
Abstract
Preclinical studies have identified numerous neuroprotective drugs that attenuate brain damage and improve functional outcome after cerebral ischemia. Despite this success in animal models, neuroprotective therapies in the clinical setting have been unsuccessful. Identification of biochemical markers common to preclinical and clinical cerebral ischemia will provide a more sensitive and objective measure of injury severity and outcome to facilitate clinical management and treatment. However, there are currently no effective biomarkers available for assessment of stroke. Nonerythroid αII-spectrin is a cytoskeletal protein that is cleaved by calpain and caspase-3 proteases to signature αII-spectrin breakdown products (αII-SBDPs) after cerebral ischemia in rodents. This investigation examined accumulation of calpain- and caspase-3-cleaved αII-SBDPs in cerebrospinal fluid (CSF) of rodents subjected to 2 hours of transient focal cerebral ischemia produced by middle cerebral artery occlusion (MCAO) followed by reperfusion. After MCAO injury, full-length αII-spectrin protein was decreased in brain tissue and increased in CSF from 24 to 72 hours after injury. Whereas αII-SBDPs were undetectable in sham-injured control animals, calpain but not caspase-3 specific αII-SBDPs were significantly increased in CSF after injury. However, caspase-3 αII-SBDPS were observed in CSF of some injured animals. These results indicate that αII-SBDPs detected in CSF after injury, particularly those mediated by calpain, may be useful diagnostic indicators of cerebral infarction that can provide important information about specific neurochemical events that have occurred in the brain after acute stroke.
Acute ischemic stroke is a significant international health concern representing a potentially catastrophic debilitating medical emergency with poor prognosis for long-term disability. With the exception of diuretics, supportive measures, and when appropriate, thrombolytic therapy with recombinant tissue plasminogen activator (tPA), there are currently no approved drug treatments for ischemic brain injury (Broderick and Hacke, 2002; Grotta, 2002; Lees, 2002). Although a number of neuroprotective drugs have proven effective in reducing infarct size or improving functional outcome in preclinical testing, none have proven successful in clinical trials (Gladstone et al., 2002; Kidwell et al., 2001). Differences between preclinical and clinical trial outcome with neuroprotective drugs in acute ischemic stroke may be caused by a variety of pitfalls that arise when attempting to extrapolate from animal to human investigations. These pitfalls may include differences in drug concentration and duration, differences in the window for therapeutic efficacy, differences in preclinical versus clinical trial design, and the lack of standardized and sensitive outcome measures (Gladstone et al., 2002; STAIR, 1999). For example, preclinical studies (typically in rodents) have traditionally used reduction of acute infarct volume as the primary measure of treatment efficacy, whereas clinical trials typically gauge treatment efficacy based upon neurologic or functional outcomes (Gladstone et al., 2002). One approach to address these discrepancies in outcome measures is for preclinical and clinical trial designs to use outcome measures that are common to both human acute ischemic stroke and to preclinical animal models of ischemia. The use of common biochemical markers may provide such an approach.
Unlike other organ-based diseases where rapid diagnosis using biomarkers (usually involving blood tests) proves invaluable to guide treatment of the disease, no such rapid and definitive diagnostic tests exist for acute ischemic brain injury. Biomarkers would have important applications in diagnosis, prognosis, and clinical research of ischemic brain injuries. Simple and rapid diagnostic tools will immensely facilitate allocation of the major medical resources required to treat acute ischemic brain injuries. Accurate diagnosis in acute care environments can significantly enhance decisions about patient management, including decisions about whether to admit or discharge patients or to administer other timeconsuming and expensive tests, including computed tomography (CT) and magnetic resonance imaging (MRI) scans. Biomarkers could provide major opportunities for the conduct of clinical research including confirmation of injury mechanism(s) and drug target identification. The temporal profile of changes in biomarkers could guide timing of treatment. Finally, biomarkers could provide a robust and sensitive clinical trial outcome measure that is obtainable more readily and with less expense than conventional neurologic assessments, thereby significantly reducing the risks and costs of human clinical trials.
Previously reported biomarkers of cerebral ischemia include neuron-specific enolase (NSE), brain specific creatine kinase enzyme (CPK-BB), S-100ß, and inflammatory cytokines such as IL-6 (Laskowitz et al., 1998). Of these, NSE and S-100ß have been the most studied. After cardiac arrest, NSE elevations in serum and cerebrospinal fluid (CSF) have been correlated with neurologic recovery (Dauberschmidt et al., 1991; Martens, 1996; Roine et al., 1989). Serum and CSF NSE values are reported to be elevated in rodent models of focal ischemia in proportion to the eventual infarct volume (Cunningham et al., 1991, 1996; Horn et al., 1995). In clinical trials, peak serum NSE values also predicted infarct volumes as shown by CT. However, correlating serum NSE values with functional outcome was less successful (Cunningham et al., 1991, 1996; Missler et al., 1997). Serum S-100ß protein levels are correlated with infracted brain volume in patients with stroke (Missler et al., 1997; Persson et al., 1987). However, recent studies indicate that serum levels of S-100ß may provide a better marker of blood-brain barrier (BBB) compromise than of parenchymal brain damage (Kanner et al., 1997). Similarly, S-100ß protein has been studied most extensively for characterization of ischemic injuries after cardiac surgery and several reports have documented postoperative serum elevations (Sellman et al., 1992; Westaby et al., 1996). However, many of these reports do not include careful studies of neurologic outcome, and several investigators have recently criticized the diagnostic utility of S-100ß during cardiac surgery (Anderson et al., 2001). Thus there is clearly a need for development of better biochemical markers for use in evaluating ischemic brain injury.
Our research efforts to develop biomarkers for traumatic brain injury (TBI) and acute ischemic brain injury have focused on αII-spectrin metabolic products as prototypical biochemical markers (Pike et al., 2001; Ringger et al., 2002). αII-spectrin is the major structural component of the cortical membrane cytoskeleton and is particularly abundant in axons and presynaptic terminals (Goodman et al., 1995; Riederer et al., 1986). It is important to note that αII-spectrin is a major substrate for both calpain and caspase-3 cysteine proteases (see Fig. 1), and the major calpain and caspase-3 cleavage sites of αII-spectrin have been well documented (Harris et al., 1988; Wang et al., 1998). Our laboratory has provided considerable evidence that αII-spectrin is processed by calpains or caspase-3 to signature cleavage products in vivo after TBI (Beer et al., 2000; Newcomb et al., 1997; Pike et al., 1998a, 2001) and in in vitro models of mechanical stretch injury (Pike et al., 2000), necrotic cell death (Zhao et al., 1999), apoptotic cell death (Pike et al., 1998b), and oxygen-glucose deprivation (Newcomb-Fernandez et al., 2001). Calpain and caspase-3 proteases also cleave αII-spectrin to signature proteolytic fragments in the brain in a rodent model of transient forebrain ischemia (Zhang et al., 2002). Although we have generated considerable laboratory data on the usefulness of αII-spectrin degradation as a biomarker for TBI in rodents (Pike et al., 2001), and more recently with preliminary data in human TBI patients (d'Avella et al., 2002), the present investigation is the first to provide evidence that calpain- and caspase-3-mediated αII-spectrin breakdown products (αII-SBDPs) can be detected in CSF after ischemic-reperfusion brain injury and can be used as biochemical markers in a rodent model of transient focal stroke in rats.
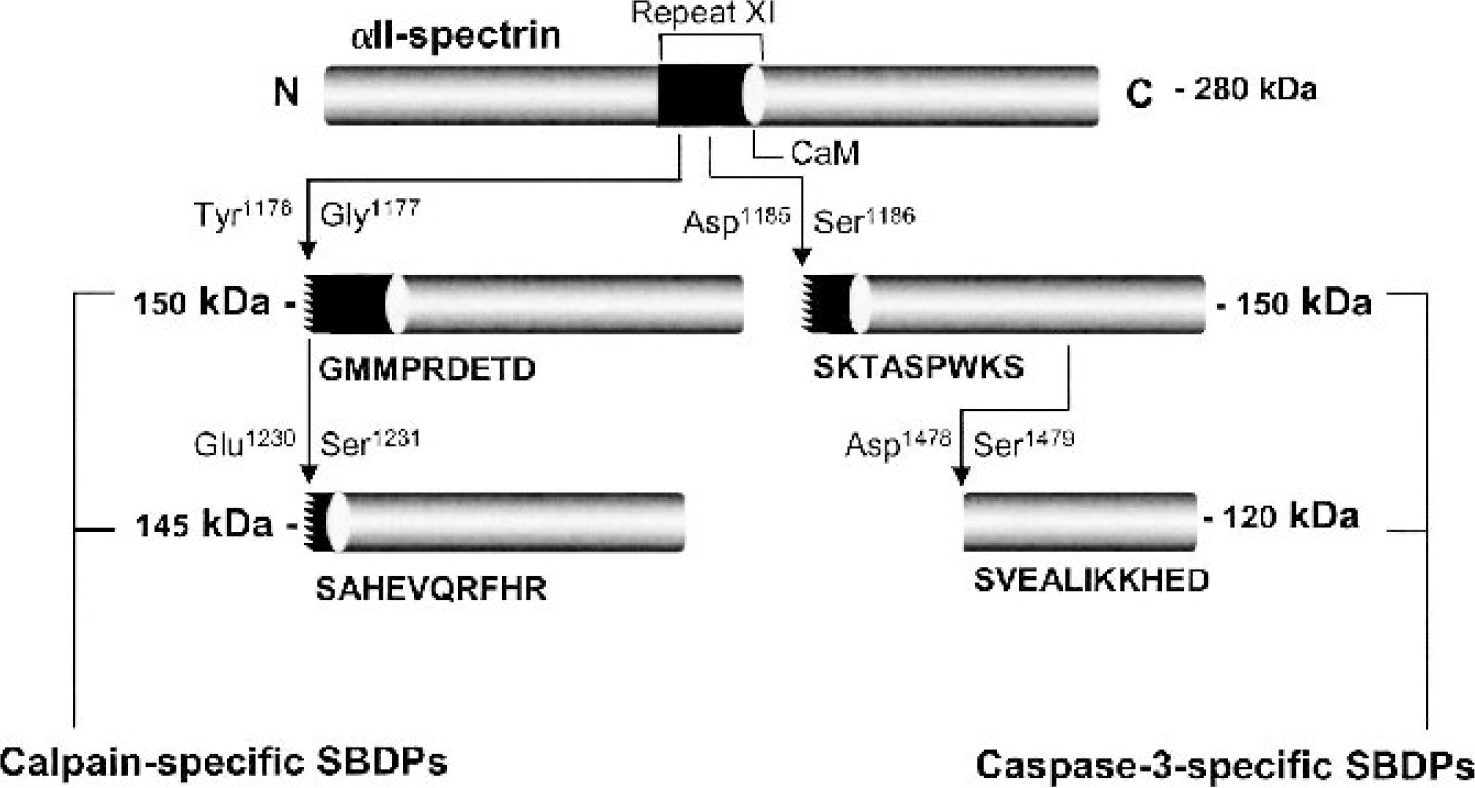
Calpain and caspase-3 cleavage of non-erythroid αII-spectrin to protease-specific αII-spectrin breakdown products (SBDPs). Shown here are the calpain cleavages (left) in αII-spectrin that result in calpain-specific SBDPs (150 and 145 kDa) and the caspase-3 cleavages (right) in αII-spectrin that result in caspase-3-specific αII-SBDPS (150 and 120 kDa). Both proteases cleave αII-spectrin in repeat 11 near the calmodulin-binding domain (CaM) to produce 150 kDa αII-SBDPs with unique N-terminal regions. A second cleavage by calpain in repeat 11 results in a calpain-specific 145 kDa αII-SBDP, whereas caspase-3 cleaves the protein in repeat 13 to produce a unique, apoptotic-specific 120 kDa fragment. For definitively identified cleavages, the flanking amino acids and initial N-terminal amino acid sequence is given.
METHODS
Surgical procedures and middle cerebral artery occlusion
A “noninvasive” filament method of MCAO occlusion used extensively by our laboratories (Berti et al., 2002; Williams et al., 2003) was used to produce cerebral ischemia in rats. The method described by Longa et al. (1989) and later modified in our laboratory by Britton et al. (1997) consists of blocking blood flow into the MCA with an intraluminal 3-0 monofilament nylon sterile suture with rounded tip introduced through an incision in the external carotid artery (ECA).
Under halothane anesthesia (5% halothane via induction chamber followed by 2% halothane via nose cone), the common carotid artery (CCA) was exposed at the level of external and internal carotid artery bifurcation with a midline neck incision. The internal carotid artery (ICA) was followed rostrally to the pterygopalatine branch and the ECA was ligated and cut at its lingual and maxillary branches. To prevent bleeding during suture insertion, the CCA and ICA were temporarily clamped with microaneurysm clips. The nylon suture was then introduced into the ICA via an incision on the ECA stump (the path of the suture can be monitored visually through the vessel wall) and advanced through the carotid canal approximately 20 mm from the carotid bifurcation until it became lodged in the narrowing of the anterior cerebral artery blocking the origin of the MCA. The skin incision was then closed using sterile autoclips. The endovascular suture remained in place for 2 hours, at which time the rat was briefly reanesthetized, and the suture filament was retracted to allow reperfusion. For sham MCAO surgeries, the same procedure was followed, but the filament was advanced only 10 mm beyond the internal-external carotid bifurcation and was left in place until the rat was killed. During all surgical procedures, animals were maintained at 37.0°C by a homeothermic heating blanket (Harvard Apparatus, Holliston, MA, U.S.A.).
After surgery, animals were placed in recovery cages with air temperature maintained at 22°C. During the 2-hour ischemia period and the initial 4-hour postreperfusion period, 75-watt warming lamps were positioned directly over the top of each cage to assist in maintaining normothermic body temperature throughout the experiment. It is important to note that at the conclusion of each experiment, rat brains showing pathologic evidence of subarachnoid hemorrhage upon necropsy were excluded from the study. Also, all rats exhibiting convulsant behaviors at any time after MCAO were excluded from the experiment.
Brain tissue and CSF collection
Brain (cortex and hippocampus) and CSF was collected from animals at various intervals after sham-injury or MCAO as previously described by our laboratory (Pike et al., 2001). At the appropriate time-points, MCAO or sham-injured animals were anesthetized as described above and secured in a stereotactic frame with the head allowed to move freely along the longitudinal axis. The head was flexed so that the external occipital protuberance in the neck was prominent and a dorsal midline incision was made over the cervical vertebrae and occiput. The atlanto-occipital membrane was exposed by blunt dissection and a 25G needle attached to polyethylene tubing was carefully lowered into the cisterna magna. Approximately 0.1 to 0.15 mL of CSF was collected from each rat. After CSF collection, animals were removed from the stereotactic frame and immediately killed by decapitation. Ipsilateral and contralateral (to the site of infarct) cortices were then rapidly dissected, rinsed in ice cold phosphate buffered saline (PBS), and snap frozen in liquid nitrogen. Cortices were excised to the level of the white matter and extended approximately 4 mm laterally and approximately 7 mm rostrocaudally. CSF samples were centrifuged at 4000 g for 4 min. at 4°C to clear any contaminating erythrocytes. Cleared CSF and frozen tissue samples were stored at −80°C until ready for use. Cortices were homogenized in a glass tube with a Teflon dounce pestle in 15 volumes of an ice-cold triple detergent lysis buffer (20 mM Hepes, 1 mM EDTA, 2 mM EGTA, 150 mM NaCl, 0.1% SDS, 1.0% IGEPAL 40, 0.5% deoxycholic acid, pH 7.5) containing a broad range protease inhibitor cocktail (Roche Molecular Biochemicals, cat. #1-836-145).
Immunoblot analyses of CSF and cortical tissues
Protein concentrations of tissue homogenates and CSF were determined by bicinchoninic acid microprotein assays (Pierce Inc., Rockford, IL, U.S.A.) with albumin standards. Proteinbalanced samples were prepared for sodium dodecyl sulfatepolyacrylamide gel electrophoresis (SDS-PAGE) in twofold loading buffer containing 0.25 M Tris (pH 6.8), 0.2 M DTT, 8% SDS, 0.02% bromophenol blue, and 20% glycerol in distilled H2O. Samples were heated for 10 minutes at 100°C and centrifuged for 1 minute at 8160g in a microcentrifuge at ambient temperature. Twenty micrograms of protein per lane were routinely resolved by SDS-PAGE on 6.5% Tris/glycine gels for 1 hour at 200 V. After electrophoresis, separated proteins were laterally transferred to polyvinylidene fluoride (PVDF) membranes in a transfer buffer containing 0.192 M glycine and 0.025 M Tris (pH 8.3) with 10% methanol at a constant voltage of 100 V for 1 hour at 4°C. Blots were blocked for 1 hour at ambient temperature in 5% nonfat milk in TBS and 0.05% Tween-20. Panceau Red (Sigma, St. Louis, MO, U.S.A.) was used to stain membranes to confirm successful transfer of protein and to insure that an equal amount of protein was loaded in each lane.
Antibodies and immunolabeling of PVDF membranes
Immunoblots containing brain or CSF protein were probed with an anti-αII-spectrin (fodrin) monoclonal antibody (FG 6090 Ab; clone AA6; cat. # FG 6090; Affiniti Research Products Limited, UK) that detects intact non erythroid αII-spectrin (280 kDa) and 150, 145, and 120 kDa cleavage fragments to αII-spectrin. A cleavage product of 150 kDa is initially produced by calpains or caspase-3 proteases (each proteolytic cleavage yields a unique amino-terminal region) (Nath et al., 1996; Wang et al., 1998) (Fig. 1). The calpain-generated 150 kDa product is further cleaved by calpain to yield a specific calpain signature product of 145 kDa (Harris et al., 1988; Nath et al., 1996), whereas the caspase-3 generated 150 kDa product is further cleaved by caspase-3 to yield an apoptotic-specific caspase-3 signature product of 120 kDa (Nath et al., 1998; Wang, 2000; Wang et al., 1998). After an overnight incubation at 4°C with the primary antibody (FG 6090 Ab, 1:4000 for brain tissue and 1:2000 for CSF), blots were incubated for 1 hour at ambient temperature in 3% nonfat milk that contained a horseradish peroxidase-conjugated goat anti-mouse IgG (1:10,000 dilution). Enhanced chemiluminescence (ECL, Amersham) reagents were used to visualize immunolabeling on Kodak Biomax ML chemiluminescent film.
Statistical analyses
Based upon our experience with the injury model and injury outcome, the sample size (n = 5 rats per group) was derived by power analysis (Kirk, 1982) for a four group comparison with α = 0.05, power of 1-β = 0.80, and a mean group difference of 2.5 standard deviation. Semiquantitative evaluation of protein levels detected by immunoblotting was performed by computer-assisted densitometric scanning (AlphaImager 2000 Digital Imaging System, San Leandro, CA, U.S.A.). Data were acquired as integrated densitometric values and transformed to percentages of the densitometric levels obtained on scans from sham-injured animals visualized on the same blot. To minimize between-film variability in exposure time, each immunoblot was run with two sham-injured control animals and two injured animals from each time point. Transformed data were evaluated by analysis of variance (ANOVA) and post hoc tests where mean density values for injured animals at each time point were compared to sham-injured control groups using Dunnett's Multiple Comparison Test. All values are given as mean ± SD. Differences were considered significant if P < 0.05. Note that multiple exposures that ranged in signal intensity level were used for every immunoblot. For quantification, immunoblots that provided the best separation between the 150 and 145 kDa bands were used. However, the blots used for quantification did not reproduce well when printed, and immunoblots with longer exposure times were used for illustrative purposes.
RESULTS
Proteolysis of αII-spectrin in the ipsilateral cortex by calpains and caspase-3 after MCAO injury
In the ipsilateral cortex, MCAO injury caused significant (P < 0.05) accumulation of the nonspecific 150 kDa αII-SBDP (generated by calpain and/or caspase-3) and of the calpain-specific 145 kDa αII-SBDP at all postinjury time points as compared with sham-injured control rats (Fig. 2). Levels of the calpain-specific 145 kDa αII-SBDP were 304%, 282%, and 301% of sham-injured control values at 24, 48, and 72 hours postinjury, respectively (Fig. 3). Levels of the nonspecific 150 kDa αII-SBDP closely matched levels of the 145 kDa fragment and were 276%, 275%, and 268% of sham-injured control values at 24, 48, and 72 hours, respectively (Fig. 3). As we and others have previously reported (Pike et al., 1998a; Zhang et al., 2002), background labeling of the 120 kDa band was detected in sham-injured control animals. This is likely caused by basal processing of αII-spectrin by caspase-3. MCAO injury resulted in a modest but significant (P < 0.05 to P < 0.001) increase above background levels of caspase-3-specific 120 kDa αII-SBDPs at all postinjury time points (Fig. 2). Levels of the caspase-3-specific 120 kDa fragment were 131%, 132%, and 140% of sham-injured control values at 24, 48, and 72 hours postinjury, respectively (Fig. 3). Although levels of the caspase-3-specific 120 kDa αII-SBDP were smaller than those produced by calpains, between-animal variability was much lower for levels of caspse-3 αII-SBDPs compared with the variability for levels of calpain αII-SBDPs.
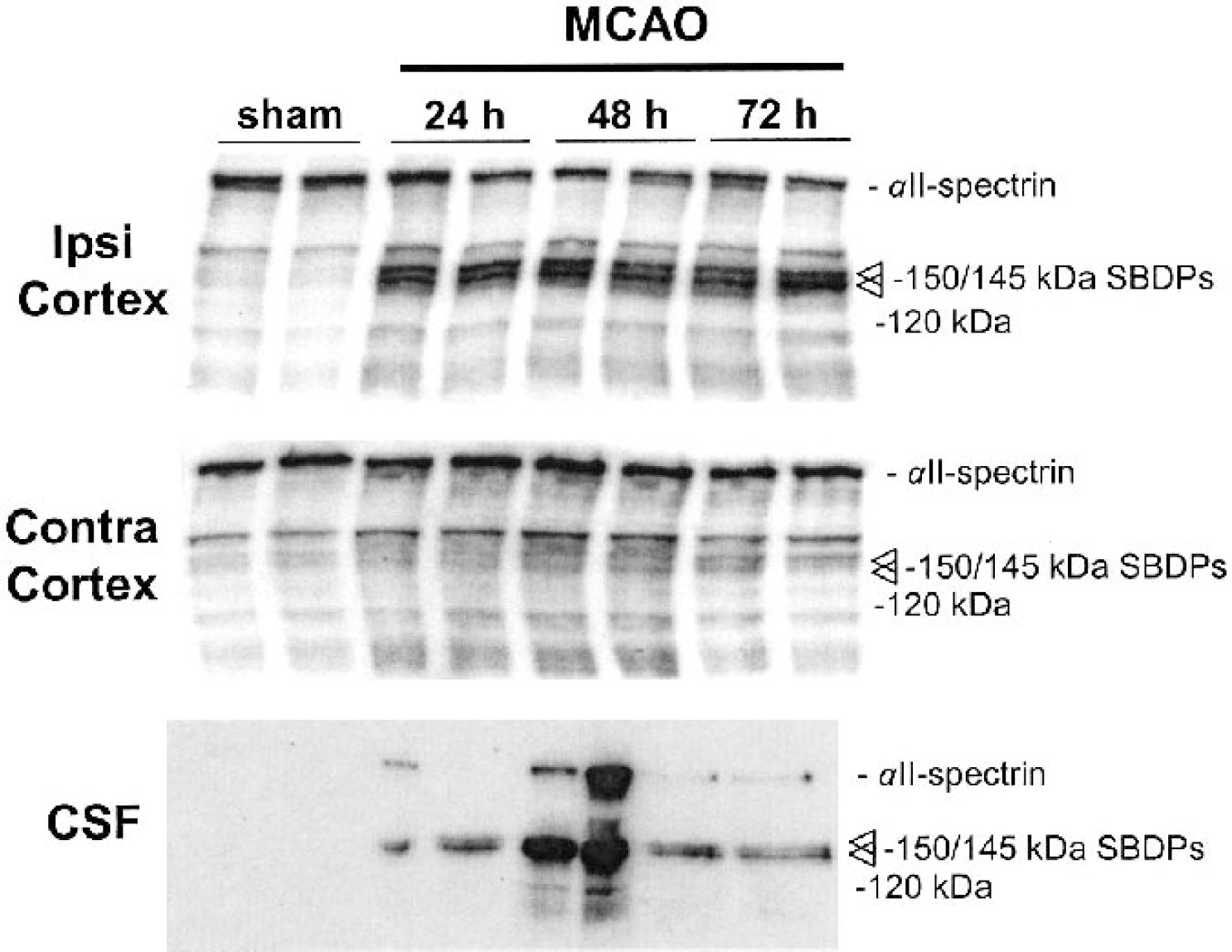
MCAO injury causes accumulation of full-length (II-spectrin (280 kDa) protein and calpain-mediated 145 kDa and caspase-3 mediated 120 kDa (II-SBDPs in CSF. MCAO resulted in proteolysis of constitutively expressed brain (II-spectrin (280 kDa) in ipsilateral but not contralateral cortex. The caspase-3-mediated, apoptotic-specific 120 kDa (II-SBDP was also increased in ipsilateral cortex after ischemia compared with sham-injured controls. Marked increases in the calpain-specific 145 kDa (II-SBDP were detected in brain and CSF of MCAO animals but not in sham-injured animals at all time points. It is of interest that whereas increased levels of the caspase-3-specific 120 kDa (II-SBDP were detected at all postinjury time points in the ipsilateral cortex, CSF levels were only detected at 48 hours postinjury. MCAO, middle cerebral artery occlusion.
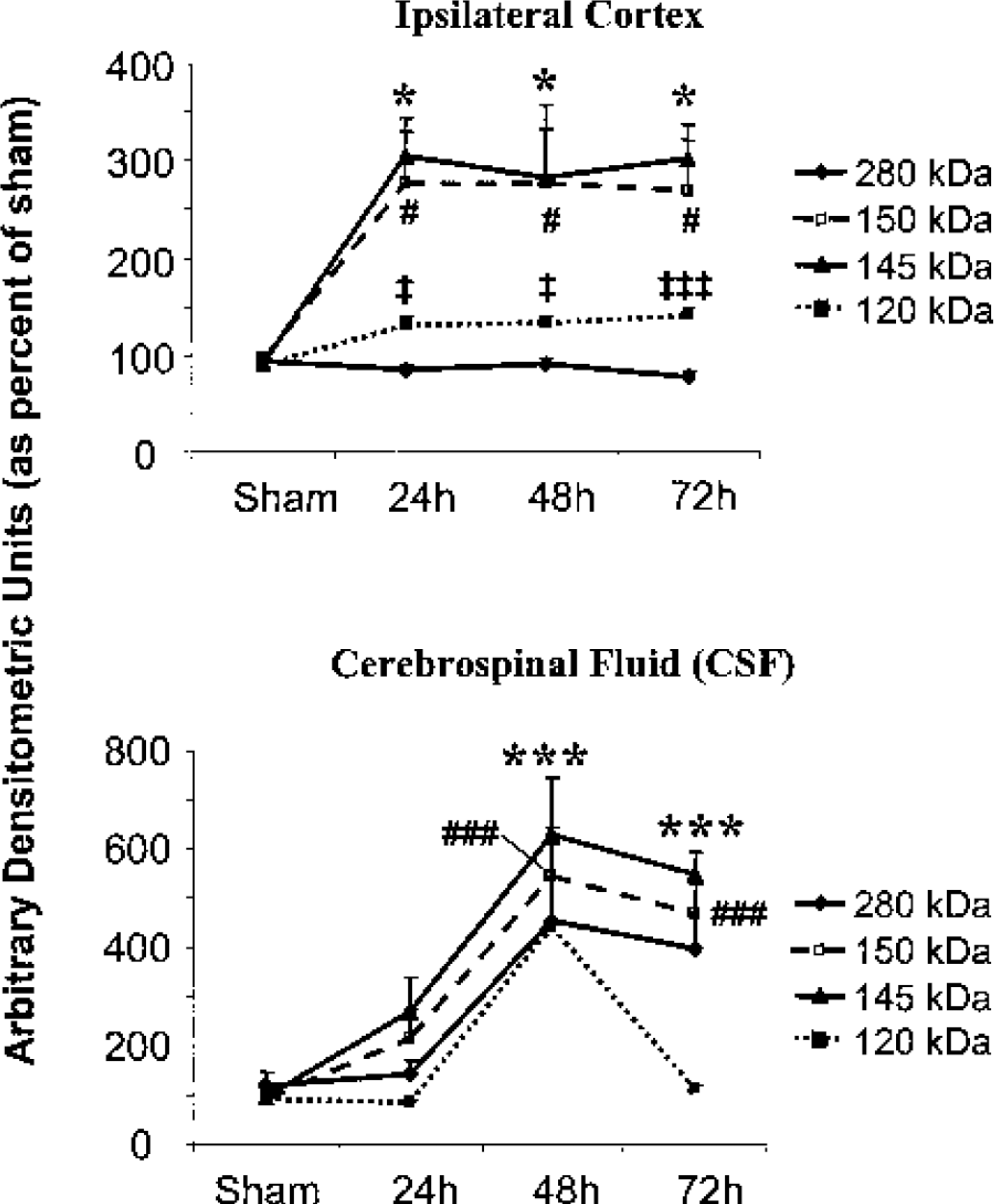
Mean (± SD) arbitrary densitometric units obtained from full-length 280 kDa (II-spectrin protein and the 150 kDa, 145 kDa, and 120 kDa (II-SBDPs. Densitometric units were converted to percent of sham-injured values. Decreases in 280 kDa (II-spectrin and increases in 150 kDa, 145 kDa, and 120 kDa (II-SBDPs (ipsilateral cortex) were associated with concomitant increases of these proteins in the CSF. Note that whereas 150 kDa and 145 kDa SBDPs were visibly detectable in CSF on western blot at 24 hours postinjury, densitometric levels were not statistically significant because of greater variability at this time point. Similarly, although the full-length 280 kDa spectrin protein and the 120 kDa (II-SBDPs were visibly detectable in CSF, large variability in protein levels between animals resulted in inability to detect statistical significance. #P < 0.05 and ###P < 0.001 for 150 kDa αII-SBDP, *P < 0.05 and ***P < 0.001 for 145 kDa αII-SBDP, ‡P < 0.05 and ‡‡‡P < 0.001 for 120 kDa αII-SBDP.
In the contralateral cortex, MCAO injury caused no significant accumulation of calpain-specific αII-SBDPs at any postinjury time point as compared with sham-injured control rats (Fig. 2). There was a visually apparent increase in 120 kDa αII-SBDPs in contralateral cortex after MCAO injury compared with sham-injured control animals, but this increase was not statistically significant. There was no significant difference between ipsilateral and contralateral levels. This result indicates that contralateral levels, although overtly elevated, were not statistically significant.
Accumulation of calpain and caspase-3 mediated αII-SBDPs in CSF after MCAO injury
Cerebrospinal fluid levels of αII-spectrin and αII-SBDPs were undetectable in sham-injured control animals (Fig. 2). However, after MCAO injury, accumulation of full-length αII-spectrin (280 kDa) and the 150, 145, and 120 kDa αII-SBDPs were overtly apparent on immunoblots at various postinjury time points (Fig. 2). Levels of the full-length αII-spectrin protein were increased in CSF of MCAO injured animals and were 144%, 453%, and 395% of sham-injured control levels at 24, 48, and 72 hours postinjury. However, there was considerable between-animal variability in levels of the full-length protein; thus quantitative analysis failed to reach statistical significance (Fig. 3). Levels of the nonspecific 150 kDa αII-SBDP and of the calpain-specific 145 kDa αII-SBDP were apparent on immunoblot at all postinjury time points, but only levels at 48 and 72 hours postinjury reached statistical significance (P < 0.001). Levels of the nonspecific 150 kDa αII-SBDP were 216%, 523%, and 467% of sham-injured control animals, and levels of the calpain-specific 145 kDa αII-SBDP were 268%, 626%, and 546% of sham-injured control values at 24, 48, and 72 hours postinjury, respectively (Fig. 3). Levels of the caspase-3-specific 120 kDa αII-SBDP were 84%, 439%, and 110% of sham-injured control levels at 24, 48, and 72 hours postinjury, respectively (Fig. 3). Although levels of caspase-3-specific 120 kDa αII-SBDPs were 439% of sham-injured control values at 48 hours postinjury, large between-animal variability precluded statistical significance.
DISCUSSION
This report provides further evidence supporting the use of calpain specific αII-SBDPs as surrogate neurochemical markers of CNS injury. Whereas some animals also had elevated CSF levels of caspase-3 specific αII-SBDPs, the large between-animal variability failed to reach statistical significance. Previous data from our laboratory demonstrate that TBI causes robust and detectable accumulation of calpain-mediated αII-SBDPs (and to a lesser extent, caspase-3-mediated αII-SBDPs) in CSF of brain-injured rodents (Pike et al., 2001). We now demonstrate that a rodent stroke model of focal ischemic injury also results in increased levels of calpain (and in some animals, caspase-3) αII-SBDPs in postinjury CSF. The results of these two studies are important in that they provide the first evidence that extraparenchymal detection of specific protein metabolic products can be used as unequivocal biochemical markers for specific neurochemical events (i.e., calpain and caspase-3 activation) that have occurred in the injured brain in at least two preclinical models of brain injury (traumatic and ischemic). It is important to note that recent preliminary clinical studies in patients with severe TBI also indicate robust levels of calpain and caspase-3 mediated αII-SBDPs in CSF (d'Avella et al., 2002).
Analysis of specific biochemical markers is a mandatory component of diagnosing dysfunction in a number of organs, including the use of troponin assays in patients with acute coronary syndromes (Newby et al., 2003). Indeed, troponin testing has rapidly evolved from its initial role in aiding diagnosis of myocardial infarction to a more complex role for risk stratification and guidance of treatment strategies (Newby et al., 2003). However, there are no such biomarkers of proven clinical usefulness for TBI and cerebral ischemia. In the case of TBI, this may be caused in part by the fact that TBI is difficult to assess, and clinical examinations are of restricted value during the first hours and days after injury. For instance, conventional diagnoses of TBI are based upon neuroimaging techniques such as CT scanning, MRI, and single-photon emission CT scanning (Jacobs et al., 1996; Kant et al., 1997; Mitchener et al., 1997). CT scanning has low sensitivity to diffuse brain damage, and the availability of MRI is limited (Kesler et al., 2000; Levi et al., 1900). In addition, single-photon emission CT scanning detects regional blood-flow abnormalities not necessarily related to structural damage. In the case of stroke, investigators have also generally recognized the need for more objective assessments of outcome, including the use of biochemical markers (Dirnagl et al., 1999; Zaremba et al., 2001). For example, diagnosis of stroke is relatively straightforward when patients present with typical symptoms; however, often symptoms of stroke are more subtle and can delay diagnosis by hours or days (Elkind, 2003). Additionally, other causes of neurologic symptoms, such as seizure, migraine, vasospasm, syncope, and peripheral vestibulopathy, can be indistinguishable from symptoms of thromboembolic transient ischemic attacks (Johnston et al., 2003). Thus a rapid and reliable biochemical marker of stroke will facilitate diagnosis and might give assurance to physicians considering administering thrombolytic agents for treatment of acute ischemic stroke.
Our laboratories' assessment of αII-SBDPs as biochemical markers in models of TBI and focal cerebral ischemia may result in considerable improvement over currently existing biochemical markers of CNS injury. For instance, other putative biomarkers of CNS injury (e.g., CPK-BB, NSE, S-100ß, and lactate dehydrogenase) are of limited value because of a lack of specificity to CNS tissues, unreliability in predicting outcome, and because they provide no specific information regarding neurochemical pathology of injured CNS tissue. Recent studies have also examined the usefulness of cleaved tau protein (cτP) as a predictor of outcome. However, whereas cτP is axonal specific, it also provides no information about specific neurochemical events that have occurred in the injured CNS. Furthermore, recent studies have presented conflicting evidence as to the usefulness of cτP as a predictor of outcome after TBI in humans (Chatfield et al., 2002; Zemlan et al., 1999). In contrast, αII-SBDPs offer several advantages as compared with the putative biomarkers just described. For instance, αII-SBDPs provide concurrent information on postinjury activity of two important proteolytic enzymes (calpain and caspase-3). Low basal levels of these proteases further optimize their usefulness as markers of cell injury. Another important characteristic is that αII-spectrin protein is not localized in erythrocytes (Goodman et al., 1995; Riederer et al., 1986). Blood is a major source of CSF contamination after TBI and hemorrhagic ischemia. Results from our previously published studies in TBI clearly demonstrate that αII-spectrin and αII-SBDPs are not detectable in whole blood samples. In contrast, the erythroid isoform of spectrin, αI-spectrin, is detectable in both blood and brain tissues (Pike et al., 2001).
However, one disadvantage is that whereas αII-spectrin is highly enriched in brain, it is not specific to brain tissue. Whereas this is not a concern for CSF detection of αII-SBDPs, it could be problematic for detection of αII-SBDPs in serum as human head-injured patients often present with multiorgan trauma. Additional studies in preclinical models and in human patients are needed to clarify this issue.
An ideal biomarker for a particular neurologic disease is one that is 100% specific and sensitive for that particular disease. However, with TBI or stroke, it is not critical that a biomarker be specific to one or the other disorder, rather, the biomarker need only indicate, with as much sensitivity as possible, the severity of brain damage that has occurred as a result of brain trauma or cerebral infarction (although a biomarker that can rapidly and accurately discriminate between hemorrhagic and thrombolytic stroke would certainly be useful). The use of calpain- and caspase-3-mediated αII-SBDPs could provide a powerful approach for determining the severity of brain damage caused by a TBI or stroke, and could also provide a clinical tool for monitoring the duration of the acute injury response and the effects of emergency or therapeutic interventions. For instance, calpain and caspase-3 are potent mediators of cell death that can be rapidly activated in response to traumatic (Beer et al., 2000; Pike et al., 1998a; Sullivan et al., 2002) or ischemic brain injury (Davoli et al., 2002; Zhang et al., 2002), and brain regions with the highest accumulation of αII-SBDPs have the highest level of neuronal cell death (Newcomb et al., 1997; Roberts-Lewis et al., 1994). It is important to note that calpain and caspsase-3 can be concurrently or independently activated after TBI (Pike et al., 1998a) or cerebral ischemia (Zhang et al., 2002), and the temporal duration of activity can vary for each protease. Thus the ability to monitor both calpain and caspase-3 activation during the acute period of CNS injury is a major advantage of αII-SBDPs over other biomarkers. Indeed, recent preliminary data obtained from CSF of severely injured TBI patients indicate that temporal accumulation of calpain- and caspase-3-mediated αII-SBDPs show different patterns of temporal expression that vary in each patient (d'Avella et al., 2002). This result is similar to our preclinical TBI and ischemic injury models in which accumulation of calpain or caspase-3 αII-SBDPs also varies between individual animals. This variability emphasizes the heterogeneous nature of TBI and ischemic pathology and points to important implications for individualized treatment of human patients with brain injury that is tailored to specific neurochemical cascades operative in the injured brain.
It is important to note that there was more variability in CSF levels of αII-SBDPs than there was in brain levels of αII-SBDPs. This variability is indicated by the smaller error bars in Fig. 3 for brain as compared with CSF levels of αII-SBDPs, and these differences in variability can be observed in individual animals in Fig. 2. The reason for the larger variability in CSF protein accumulation is unknown but may reflect differences in an individual animal's CSF circulation after ischemia. For example, differences in increased intracranial pressure after ischemia may restrict passage of CSF through various foramina that may preclude detection of secreted proteins into the cisterna magna (source of CSF in the present study). Additional studies should examine differences in intraventricular versus intracisternal levels of accumulated αII-SBDPs. In addition, it will be necessary to correlate CSF levels of αII-SBDPs with traditional outcome measures such as infarct size and behavioral function after ischemia.
Although calpain and caspase-3 cysteine proteases are known to be important mediators of cell death and dysfunction in numerous CNS diseases and injuries including cerebral ischemia, the results of this investigation clearly indicate much more calpain-mediated cleavage of αII-spectrin in both brain and CSF than was observed for caspase-3. Using αII-spectrin as a substrate marker, we have routinely observed this more pronounced calpain cleavage pattern after TBI as well (Pike et al., 1998a, 2001). In addition, a recent study by Zhang and coworkers (2002) also observed evidence of more predominant calpain versus caspase-3 protease activity following transient forebrain ischemia in the rat. However, because calpain and caspase-3 compete for many of the same substrates (Wang et al., 2000), and the interaction between calpain and caspase proteases is complex (Newcomb-Fernandez et al., 2001; Zhang et al., 2002), it is difficult to determine the true extent of proteolytic activity by using a single substrate marker. Moreover, detection of caspase-3 mediated αII-SBDPs in CSF or serum after CNS injury may prove difficult because of the rupture of plasma membranes during necrotic cell death (in which calpain cleavage and substrate release into the extracellular milieu will predominate) compared with caspase-3 activation that is more prominent during apoptotic cell death (in which plasma membranes are left largely intact). These caveats should be taken into consideration when developing biochemical markers of CNS injury and in interpreting the results of markers of calpain and caspase-3 proteases.
CONCLUSIONS
This report provides further evidence supporting the use of calpain- and caspase-3-mediated αII-SBDPs as neurochemical markers of CNS injury. Although numerous other proteins, peptides, amino acids, etc., have been identified in CSF after TBI and acute cerebral ischemia, no such surrogate marker of CNS injury has yet provided a window of insight into specific neurochemical events that have occurred as a result of traumatic or ischemic brain injury. The use of protease-specific αII-SBDPs as biomarkers offers several advantages over existing biomarkers of traumatic or ischemic brain injury, including the ability to provide concurrent information about the activity of two major proteolytic effectors of cell death. Additional studies to further characterize the sensitivity of αII-SBDPs (e.g., in serum and across injury magnitudes) are ongoing. In addition, it is thought that the development of other CNS-specific biomarkers used in conjunction with αII-SBDPs will provide researchers and clinicians with powerful tools for diagnosing and assessing CNS injury, for monitoring recovery, and for guiding appropriate administration of therapeutic compounds. Finally, it is thought that recent advancements in antibody-based specific identification technologies will facilitate development of rapid, sensitive, and easy-to-use kits for research and clinical environments.