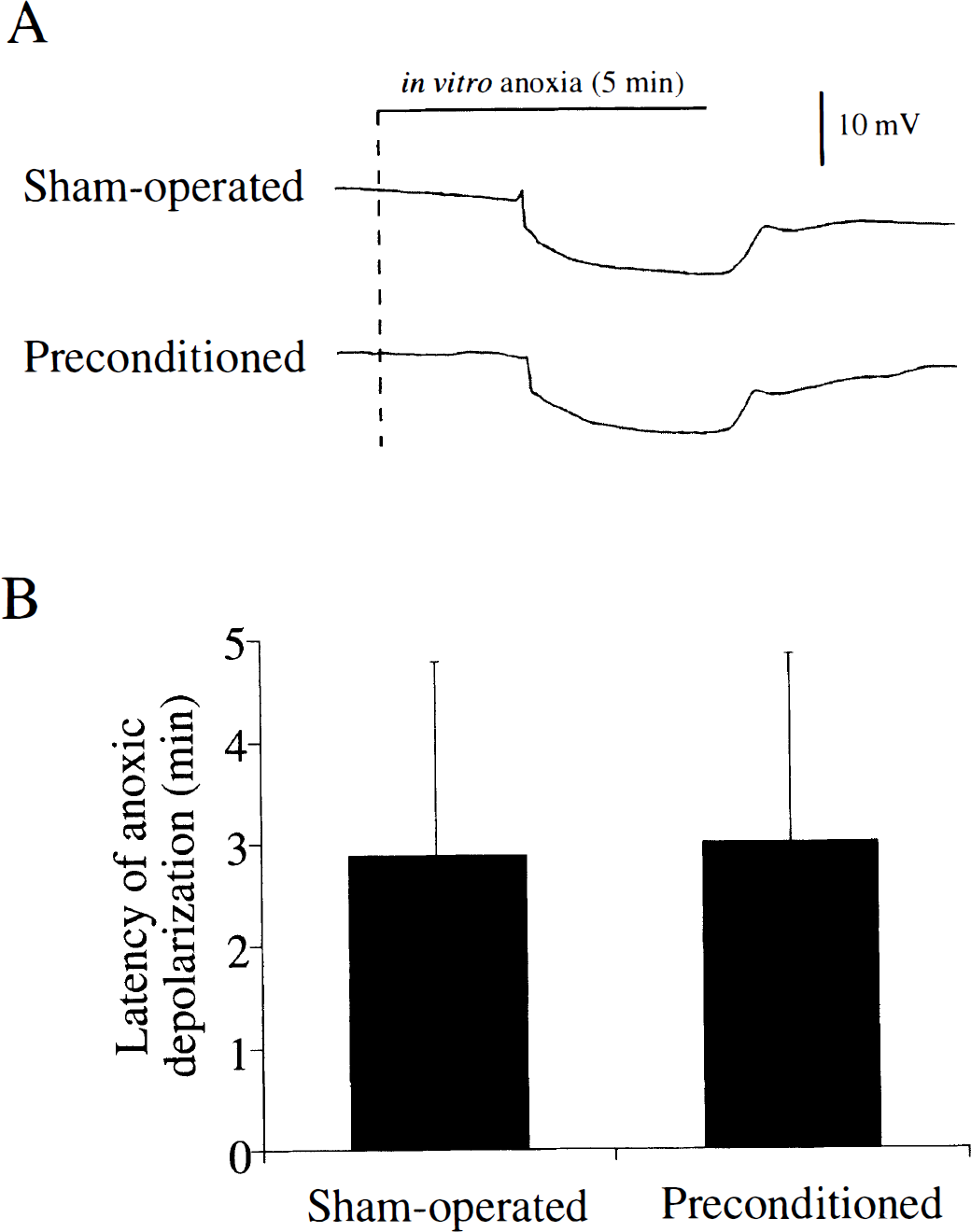
(
Preconditioning ischemia inhibits anoxic long-term potentiation
Changing the perfusate from the normal ACSF to the modified ACSF containing CNQX and low Mg2+ resulted in NMDA-mediated fEPSP that were characterized by a long duration and a slower decay. It was reported that a brief period of in vitro anoxia resulted in subsequent long-lasting potentiation of NMDA-mediated fEPSP (Crepel et al., 1993b). We observed this type of anoxic LTP in the slices from normal control gerbils. A brief period of in vitro anoxia induced a sudden decrease and then disappearance of fEPSP. Changing the medium back to the oxygenated modified ACSF resulted in a gradual recovery followed by long-lasting potentiation of fEPSP (anoxic LTP) (Fig. 4A). However, it was somewhat more difficult to observe anoxic LTP in the modified ACSF than to observe tetanic LTP in normal ACSF. In normal ACSF, recording of fEPSP and induction of tetanic LTP was possible as long as 10 hours after preparation of slices. In modified ACSF, it was difficult to obtain a stable series of fEPSP if slices were incubated longer than 6 hours. Therefore, we examined only slices with an incubation time shorter than 6 hours (maximal 4 slices from one gerbil). Anoxic LTP was observed in 51.4% of the examined slices prepared from normal control gerbils. Similarly, anoxic LTP was observed in 60.9% of slices prepared from gerbils 2 to 3 days after the sham operation (typical recording shown in Fig. 4B). However, anoxic LTP was rarely observed in slices prepared from gerbils 2 to 3 days after the preconditioning ischemia. Although fEPSP gradually recovered after in vitro anoxia in these slices, they never reached the level observed before in vitro anoxia (Fig. 4C). Only 11.1% of slices prepared from gerbils 2 to 3 days after the preconditioning ischemia exhibited anoxic LTP. In contrast, slices prepared from gerbils 9 days after preconditioning ischemia again showed anoxic LTP (Fig. 4D). Anoxic LTP was observed in 53.8% of slices from gerbils in this group. The percentage of slices that exhibited anoxic LTP in gerbils 2 to 3 days after preconditioning was significantly smaller than those in normal control gerbils and in sham-operated gerbils (Fig. 5). The ratio of LTP-positive slices to total slices in gerbils 9 days after preconditioning was greater than that in gerbils 2 to 3 days after preconditioning (χ2 test for independence, P = 0.004).
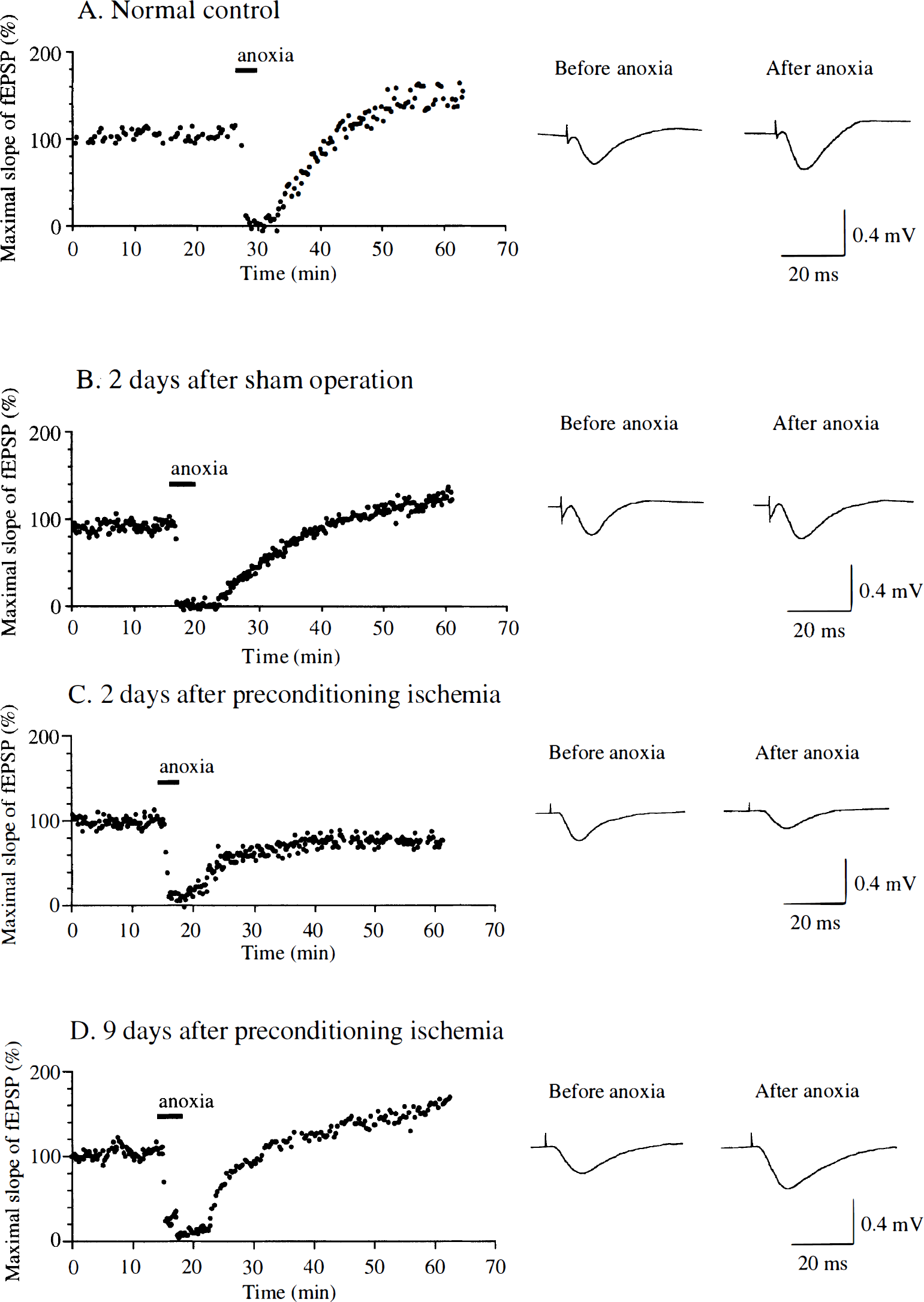
Representative recordings of continuous monitoring of the initial slope of fEPSP extracellularly recorded from the stratum radiatum of the CA1 region in hippocampal slices prepared from normal control gerbils (
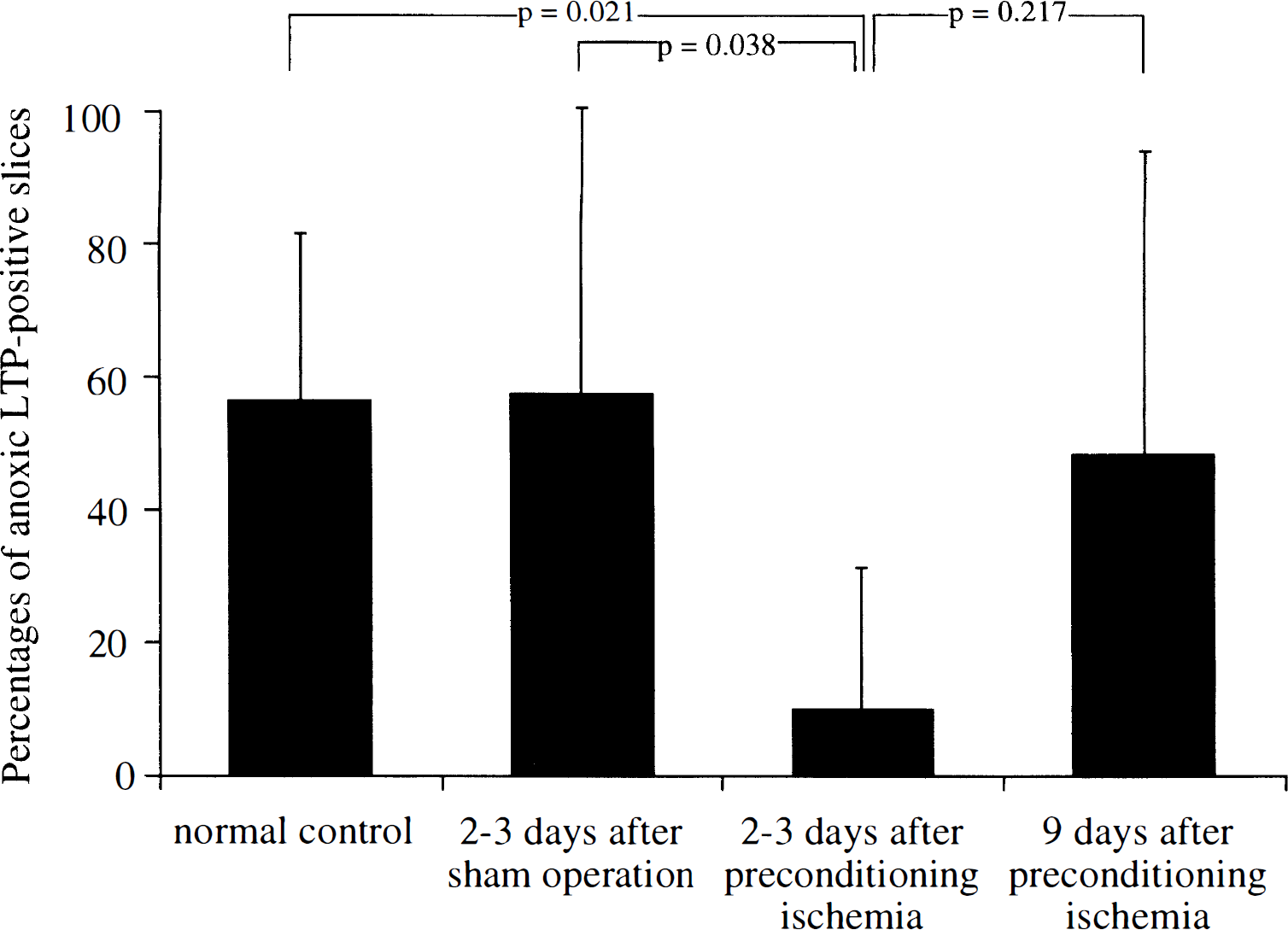
Bar graphs showing the percentages of hippocampal slices that exhibited anoxic LTP. Hippocampal slices were prepared from 13 normal control gerbils (35 slices), 8 gerbils 2 to 3 days after the sham operation (23 slices), 11 gerbils 2 to 3 days after the preconditioning ischemia (27 slices), and 5 gerbils 9 days after the preconditioning ischemia (13 slices). The percentage of slices that exhibited anoxic LTP in gerbils 2 to 3 days after preconditioning was significantly smaller than those in normal control gerbils and in sham-operated gerbils. The difference in the percentages of anoxic LTP-positive slices between gerbils 2 to 3 days after preconditioning and gerbils 9 days after preconditioning was not statistically significant although it was significant when the percentages of anoxic LTP-positive slices to all slices in each group were compared (χ2 test for independence, P = 0.004).
DISCUSSION
We previously reported that a majority of CA1 pyramidal neurons lost the capacity for tetanic LTP at 7 to 30 hours after a single 5 minutes of ischemia. Then 4 to 5 days after ischemia, no synaptic potential could be recorded from CA1 neurons (Kirino, et al., 1992), concurrent with histologic observation of neuronal loss (Kirino, 1982). In contrast, we found that the capacity for tetanic LTP in CA1 neurons was preserved 7 to 8 days after 5 minutes of ischemia when gerbils had been preconditioned with 2 minutes of ischemia. It is remarkable that the capacity for LTP in CA1 pyramidal neurons decreased at 1 to 2 days after the double ischemia but it recovered several days later (Fig. 1 and Fig. 2). The electrophysiologic signs of recovery are in line with the results of our previous morphologic study using the same gerbil model of ischemic tolerance (Kirino, et al., 1991). Electrophysiologic experiments using brain slices may not completely reflect the in vivo environment. This is especially true in recirculation phases after ischemia. The capacity for LTP should ultimately be examined in an in vivo study by electrophysiologic method. However, comparison of our previous and present findings in hippocampal slices suggested that ischemic tolerance acquired by preconditioning ischemia results in not only morphologic preservation of CA1 neurons but also functional recovery after subsequent lethal ischemia.
The role of intracellular signal transduction has been emphasized in the mechanism of LTP. The protease calpain, phosphatases such as calcineurin, phospholipases, and protein kinases have been implicated in a process of expression of LTP (Oliver et al., 1989; Ikegami et al., 1996; Malenka et al., 1989). These calcium-dependent enzymes are activated by elevation of intracellular Ca2+ via not only activation of NMDA channels but also calcium release from internal stores triggered by inositol 1,4,5-triphosphate (IP3). Interestingly, IP3 binding in the CA1 area showed a transient reduction at 1 day and then recovered to a control level at 7 days after the double ischemia (Kato et al., 1992b). The similar postischemic time course of the changes in IP3 binding and the capacity for LTP, along with the essential role of IP3 in expression of LTP, suggests that transient inhibition and subsequent recovery of the capacity for LTP after the double ischemia was mediated by postischemic alterations in second-messenger systems.
The mechanism by which acquired tolerance protects neurons is still controversial. Involvement of a variety of potential neuroprotectants has been suggested, such as HSP70 (Kirino et al., 1991; Aoki et al., 1993a; Aoki et al., 1993b; Liu et al., 1993; Nishi et al., 1993; Glazier et al., 1994), ubiquitin (Kato et al., 1993), superoxide dismutase (Ohtsuki et al., 1992; Kato et al., 1995), the oncogene bcl-2 (Shimazaki et al., 1994), and interleukin-1 (Ohtsuki et al., 1996). It appeared that those neuroprotectants induced by the preconditioning ischemia do not reduce the severity of the second lethal ischemia per se but exert their protective effects by acting on the subsequent cascade of degenerative processes. This is supported by the observation that the preconditioning ischemia did not alter the amount of excitatory and inhibitory neurotransmitters released during the second ischemia (Nakata et al., 1992).
The initial major event in the processes of ischemic neuronal injury is a rapid and large depolarization concomitant with breakdown of ion homeostasis (Haddad and Jiang, 1993). Latency of anoxic depolarization generally correlates inversely with intensity of neuronal damage because the earlier depolarization of neurons results in the longer exposure to the disturbed cellular environment (Somjen et al., 1990). Indeed, hypothermia, which has a strong protective effect on ischemic neurons (Busto et al., 1987), delays anoxic depolarization (Katsura et al., 1992). In contrast, in the ischemic tolerance paradigm, we found that the preconditioning ischemia did not change latency of anoxic depolarization. Again, our observation in hippocampal slices may not reflect the in vivo situation because latency of anoxic depolarization may be affected by extracellular concentration of potassium and glucose, which is different from the in vivo condition. However, latency ischemic depolarization recorded in vivo during the second ischemia was similarly not affected by preconditioning ischemia in the same gerbil model (Abe and Nowak, personal communication). Thus, it was indicated that acquired tolerance does not reduce the impact in the very initial phase of the second lethal ischemia per se.
A recent pharmacologic study revealed that activation of ATP-sensitive K+ channels (KATP channels) is necessary (but not sufficient) for the expression of ischemic tolerance (Heurteaux et al., 1995). These channels are normally inhibited by physiologic levels of ATP. A decrease in cytosolic ATP concentration opens these channels and leads to membrane hyperpolarization. The authors speculated that adenosine released during the preconditioning ischemia activates KATP channels via adenosine A1 receptors and protects against the second ischemia by either inhibiting presynaptic glutamate release (Amoroso et al., 1990; Ben-Ari et al., 1990) or decreasing glutamate toxicity because of postsynaptic hyperpolarization (Mourre et al., 1989). However, it seems difficult to explain with this hypothesis the delayed time course of tolerance induction in the brain (Kato, et al., 1991), and is unlikely in ischemic tolerance observed in cardiac myocytes (Murry et al., 1986; Gross and Auchampach, 1992). It also cannot explain the observations of Nakata et al. (1992) and of our present study that the preconditioning ischemia did not change latency of anoxic depolarization and neurotransmitter release. The exact role of KATP channels in ischemic tolerance in the brain remains to be elucidated.
Although the preconditioning ischemia did not change latency of anoxic depolarization, it inhibited anoxic LTP in hippocampal slices prepared during the period of tolerance expression. The observation indicates that the preconditioning ischemia affected the response of NMDA receptors to anoxia in a delayed fashion. In view of the significant roles of NMDA receptors and elevation of intracellular calcium in ischemic neuronal injury (Choi, 1990), it is speculated that inhibition of postanoxic long-lasting overactivation of NMDA receptor-mediated responses may attenuate subsequent neuronal damage. Although the exact role of anoxic LTP in in vivo ischemia has not been fully determined, similar increases in NMDA receptor-mediated responses and calcium uptake were observed in the in vivo condition (Andiné et al., 1988, 1992) or in hippocampal slices prepared after in vivo ischemia (Urban et al., 1990; Hori and Carpenter, 1994). Thus, induced tolerance may protect neurons by preventing postischemic overactivation of NMDA receptors and reducing elevation of intracellular calcium concentration. Experiments with direct visualization of intracellular calcium concentration in hippocampal slices with or without prior in vivo preconditioning ischemia may be a possible experimental paradigm to further support the speculation. The role of Ca2+-permeable AMPA receptors in the process of neuronal injury after a single 5 minutes of ischemia has also been reported (Tsubokawa et al., 1994). The possibility that receptors other than NMDA receptors are involved in the mechanisms of tolerance cannot fully be excluded. However, from our results showing that LTP induction in the tolerance-acquired hippocampus was similar to that in the control (Fig. 1), it is unlikely that AMPA receptors were modified in the process of induction of tolerance.
Although the means by which transient anoxia and ischemia induce subsequent long-lasting overactivation of NMDA receptor-mediated responses is not fully elucidated, a mechanism mediated by the redox modulation of the NMDA receptors was suggested (Gozlan et al., 1994). NMDA receptor-mediated responses were enhanced by disulfide-reducing drugs such as dithiothreitol and decreased by thiol-oxidizing reagents such as 5,5′-dithiobis-2-nitrobenzoic acid (DTNB). Thus, reducing conditions during oxygen deprivation may potentiate NMDA receptor-mediated responses via the reduction of disulfide bonds of cysteine residues of the receptors (Hammond et al., 1994; Gozlan and Ben-Ari, 1995). Interestingly, anoxic LTP could not be generated in the presence of DTNB, although DTNB did not prevent tetanic LTP in physiologic conditions (Hammond et al., 1994). We observed that the capacity for tetanic LTP was preserved (Fig. 1B and Fig. 2) although anoxic LTP was inhibited during the period of tolerance expression (Fig. 4C and Fig. 5). It was suggested that induction and expression of ischemic tolerance may be associated with redox modulation of NMDA receptors. However, this is not necessarily the only possibility, because NMDA receptors are modulated by a variety of factors, such as pH, mechanical swelling of neurons, extracellular glycine, intracellular arachidonic acid, protein kinase C activity, activity of protein tyrosine kinases, and stability of the cytoskeleton (Hammond et al., 1994).
Footnotes
Abbreviations used
Acknowledgments
The authors thank Ms. Kayoko Matsumoto, Noriko Kishino, and Tomomi Iwasawa for their excellent technical assistance, and Dr. Thaddeus S. Nowak, Jr. for critically reading the manuscript.