
Abstract
Robust ischemic preconditioning has been shown in rodent brain, but there are concerns regarding the persistence of neuron protection. This issue was examined in rat hippocampus following 4-vessel occlusion (4-VO) ischemia, using DC shifts characteristic of ischemic depolarization to reproducibly define insult severity. Preconditioning ischemia producing 2 to 3.5 mins depolarization was followed at intervals of 2, 5, or 7 days by test insults of varied duration, after which CA1 counts were obtained at 1, 2, 4, or 12 weeks. Neuron loss in naive animals increased with depolarization time longer than 4 mins regardless of postischemic survival interval. Preconditioning 2, 5, or 7 days before test insults prolonged the injury threshold evaluated at 1 week survival to 15, 9, or 6 mins, respectively, showing robust protection and a rapid decay of the protected state. However, by 2 weeks survival after preconditioning at a 2-day interval, the injury threshold dramatically regressed from 15 to 9 mins. Thereafter protection remained relatively stable through 1 month, but slight progression of neuron injury was evident at 3 months. Inflammatory responses were seen in both naive and preconditioned hippocampi throughout this interval, appropriate to the extent of neuron injury. These studies show distinct components of transient and lasting protection after ischemic preconditioning. Finally, it was found that ischemic depolarization was delayed by approximately 1 min in optimally preconditioned rat hippocampus, in contrast to previous results in the gerbil, identifying one specific mechanism by which insult severity is reduced in this model.
Introduction
Neuroprotective effects of prior insults have been widely studied in a range of ischemia models, as recently reviewed (Dirnagl et al, 2003; Kirino, 2002). Preconditioning by transient global ischemia in vivo was initially reported in gerbil hippocampus (Kirino et al, 1991; Kitagawa et al, 1990). Rat models showed somewhat more consistent and quantitatively complete protection after test insults of moderate severity (Liu et al, 1992; Shamloo and Wieloch, 1999), although reproducibility in the gerbil model could be improved by application of repeated preconditioning insults (Kitagawa et al, 1990; Ohtsuki et al, 1996). More recent studies have used quantitative monitoring of DC potential shifts characteristic of ischemic depolarization to define insult severity, establishing optimal conditions for ischemic preconditioning in gerbil and rat (Abe and Nowak, 2004; Halaby et al, 2004).
Some studies have raised a fundamental concern that the protection found after ischemic preconditioning may not be permanent (Corbett and Crooks, 1997; Dowden and Corbett, 1999), although lasting protection has been specifically noted in other reports (Ohta et al, 1996; Shamloo and Wieloch, 1999). Results in a depolarization-monitored gerbil model provided preliminary evidence of striking injury progression between 1 week and 2 months after severe test insults (Abe and Nowak, 2004). In the present study, the relationship between post-priming delay and long-term CA1 neuron protection was evaluated with quantitative depolarization monitoring in the rat 4-vessel occlusion (4-VO) model. A detailed characterization of depolarization kinetics is also presented, identifying a significant species difference in the mechanisms contributing to induced tolerance that appears to account for the more robust protection typically reported for ischemic preconditioning in the rat.
Methods
Transient Forebrain Ischemia and Electrophysiologic Monitoring
Protocols were approved by the Animal Care and Use Committee, University of Tennessee, and performed in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals. Male Wistar rats (n = 138) (Hilltop Lab Animals, Scottdale, PA, USA) weighing 250 to 300 g were subjected to transient forebrain ischemia by 4-VO (Pulsinelli and Brierley, 1979; Pulsinelli and Duffy, 1983) with DC potential monitoring essentially as previously described (Abe and Nowak, 1996, 2004; Halaby et al, 2004). All surgical procedures were carried out under anesthesia with 1% to 2% halothane in 70% N2 and 30% O2. In brief, common carotid arteries were fitted with a teflon/silastic occluding device, vertebral arteries were electrocauterized at the level of the first vertebrae, and animals were allowed to recover from anesthesia. Following an overnight fast, the animals were again anesthetized and placed in a stereotaxic frame. Epidural and rectal temperature probes were positioned, and both were maintained at 37°C. Glass electrodes filled with 2 mol/L NaCl (1 to 10 MΩ) were placed in hippocampus (4 mm caudal, 3 mm lateral to bregma, 2.5 mm below the cortical surface) (Paxinos and Watson, 1982), and a chloridized silver reference electrode was placed subcutaneously. DC potential was amplified using an Electro 750 electrometer (World Precision Instruments, Sarasota, FL, USA), and recorded using a digital interface. Occluding devices were tightened to induce forebrain ischemia, and the duration of ischemic depolarization was determined in each hippocampus as an index of insult severity (Abe and Nowak, 1996, 2004; Gao et al, 1998). Based on prior experience with repolarization kinetics, occlusions were released approximately 90 secs before the targeted repolarization time in the hippocampus that had first depolarized. After recirculation, the electrodes were removed, surgical incisions were closed, and anesthesia was discontinued, after which rectal temperature continued to be controlled for 1.5 h. The identical procedure was used for second occlusions in studies involving repeated ischemic insults, at intervals of 2, 5, or 7 days as specified for individual experiments. Sham-operated animals underwent the same surgical procedures, including vertebral artery cauterization and electrode insertion, but without carotid artery occlusion.
Previous rat studies have rigorously investigated the depolarization requirements for optimal CA1 neuron protection by preconditioning insults at a survival interval of 1 week (Halaby et al, 2004) (EM Howard, M Ueda and TS Nowak Jr, manuscripts in preparation). These identified maximal protection after priming insults of 2 to 3.5 mins depolarization, consistent with the 2 to 3 mins window identified in a gerbil model (Abe and Nowak, 2004). Hippocampi that did not experience initial insults within this window were excluded from subsequent histopathological analysis.
Histopathology
Rats were anesthetized with ketamine/xylazine (87/13 mg/kg) and transcardially perfused with heparinized saline, followed by a fixative consisting of 37% formaldehyde: glacial acetic acid:methanol (1:1:8). Brains were postfixed for several days and embedded in paraffin, after which 8 μm sections were cut at the level of the dorsal hippocampus and stained with hematoxylin/eosin. Neurons in a 200 μm length of CA1 arc dorsal to the dentate gyrus at the level of medial habenula were counted under a × 50 objective by a masked investigator. Pyramidal layer densities (cells/mm) were calculated from counts of triplicate sections for each hippocampus. Phagocytic ‘rod’ cell transformation of microglia was graded in the same region of CA1 as follows: 0—none; 1—scattered foci; 2—diffusely evident; 3—generalized and prominent; 4—gross inflammatory infiltrate.
Statistical Analysis
Because hippocampal depolarization kinetics varied between hemispheres in the same animal, individual hippocampi were treated independently for statistical purposes. Comparisons of neuron survival and depolarization parameters for multiple groups involved analysis of variance followed by Scheffé F-test, with P<0.05 considered significant. Comparisons of depolarization kinetics in the same hippocampus during repeated occlusions used a paired t-test. Both tests were implemented in the program StatView 5.0 (SAS Institute, Inc., Cary, NC, USA).
RESULTS
Preconditioning at Long Survival Intervals
Depolarization thresholds for CA1 injury are compared for naive and preconditioned hippocampi at various survival intervals in Figure 1. When examined 1 week after the test occlusions, optimal preconditioning at an interval of 2 days resulted in a several-fold increase in injury threshold relative to that of naive hippocampi, but this protection was markedly reduced with a 5-day interval between priming and test insults. Protection was further attenuated with an interval of 7 days between priming and test insults. Sham preconditioning produced no detectable protection. The depolarization threshold for CA1 loss in naive hippocampi remained unchanged whether examined at 1, 2, 4, or 12 weeks after an ischemic insult, with essentially complete neuron loss after depolarizations of 7 mins or longer. However, a striking regression in injury threshold occurred in preconditioned hippocampi between 1 week and 2 weeks survival, particularly evident for the optimal 2-day preconditioning interval. The efficacy of preconditioning was compared quantitatively (Table 1) for those hippocampi in each group that experienced test insults in the range of 7 to 9 mins that produced consistent loss in naive animals (shaded bars in Figure 1). Groups of naive animals subjected to sham test insults were also evaluated at each survival interval. At 1 week there was a trend toward decreased cell number in all preconditioned groups, but this only reached statistical significance for the 7-day interval between priming and test insults. At 2 weeks survival, there was a significant decline in cell number in those animals tested 5 days after preconditioning, and such hippocampi showed further neuron loss at 1 month and 3 months that was again statistically significant at the later time point. A modest but significant loss of CA1 neurons was detected at 3 months in hippocampi optimally preconditioned with a 2-day interval between priming and test insults.
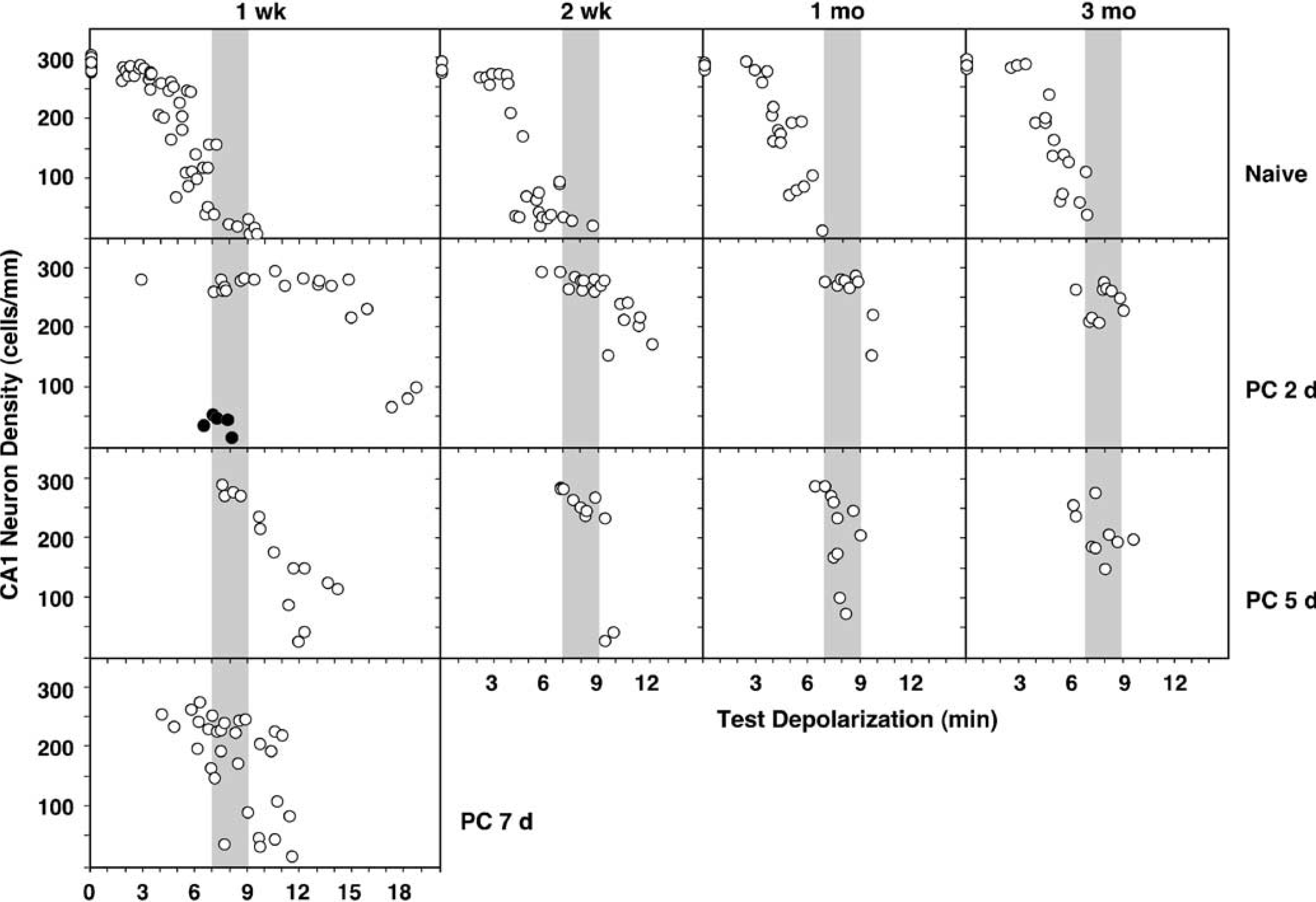
Depolarization thresholds for CA1 injury in naive and preconditioned rat hippocampus evaluated at prolonged survival intervals. Rats were initially subjected to occlusions producing 2 to 3.5 mins depolarizations that achieve optimal preconditioning (PC), to a sham procedure, or were used as untreated naive animals. Thresholds for CA1 injury were then determined after test insults of varied durations to naive animals or at intervals of 2, 5, or 7 days after preconditioning (open circles), or 2 days after sham pretreatment (closed circles), with subsequent histopathologic evaluation at intervals of 1, 2, 4, and 12 weeks. The injury threshold increased 2- to 3-fold 1 week after optimal preconditioning at a 2-day interval between priming and test insults, but became strikingly less effective as this interval was increased to 5 and 7 days. The injury threshold remained unchanged in naive animals even at long survival times, but a striking regression in the preconditioning effect was evident by 2 weeks in the preconditioned groups. There was some evidence of continuing cell loss at later intervals, as documented further in Table 1 for hippocampi that experienced insults in a defined range of 7 to 9 mins (shaded bars).
Effect of priming-challenge delay and survival interval on CA1 neuron preservation by ischemic preconditioning
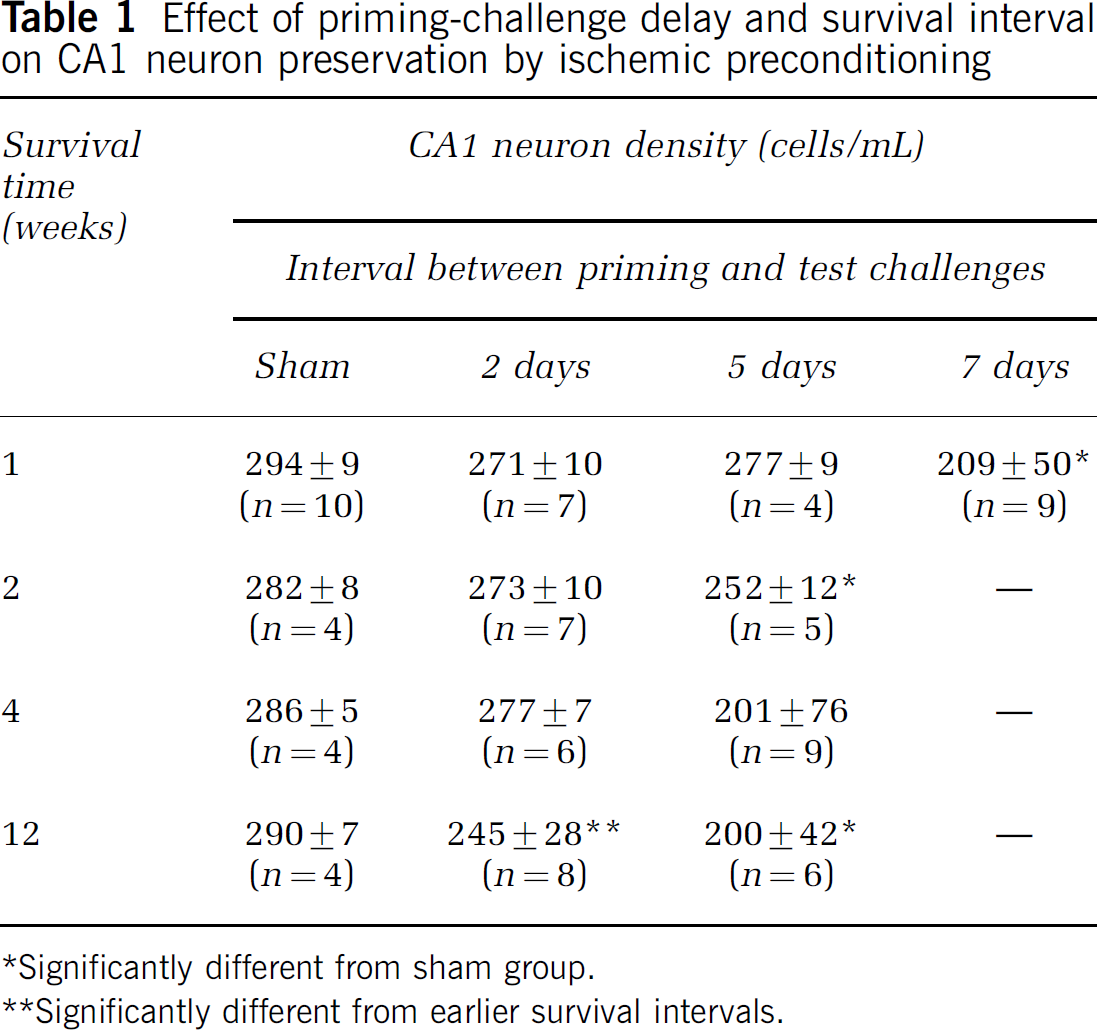
*Significantly different from sham group.
**Significantly different from earlier survival intervals.
Depolarization Kinetics During Priming and Test Insults
Representative records illustrate priming and test depolarizations for one hippocampus (Figure 4A), defining the parameters measured in these studies, and showing the characteristic lags between occlusion and depolarization, and between release and repolarization, respectively. Comparison of depolarization kinetics during priming and test insults revealed a significant shift in mean lag to depolarization onset (Figure 4B), and the delays in depolarization were comparable in groups evaluated at 2, 5 and 7 days intervals between insults (Figure 4C). Mean time to depolarization onset was 134±60 secs during priming occlusions and 203±102 secs during test occlusions (P<0.0001, n = 157, paired t-test). A separate concurrent study showed a mean lag to depolarization onset of 139±48 secs (n = 122) for naive hippocampi, 214±55 secs (n = 26) for a preconditioned group evaluated at 5 days (P<0.0001 versus naive, Scheffé F-test), and 148±46 secs (n = 30) for a sham-preconditioned group subjected to test ischemia at the same interval. The depolarization lag in the sham group did not differ from that of naive hippocampi (P = 0.655) and was significantly shorter than that observed for the preconditioned group (P<0.0001). This shows the consistency of depolarization delay after preconditioning, and establishes that prior exposure to the sham procedures of anesthesia, vertebral artery cauterization, carotid manipulation, and electrode insertion do not significantly impact the response to subsequent carotid artery occlusion.
Morphologic comparisons of selected hippocampi are illustrated in Figure 2. Preconditioned hippocampi subjected 2 days later to 9 and 13 mins test depolarizations, both showed well-preserved CA1 neurons at 1 week survival. A modest inflammatory response was detected after longer insults, evident as elongate ‘rod’ cells arrayed parallel to the apical dendrites of pyramidal neurons especially in the more vulnerable medial CA1 region, apparently secondary to subtle neuron loss. These were also seen in naive animals after single depolarizing insults corresponding to the longer end of the preconditioning range (3 to 3.5 mins), and became more prominent in both naive and preconditioned animals with increasing insult severity and associated neuron loss at all survival intervals through 1 month (Figure 3). Some attenuation of the response was evident by 3 months (not shown), and an optimally preconditioned hippocampus evaluated 12 weeks after a 9-min insult showed a normal-appearing pyramidal cell layer (Figure 2), although measured cell density was in the illustrated case reduced by 15%.
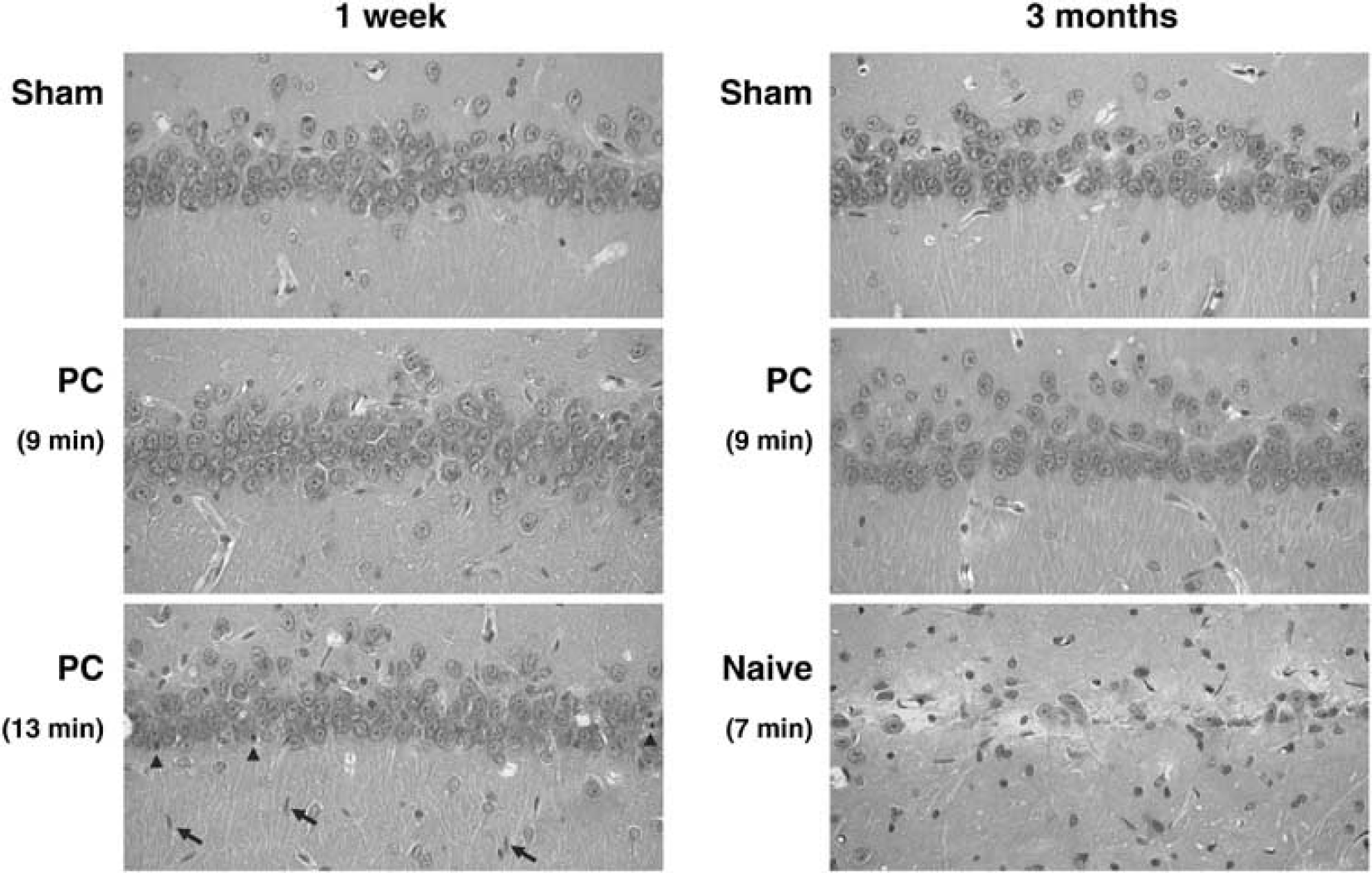
Morphologic evaluations of optimally preconditioned hippocampi after subsequent test insults. Rats were subjected to sham surgery, or to preconditioning ischemia (PC) followed 2 days later by test insults producing ischemic depolarizations of the durations indicated. Animals were perfusion fixed at 1 week or 3 months and paraffin sections were cut and stained with hematoxylin-eosin as described in the text. CA1 neurons were well preserved after 9 mins depolarizations at both survival intervals, illustrating the persistence of protection after test insults of moderate severity. Although neuron number was generally well preserved 1 week after the longer 13 mins insult, a small number of eosinophilic neurons could be detected (arrowheads) accompanied by activated microglia with rod cell morphology (arrows). Optimally preconditioned hippocampi exhibited normal CA1 morphology 3 months after test insults of moderate severity, under conditions of slight but significant decreases in pyramidal cell layer density (Table 1). Severe injury was evident in naive hippocampus subjected to comparable test insults.
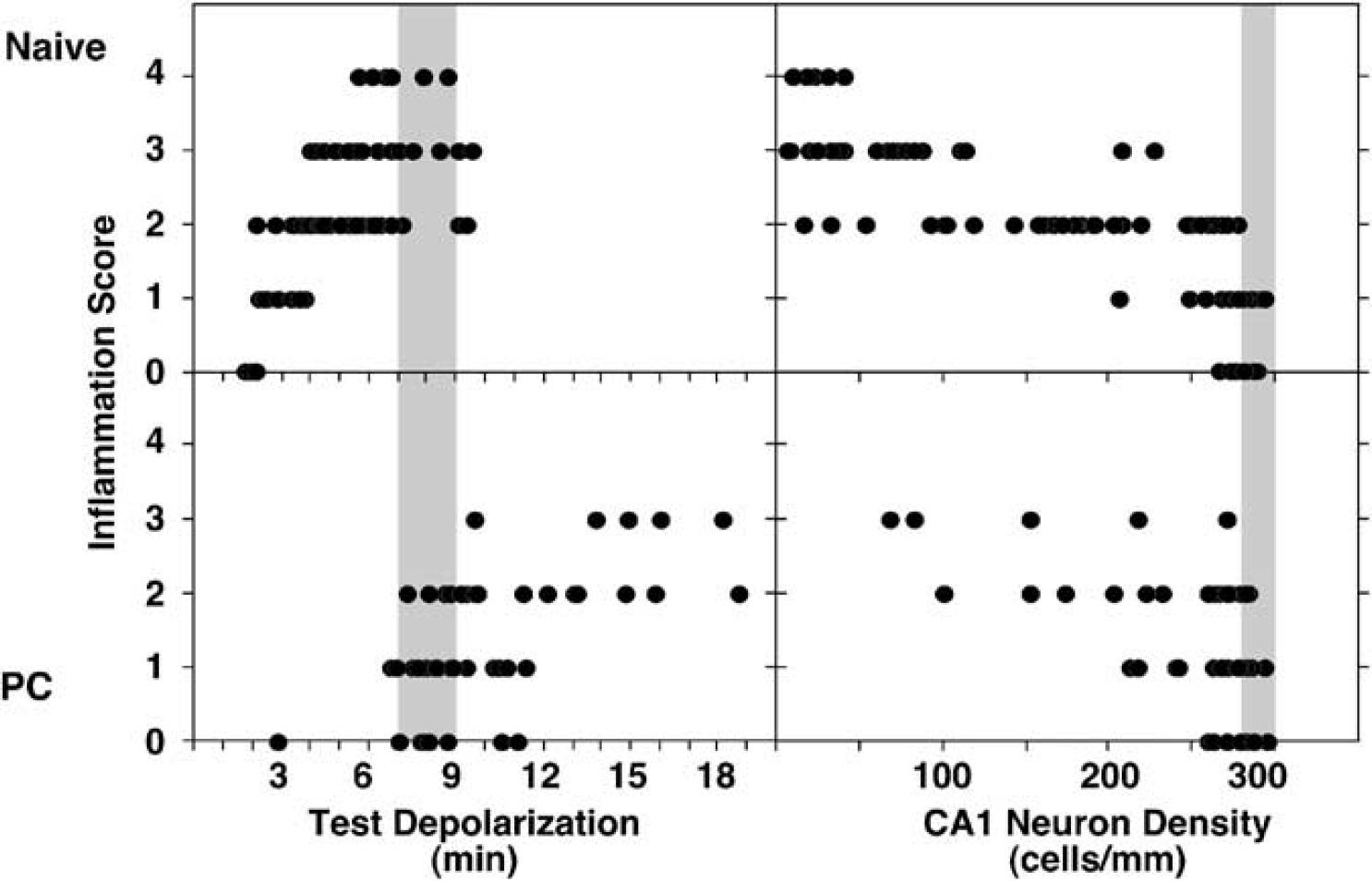
Inflammatory responses in naive and preconditioned hippocampi. (Left panels) Naive rats and animals subjected to optimal preconditioning at a 2-day interval (PC) were subjected to test depolarization of the indicated durations, and inflammation in the CA1 region was graded at 1, 2, and 4 weeks. Results were similar at these survival intervals and were pooled for presentation. Modest inflammation was consistently observed after 3 to 4 mins depolarizations in naive hippocampi and became maximal after insults that produced severe neuron loss (shaded bar). In contrast, preconditioned hippocampi showed increasing inflammation only as insult severity increased above the latter threshold. (Right panels) Inflammation in naive and preconditioned hippocampi showed a consistent relationship to CA1 neuron loss, with microglial activation becoming evident with even slight decreases in cell number below the control range (shaded bar).
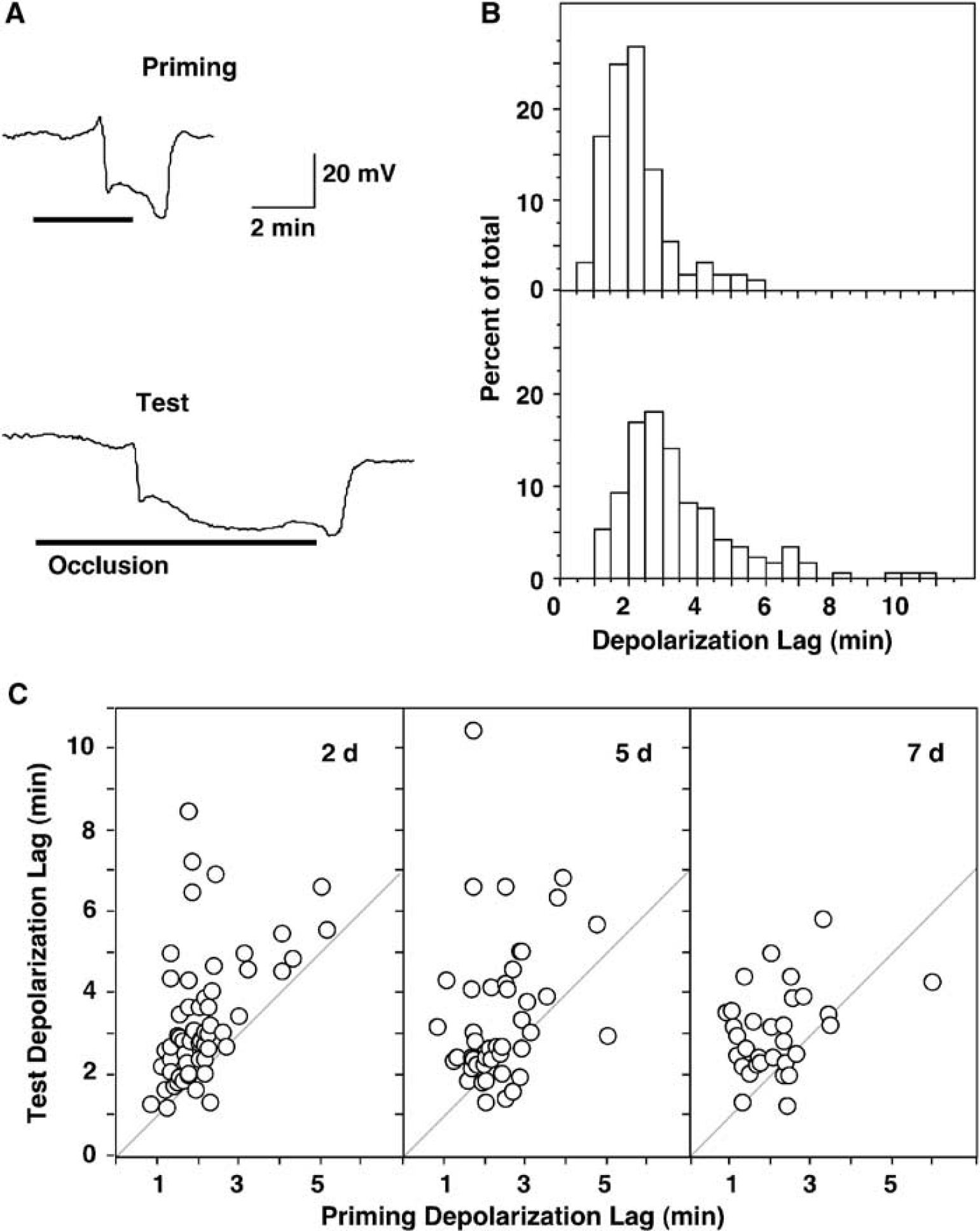
Depolarization kinetics in naive and preconditioned hippocampi. (
DISCUSSION
These studies show a clear component of lasting protection after optimal ischemic preconditioning in rat hippocampus, with an approximate doubling of the CA1 injury threshold persisting through 3 months. However, this represents a striking attenuation of protection relative to that observed at 1 week survival. Further, delayed depolarization in preconditioned hippocampus is concluded to contribute substantially to the protection observed in previous studies in which test insults were produced without depolarization monitoring.
Temporal Factors in Ischemic Injury and Preconditioning
Sham surgery and electrode placement alone have no long-term impact on neuron survival that could complicate the interpretation of the response to insults (Table 1). Naive animals subjected to transient ischemia exhibited a stable depolarization threshold for CA1 loss at all survival intervals evaluated in this study (Figure 1). The rate of ‘maturation’ of ischemic injury has been long recognized to vary with insult severity (Ito et al, 1975). However, the present results indicate that there is an absolute threshold for CA1 neuron loss of 3 to 4 mins depolarization, below which no appreciable damage will be evident even at prolonged recirculation intervals. Somewhat lower thresholds may be expected for medial CA1/subiculum (Iwai et al, 1995), while injury grades that incorporate less vulnerable lateral CA1 contribute to a flattening of the threshold relationship (Xu et al, 1999).
The most robust protection was evident at a 2-day interval between priming and test insults, with decreasing effect at 5 and 7 days (Figure 1, Table 1). This is generally consistent with the time course of preconditioning decay noted in previous rodent studies (Abe and Nowak, 2004; Kirino et al, 1991; Nishino and Nowak, 2004; Shamloo and Wieloch, 1999). However, the remarkable increase in the depolarization threshold for CA1 neuron loss in preconditioned hippocampi at 1 week showed a dramatic regression by 2 weeks survival after test challenges (Figure 1). This establishes that the more robust component of protection represents primarily a delay in cell loss, replicating preliminary observations obtained with identical methods in a gerbil model (Abe and Nowak, 2004), and rigorously confirming earlier indications of delayed insult progression after preconditioning (Corbett and Crooks, 1997; Dowden and Corbett, 1999).
These results also identify a component of relatively stable protection. For example, although the injury threshold detectably regressed beyond 2 weeks for test insults administered 5 days after priming ischemia, mean neuron counts did not differ at 1 and 3 months (Table 1). More importantly, nearly maximal CA1 preservation was observed through 3 months in hippocampi subjected to standard 7 to 9 mins test depolarizations administered 2 days after preconditioning. This is consistent with the persistent protection reported 2 months after test insults of comparable duration in other rat studies (Shamloo and Wieloch, 1999), and stable protection also has been noted after short test insults in the gerbil model (Abe and Nowak, 2004; Ohta et al, 1996). Nevertheless, the slight but significant decrement in CA1 survival at the longest interval examined (Table 1) raises concerns that even optimal preconditioning may not assure ‘permanent’ protection, and still longer term studies are required to address this issue.
Inflammation in Preconditioned Hippocampus
Scattered microglial activation to typical ‘rod’ cell morphology was seen after longer test insults to preconditioned hippocampi, in which gross CA1 neuron number was initially preserved (Figure 2). Well-recognized as a late stage in microglial reactivity (Brierley and Brown, 1982), this phagocytic transformation is typically observed after the onset of overt neuron loss after single ischemic insults (Kato et al, 1995; Morioka et al, 1993). In naive animals there was a common threshold of 3 to 4 mins depolarization for both inflammation and neuron loss (Figures 1 and 3). In contrast, the threshold for producing detectable inflammation was shifted to 7 to 8 mins after optimal preconditioning, and the relationship between inflammation and CA1 injury remained constant in naive and preconditioned hippocampi (Figure 3). Although arguments have been made for the potential contribution of inflammatory processes to injury progression after global ischemia (Yrjänheikki et al, 1998), the present results can be most straightforwardly interpreted to reflect secondary responses to subtle neuron loss. Mean CA1 neuron counts in optimally preconditioned hippocampi after moderate test insults (7 to 9 mins) were always lower than those of sham-operated hippocampi (Table 1), consistent with the slight but statistically significant decrease in neuron survival noted in gerbil hippocampus after optimal preconditioning (Abe and Nowak, 2004).
Changes in Depolarization Kinetics Associated with Preconditioning
Shorter depolarizations during test occlusions clearly favor more complete and more lasting protection (Figure 1). Delayed depolarization (Figure 4) is therefore a key contributor to ischemic tolerance in rat models using test insults defined by occlusion duration. The gerbil exhibits only slight (<10 secs) delay in the onset of depolarization during test insults after preconditioning (Abe and Nowak, 2004), and hippocampal slices from such animals likewise show no increase in depolarization latency (Kawai et al, 1998). In contrast, the rat 4-VO model shows a mean increase of more than 1 min in the lag between occlusion and depolarization. Recent results in another laboratory are in close agreement, indicating a 90-secs depolarization delay in preconditioned rat hippocampus (Y Takeda and H Harada, personal communication). For typical studies using test occlusions of 6–10 mins (Dave et al, 2001; Kato et al, 1995; Shamloo and Wieloch, 1999), delayed depolarization would therefore achieve at least 10% and perhaps as much as 25% reductions in effective insult severity. Interestingly, chemical preconditioning by in vivo pretreatment of gerbils with 3-nitropropionic acid was found to delay depolarization in hippocampal slices during subsequent hypoxia in vitro (Aketa et al, 2000), indicating the potential heterogeneity in factors contributing to protection across preconditioning treatments within a given species.
Mechanisms responsible for the observed depolarization delay remain to be identified. Potentiation of ATP-dependent potassium (KATP) channel activity would slow depolarization kinetics and be neuroprotective (Ben-Ari et al, 1990; Heurteaux et al, 1993; Lauritzen et al, 1997; Reshef et al, 1998; Sorimachi and Nowak, 2004; Wind et al, 1997), but this possibility has yet to be directly examined. Altered AMPA receptor subunit expression is debated as a factor in ischemic preconditioning (Sommer and Kiessling, 2002; Tanaka et al, 2002; Yamaguchi et al, 1999), but some evidence indicates that, in apparent contrast to the gerbil, preconditioning in the rat may increase expression of the Ca2+ impermeable, rapidly desensitizing GluR2 subunit (Alsbo et al, 2001; Kjøller and Diemer, 2000). Recent results have associated increased expression of uncoupling protein-2 (UCP-2) with neuroprotection (Diano et al, 2003; Mattiasson et al, 2003), but its potential impact on ischemic depolarization per se is unclear. Established global ischemia models are characterized by very low residual flow during occlusion (Kågström et al, 1983; Pulsinelli et al, 1982), but perfusion effects of preconditioning are also possible. Better preservation of CBF and ATP levels during global ischemia has been shown in rat cortex after several days of ipsilateral carotid artery occlusion (Bronner et al, 1998). Although primarily addressed in the context of focal ischemic insults, there is increasing appreciation of the role of nitric oxide (NO) produced by endothelial nitric oxide synthase (eNOS) to acutely maintain cerebral blood flow (CBF) during ischemia (Endres et al, 2004). Acute pharmacological manipulation of NO levels did not grossly affect the depth or duration of global ischemia under conditions of consistent blood pressure reduction in a model of 2-VO plus hypotension (Zhao et al, 1999). However, NOS inhibition has been reported to impair CBF during early reperfusion in a similar model (Yusa, 2000). There is some recent evidence for an increase in eNOS expression after preconditioning ischemia (Hashiguchi et al, 2004), but this was reported in a gerbil model, which as noted shows a negligible shift in depolarization kinetics (Abe and Nowak, 2004). Metabolic rate will clearly impact the kinetics of ischemic depolarization (Astrup et al, 1981; Nakashima et al, 1995; Xie et al, 1995), and decreased basal CBF was considered to reflect reduced metabolic rate after preconditioning by spreading depression in a rat focal ischemia model (Otori et al, 2003). Glucose utilization was attenuated by approximately 50% in rat cortex after topical KCl application that induced repeated spreading depressions, although a similar effect was also observed with NaCl treatment (Kawahara et al, 1999). Future studies should clearly consider more fully the impact of preconditioning treatments on metabolism and perfusion in rat brain.
Summary
These studies define significant limits to the protection that can be achieved in rat hippocampus after optimal ischemic preconditioning. Striking protection against even long (>10 mins) insults is evident at 1 week but rapidly degrades. Such ‘false preconditioning’ overtly confounds any study in which histopathology is evaluated earlier than several weeks after insult. Lasting protection remains evident through 3 months after test insults of moderate severity, amounting to a persistent doubling of the depolarization threshold for CA1 loss relative to naive animals. The efficacy of such ‘true preconditioning’ is relatively modest, and even this may not be permanent. The present results therefore identify a phenomenon of very delayed injury maturation in the preconditioned hippocampus, and further studies are needed to completely define the time course of protracted neuron death in such models. Finally, delayed depolarization was commonly observed in preconditioned rat hippocampus that does not occur in a comparable gerbil model. Although eliminated as a factor in the present experiments, delayed depolarization significantly confounds results based on occlusion time alone, and has undoubtedly contributed to protection in previous studies of ischemic preconditioning in rats. This may be considered a ‘pseudo-preconditioning’ since it reflects an attenuation of the effective insult severity during ischemia rather that an improved response to a consistent metabolic challenge, but if observed in human brain could nevertheless represent an important component of clinically relevant preconditioning effects. Together these findings provide a partial dissection of the multifactorial nature of neuroprotective mechanisms that operate after preconditioning ischemia.