
Abstract
The authors recently cloned a cDNA for an ICE/CED3-related cysteine protease from rat brain, which is closely related to human CPP32 (now designated caspase-3). In situ hybridization histochemistry revealed a profound developmental regulation of the caspase-3 transcript in rat brain, with relatively high levels of caspase-3 mRNA observed in neurons of the fetal and neonatal brain and low levels of mRNA in neurons of the adult brain. The authors report that transient forebrain ischemia, which results in a delayed apoptotic death of CA1 pyramidal neurons, results in prolonged expression of caspase-3 mRNA in these same pyramidal neurons. Up-regulation of caspase-3 mRNA in CA1 pyramidal neurons is prominent 24 hours after transient global ischemia, and expression is maintained at higher levels for at least 72 hours after ischemia. However, by 96 hours after ischemia, a marked decrease in caspase-3 mRNA expression is observed in CA1 pyramidal neurons, showing severe degenerative changes (e.g., nuclear condensation). By contrast, there is no change in the expression of a closely related member of caspase family, caspase-2, in CA1 pyramidal neurons after global ischemia. Instead, caspase-2 mRNA is induced in lamina layers of cerebral cortex 24 hours after the ischemia. A selective and prolonged induction of the caspase-3 gene in committed CA1 pyramidal neurons suggests that transcriptional activation of this caspase-3 gene may be involved in the apoptotic cell death cascade of CA1 neurons after transient global ischemia.
Keywords
Apoptosis is a physiologic mechanism by which a cell dies through activation of an intrinsic cell death or suicide program (for review see Vaux and Strasser, 1996; Jacobson et al., 1997). Activation of this cell death program leads to a cascade of intracellular events that includes a commitment point, followed by the appearance of characteristic ultrastructural changes in the nucleus, cell death, and eventually, phagocytosis (Kerr et al., 1972). Recent studies have provided strong evidence that apoptotic cell death not only is required for normal central nervous system development (Raff et al., 1993; Saunders, 1996), but it also is involved in the pathologic death of neurons that occurs in various neurodegenerative disorders, such as Alzheimer's disease, and ischemic (stroke) and traumatic brain injury (Cotman and Anderson, 1995; Lassmann et al., 1995; Bredesen 1995; Du et al., 1996 Chopp et al., 1996). Although the precise cellular mechanism underlying apoptosis especially in mammalian neurons is poorly understood, it has become apparent that apoptosis, like the cell cycle, is not a simple metabolic cascade but is the result of a rather complex set (programmed in many cells) of molecular interactions between several proteins.
Recently, two families of aspartyl-specific cysteine proteases have been identified in the nematode Caenorhabditis elegans and shown to be critical for the programmed cell death via apoptosis of neurons during nematode development. Mammalian homologues of these cysteine proteases (designated caspases) (Alnemri et al., 1996) also have been identified, and data are accumulating that these subserve important roles in mediating apoptosis of various mammalian cells (Gagliardini et al., 1994; Enari et al., 1995; Los et al., 1995). Moreover, the induced expression of single caspase, such as caspase-3 and mouse and human caspase-2, in transfected cell lines is sufficient to induce apoptosis (Miura et al., 1993; Kumar et al., 1994; Fernandes-Alnemri et al., 1995; Faucheu et al., 1995), which has been explained, at least partially, by the autocatalytic activity of these proteases (Wang et al., 1996). This phenomenon is also observed when the caspase-3 precursor (procaspase-3) is overexpressed in baculovirus (Xue and Horvitz, 1995).
Although several caspases have been identified in cells from the immune system and various tumor cell lines (Miura et al., 1993; Kumar et al., 1994; Fernandes-Alnemri et al., 1995), their regional and developmental expression in brain and their involvement in neuronal apoptosis for the most part have yet to be defined. Of the ten caspase genes cloned to date (Alnemri et al., 1996; Vaux and Strasser, 1996; Lippke et al., 1996; Liu et al., 1996), several have now been shown to be expressed in the central nervous system. Both the caspase-3 and caspase-2 genes, for example, are expressed in the brain and developmentally regulated (Kumar et al., 1994; Ni et al., 1997a). Only low levels of caspase-1 can be detected in the brain, principally in non-neuronal cells and in some interneurons (Bhat et al., 1996). In caspase-1–deficient mice, the central nervous system develops normally despite major defects in the caspase-1–dependent generation of mature interleukin-1 and other cytokines (Li et al., 1995; Kuida et al., 1995). In contrast, brain development is affected profoundly in caspase-3–deficient mice because of decreased neuronal apoptosis, but no discernible histologic abnormalities were observed in many peripheral tissues, such as heart, lung, liver, kidney, spleen, and testis (Kuida et al., 1996). These studies suggest that caspase-3 plays a critical role during morphogenetic cell death of mammalian neurons.
After transient global ischemia, CA1 pyramidal neurons of the hippocampus degenerate in a characteristic fashion in several species (Pulsinelli and Brierley, 1979; Pulsinelli et al., 1982). The transient global ischemia model has been used extensively to identify intrinsic mechanisms underlying delayed neuronal cell death (Kirino 1982; Kirino and Sano, 1994). Moreover, CA1 pyramidal neurons undergo selective and delayed cell death in humans after global ischemia, such as that which occurs after myocardial infarction. The delayed cell death of CA1 pyramidal neurons after transient global ischemia appears to be apoptotic in nature, based on classic criteria for defining apoptosis, including alterations in cellular and nuclear morphology by light and electron microscopy and the presence of internucleosomal DNA fragmentation as determined by in situ TUNEL labeling (MacManus et al., 1993; Kihara et al., 1994; Nitatori et al., 1995), although necrosis also is implicated (Deshpande et al., 1992). In the present study, we measured the expression of the transcript for caspase-3 in various regions of the adult rat brain after transient global forebrain ischemia. Our data suggest that a prolonged transcriptional activation of caspase-3 may be involved in the delayed degeneration of CA1 pyramidal neurons after global ischemia.
MATERIALS AND METHODS
Four-vessel occlusion
Transient forebrain ischemia was induced by four-vessel occlusion according to the method of Pulsinelli and Brierley (1979). Briefly, male Wistar rats (250–270 g; Hilltop Laboratories, Scottsdale, PA, U.S.A.) were prepared for forebrain ischemia under halothane anesthesia by electrocauterizing the vertebral arteries bilaterally and placing atraumatic carotid clasps around the common carotid arteries without interrupting the arterial blood flow. On the following day, forebrain ischemia was induced by tightening the clasps for 30 minutes. Three animals were not subjected to ischemia and served as controls. One, 2, 3, and 4 days after ischemia, animals (3–5 animals per time point) were killed and the brains processed as described in the following section. The animals were removed from anesthesia during the period of ischemia. All animal procedures were performed in compliance with and following approval of the Lilly Research Laboratories Animal Care and Use Committee.
Probe labeling
35S-labeled sense and antisense RNA probes were synthesized by in vitro transcription of caspase-3 (Ni et al., 1995) and caspase-2 cDNAs as described by Stratagene, and used for in situ hybridization, as described previously (Ni et al., 1995). RNA probes were shortened to an average length of 150 base pair by alkaline hydrolysis before hybridization (Ni et al., 1995).
Tissue preparation
Forebrains were dissected rapidly and frozen in isopentane, chilled using dry ice, and serially sectioned in the coronal plane throughout the rostrocaudal extent of the hippocampus. Twenty-micrometer thick sections were used for immunocytochemistry, and 15-μm thick sections were used for in situ hybridization. Regions evaluated included only those the same coronal plane as the hippocampus.
In situ nick end labeling
Frozen sections were postfixed in 4% buffered paraformaldehyde (pH 7.4) and subsequently labeled using the Apotag in situ apoptosis detection kit (peroxidase) according to manufacturer's recommendations (Oncor, Gaithersburg, MD, U.S.A.). Briefly, sections were equilibrated, incubated at 37°C with terminal deoxynucleotidyl transferase (Tdt) and digoxigeninnucleotide for 1 hour, followed by antidigoxigenin-peroxidase. The reaction product was visualized using diaminobenzidine/H2O2; sections were counterstained with methyl green, dehydrated, and mounted using permount. For negative controls, sections from sham-operated controls also were stained in parallel as aforementioned; moreover, sections from animals subjected to forebrain ischemia were incubated with the Tdt enzyme alone.
Immunocytochemistry
Immunocytochemistry was performed using the monoclonal antibody OX42 (Bioproducts for Science, Indianapolis, IN, U.S.A.). This antibody identifies the CR3 complement receptor expressed on activated microglia (Graeber et al., 1988; Gehrmann et al., 1992). Frozen sections were postfixed in 3.7% formalin (5 minutes), followed by acetone (50% acetone, 2 minutes; 100%, 3 minutes; 50%, 2 minutes) and then quenched for 30 minutes in 0.25% H2O2 in 10% nonbuffered formalin. Immunocytochemistry was performed using the avidin-biotin peroxidase system (ABC kit, Vector Labs, Burlingame, CA, U.S.A.).
In situ hybridization histochemistry
In situ hybridization analysis was performed as described in detail elsewhere (Ni et al., 1995 and 1997b). Briefly, frozen sections (15 μm) were prepared on a cryostat, mounted on poly-L-lysine—coated glass slides and dried at room temperature. Before hybridization, sections were warmed to 25°C, fixed in 4% formaldehyde, and immersed for 10 minutes in 0.25% acetic anhydride, 0.1 mol/L triethanolamine hydrochloride, and 0.9% sodium chloride (NaCl). The 35S-labeled riboprobes were added to a hybridization buffer composed of 50% formamide, 0.3 mol/L NaCl, 20 mmol/L TRIS-hydrochloride (pH 8), 5 mmol/L edetic acid, 500 μg tRNA/mL, 10% dextran sulfate, 10 mmol/L dithiothreitol, and 0.02% each of bovine serum albumin, ficoll, and polyvinylpyrrolidone. The slides then were incubated in a humidified chamber overnight at 55°C. Then the sections were treated with RNase A (20 μg/mL) for 30 minutes at room temperature, followed by a 15-minute wash in 0.1 xSSC at 55°C. For analysis at the cellular level, the slides were dipped in Kodak NTB3 nuclear emulsion, stored with desiccant at 4°C for 10 days, developed, and counterstained with hematoxylin/eosin-phloxine for microscopic evaluation. The hybridization signal was visualized by bright-field microscopy and densitometrically analyzed using NIH Image (Bethesda, MD, U.S.A.). Caspase-3 mRNA levels were measured in the CA1 region of CA1/CA2 borderline. Seventy-two hours after ischemia, nondegenerated CA1 regions were used to represent the caspase-3 mRNA level. The means of arbitrary density from five to ten images of three to five animals per time point are normalized by using the ratio of CA1/cortex.
RESULTS
Expression of caspase-3 mRNA in CA1 pyramidal neurons after transient forebrain ischemia
CA1 pyramidal neurons of the hippocampus are selectively vulnerable to transient ischemic injury. In agreement with previous studies (Pulsinelli and Brierley, 1979; MacManus et al., 1993; Kihara et al., 1994; Nitatori et al., 1995), 30 minutes of transient global forebrain ischemia resulted in selective degeneration (apoptosis) of neurons in the hippocampal CA1 region more than 72 hours after ischemia, based on the classic criteria for apoptosis, such as condensation of nuclear chromatin (hematoxylin-and-eosin staining), in situ labeling of DNA fragmentation (TUNEL labeling), and CA1 microglial activation (OX42 immunoreactivity), as shown in Fig. 1.
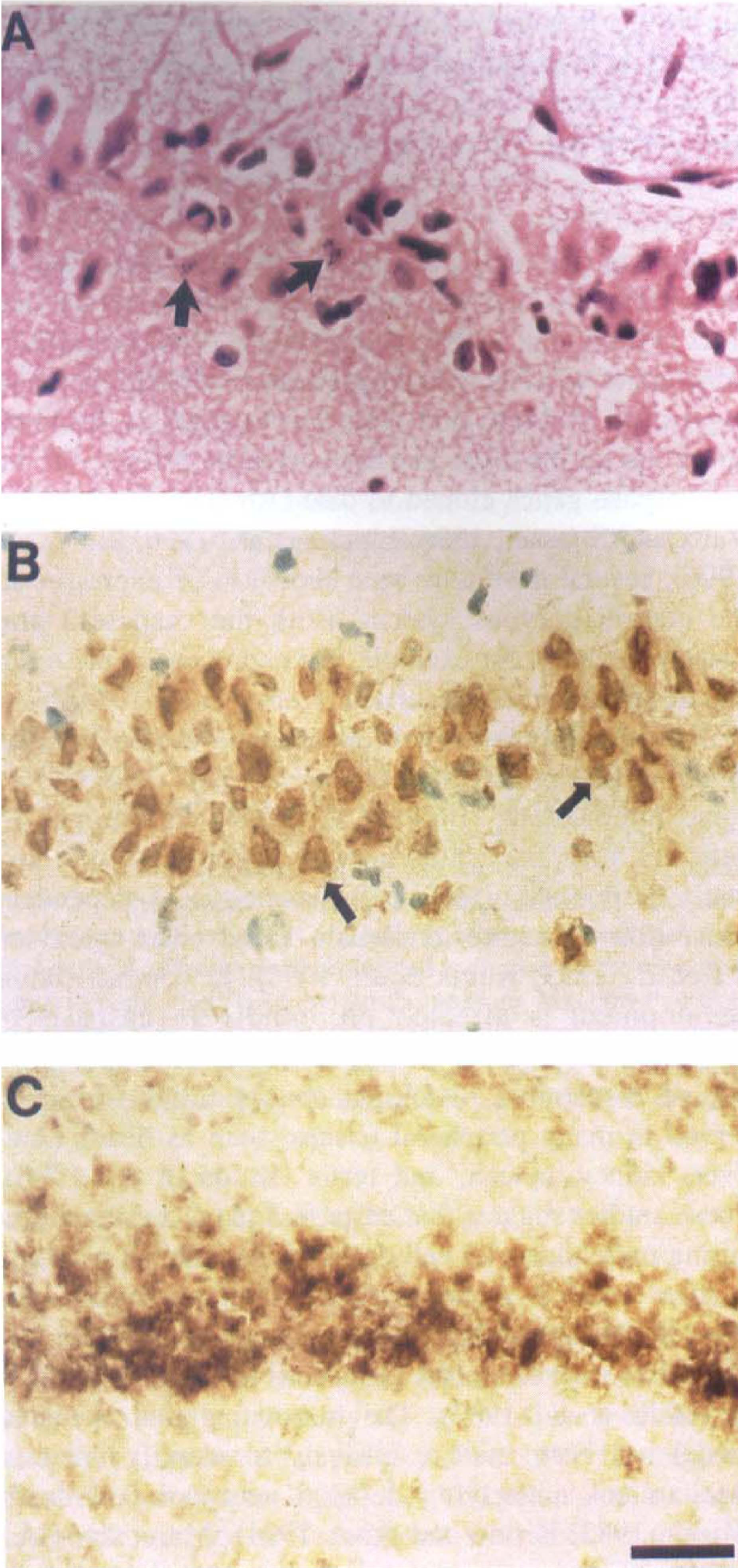
Apoptotic cell death of CA1 pyramidal neurons after transient global ischemia. Sections were from animals subjected to 30 minutes of global forebrain ischemia and examined with hematoxylin-and-eosin (H&E) staining (
To evaluate whether transient forebrain ischemia activates the transcription of caspase-3 gene, we employed in situ hybridization histochemistry to measure caspase-3 mRNA in the CA1 regions of hippocampus. As shown in Fig. 2, hybridization with the 35S-labeled caspase-3 probe revealed an induction of caspase-3 mRNA in the CA1 pyramidal cell layer of the hippocampus 24, 48, and 72 hours after transient forebrain ischemia (Fig. 2A–C). Prolonged caspase-3 mRNA expression in the CA1 region was evident because a robust and selective increase in caspase-3 mRNA was observed in CA1 pyramidal neurons 72 hours after ischemia but not in adjacent pyramidal neurons of CA2-CA4 regions or in neurons of the cerebral cortex. An anticipated reduction in caspase-3 was found in CA1 pyramidal neurons 96 hours after ischemia, when CA1 neurons show severe cell damage, suggesting that loss of caspase-3 mRNA is caused by loss of viability of caspase-3–expressing CA1 neurons. A transient increase in caspase-3 mRNA expression is observed in the CA3 region of the hippocampus 24 hours after ischemia, but levels of caspase-3 mRNA are similar to controls by 48 hours after ischemia (Fig. 2A and 2E). It appears that caspase-3 mRNA is induced in virtually all CA1 pyramidal neurons, including those in the paramedian zone 24 hours after ischemia, but it is reduced or absent in CA1 neurons of the paramedian zone, which are most sensitive to ischemic injury and which have degenerated by 72 hours after ischemia.
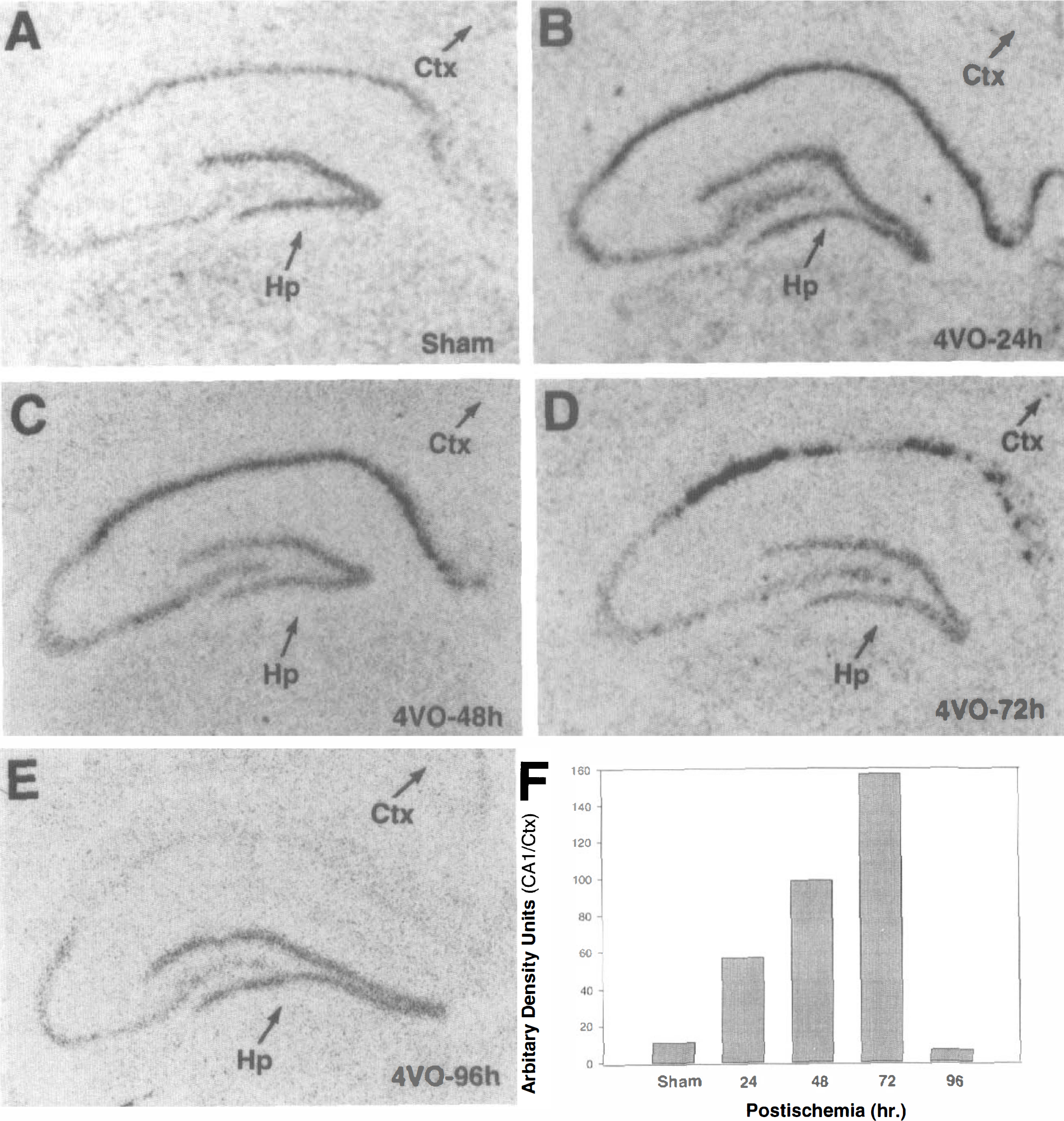
Transient forebrain ischemia induces any early and prolonged expression of caspase-3 mRNA in CA1 neurons. Cryostat-prepared tissue sections were hybridized with caspase-3 riboprobe and processed for in situ hybridization histochemistry, as described in the Materials and Methods section. In addition to the sham control (
To examine whether the loss of caspase-3 mRNA in these neurons is caused by a loss of cell viability, we examined sections of hippocampus that showed varying degrees of damage 72 hours after ischemia (i.e., when delayed cell death begins to occur). As shown in Fig. 3, caspase-3 mRNA is induced in CA1 neurons that do not yet show ischemic damage (i.e., no nuclear condensation), whereas neurons showing ischemic damage (i.e., nuclear condensation) lack caspase-3 mRNA, suggesting that caspase-3 mRNA is induced in CA1 pyramidal neurons that are committed to die but have not yet degenerated. These data are in agreement with the observation that at later time points after ischemia (e.g., 96 hours), most CA1 neurons have died and little caspase-3 mRNA can be detected (Fig. 2C).
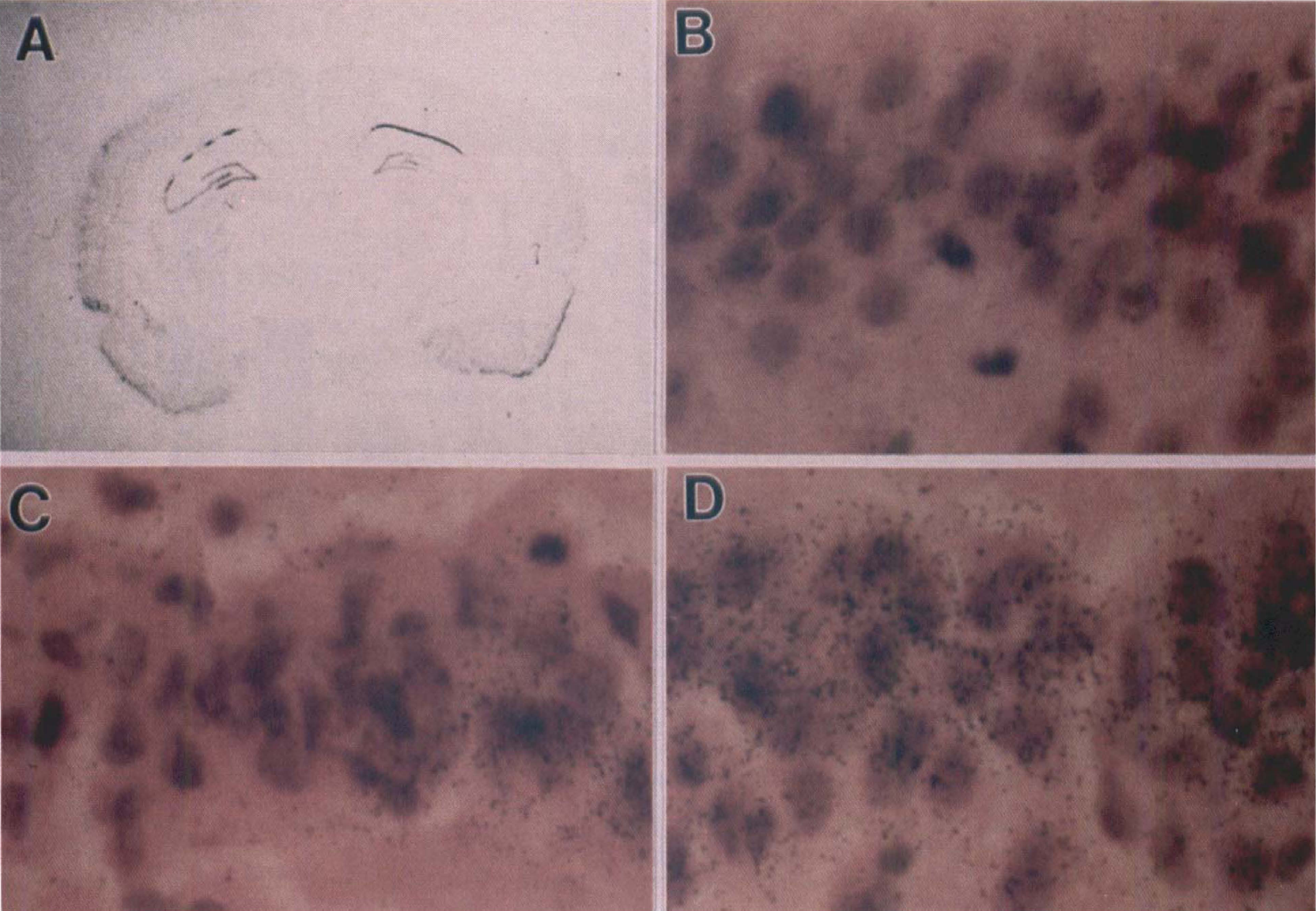
Induction of caspase-3 transcripts in committed but not dead CA1 neurons. Hybridization with caspase-3 antisense probe resulted in a strong hybridization signal in the CA1 pyramidal layer of hippocampus 72 hours after transient global ischemia (
To further confirm the induced expression of caspase-3 mRNA after transient global ischemia, we measured the expression of another gene encoding a brain specific Na+-dependent phosphate cotransporter, (rBNPI), previously shown to be expressed in CA1 hippocampal neurons (Ni et al., 1995). rBNPI mRNA is expressed abundantly in the rat hippocampus and is reduced specifically in CA1 pyramidal neurons after transient global ischemia (Ni et al., 1997b). As shown in Fig. 4, in near-adjacent sections, rBNPI mRNA is selectively reduced after global ischemia in CA1 neurons of hippocampus 72 hours after ischemia, whereas caspase-3 mRNA is dramatically induced.
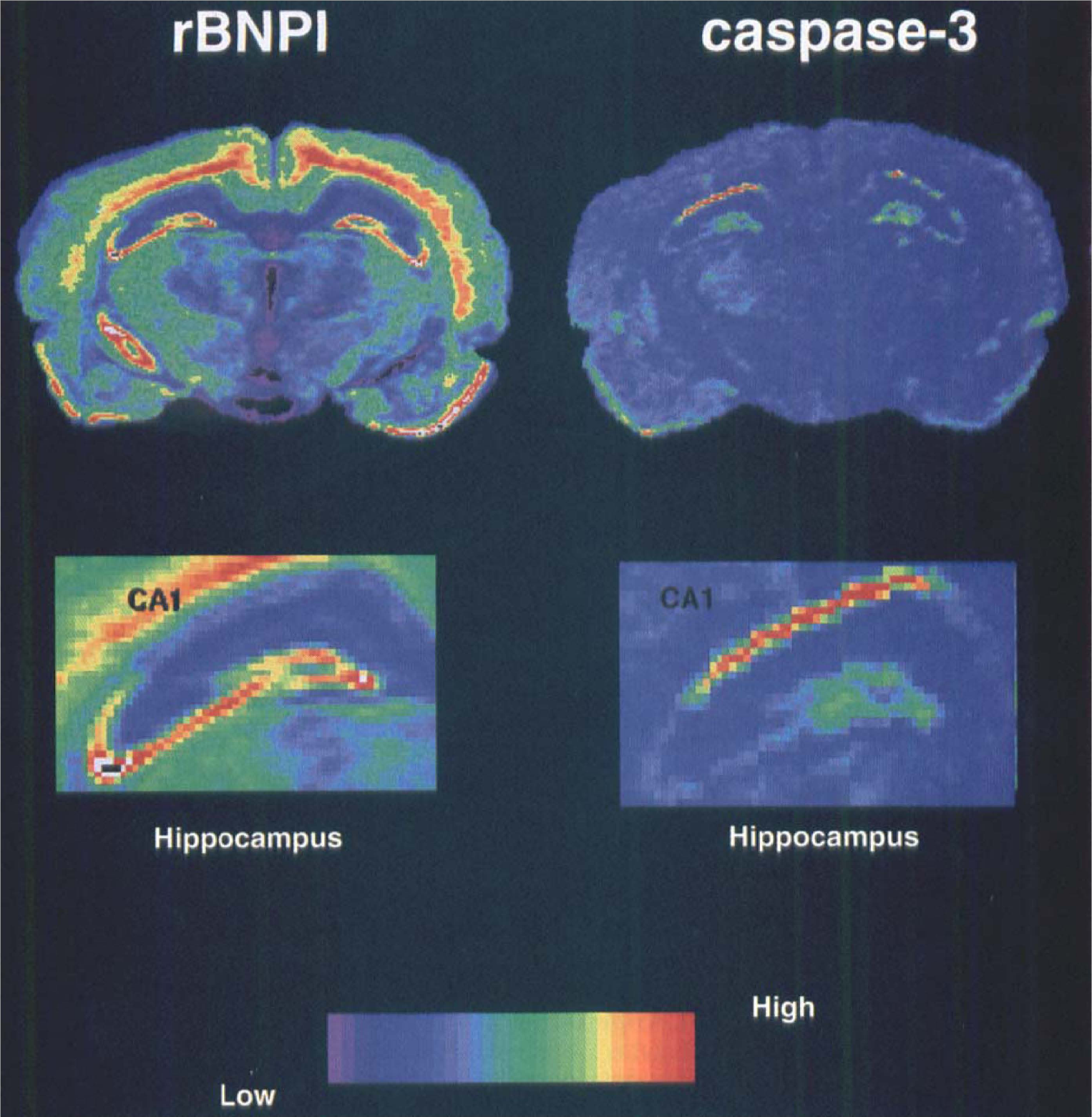
Specific induction of caspase-3 mRNA in the CA1 neurons. Ischemic insult resulted in a delayed cell death associated with a dramatic decrease in sodium-dependent Pi cotransporter (BNPI) mRNA in CA1 region (Ni et al., 1997b) (
Differential expression of the caspase-3 and caspase-2 genes after transient global ischemia
To further evaluate the specificity of caspase-3 gene expression after transient global ischemia, we examined the expression of another member of caspase family, caspase-2, using in situ hybridization on near-adjacent sections. Caspase-2 also is expressed in neurons of the rat brain and CA1 pyramidal neurons of the hippocampus in particular (Kumar et al., 1994). As shown in Fig. 5, in situ hybridization of caspase-3 and caspase-2 mRNAs revealed a markedly different pattern of expression. Although there is an increase in caspase-2 mRNA in the cerebral cortex 24 hours after ischemia, no significant change in caspase-2 mRNA (compared with controls) is observed in the CA1 cell layer of the hippocampus 24 or 72 hours after ischemia. By contrast, the levels of caspase-3 mRNA are increased in the CA1 region as measured in near-adjacent sections, suggesting that transient forebrain ischemia may selectively induce the expression of caspase-3 in this brain region.
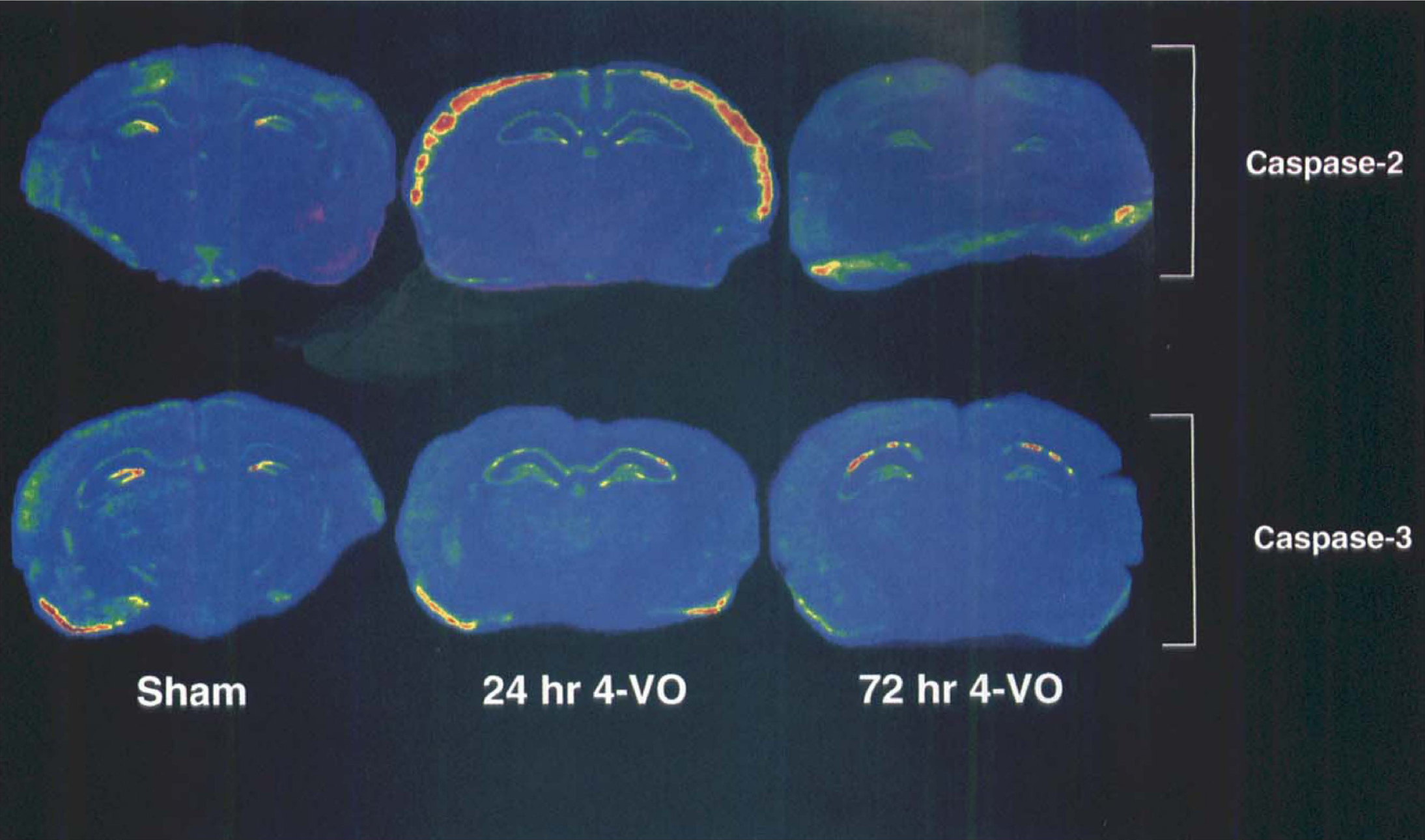
Differential expression of caspase-3 and caspase-2 mRNAs in the rat brain after ischemic insult. Expression of caspase-3 and caspase-2 mRNA, a closely related cell death cysteine protease, at various time points after ischemic insult is shown. No significant induction or loss of caspase-2 mRNA is observed in the CA1 pyramidal layer of hippocampus 24 or 72 hours after ischemia, although an induction of caspase-2 mRNA is observed in the cerebral cortex 24 hours after ischemia (top). By contrast, the caspase-3 gene is induced in the CA1 pyramidal layer of the hippocampus using near-adjacent sections (bottom).
DISCUSSION
To our knowledge, our data represent the first demonstration of a prolonged increase in the expression of caspase-3 in CA1 pyramidal neurons of the hippocampus after transient global ischemia. These neurons have been shown previously to undergo a rather selective delayed-type of degeneration that is apoptotic in nature. In contrast, there was no change in the expression of caspase-2 mRNA, a closely related member of caspase family in the CA1 pyramidal neurons, when measured in near-adjacent sections. The early and prolonged induction of caspase-3 mRNA in CA1 neurons that are committed to apoptotic cell death but that are not yet dead suggests that caspase-3 may well mediate neuronal apoptosis in this model.
Using in situ hybridization histochemistry, we characterized the cellular localization of caspase-3 mRNA expression in neurons of the cerebral cortex and hippocampus and relatively higher levels of expression in granule neurons of the cerebellum. The physiologic significance of this constitutive expression vis-à-vis the role of caspase-3 in neuronal apoptosis still is unclear. It is curious that mRNA for a cell death gene is expressed even at low levels in cells/neurons not undergoing apoptosis. Conceivably, however, not all caspase-3 mRNA is translated into functional protein or, alternatively, there may be a critical level or concentration of protein necessary for induction of apoptosis. Moreover, the caspase-3 proenzyme, like most members of caspase family, must be processed proteolytically to catalytically active subunits and therefore, other factors may regulate the processing of the caspase-3 proenzyme encoded by the mRNA measured in our experiments. Finally, native or endogenous inhibitors of caspase-3 may exist to prevent caspase-3–mediated apoptosis. It appears that other caspase-1–related proteases are expressed at low levels in certain tissues in which cells apparently are not undergoing apoptosis, suggesting that these cysteine-related proteases are involved in cellular functions other than cell death when present at “normal” levels. Caspase-1 itself usually is involved in converting prointerleukin-1β to active interleukin-1β (Black et al., 1989; Kostura et al., 1989).
Mammalian cells that overexpress genes for members of caspase family, including caspase-3, die via apoptosis (Miura et al., 1993; Kumar et al., 1994; Fernandes-Alnemri et al., 1995). Thus, increased expression of caspase-3 mRNA in CA1 hippocampal neurons—if accompanied by increased protein and catalytically active protease activity—may contribute (or mediate) their delayed death via apoptosis. Previous experiments have demonstrated that intraventricular administration of cycloheximide, a protein synthesis inhibitor, attenuates ischemia-induced cell death of hippocampal neurons (Goto et al., 1990; Shigeno et al., 1990) including CA1 pyramidal neurons, suggesting that transcriptional or translational activation of an ischemia-induced genetic program may contribute to apoptotic cell death after ischemia. Moreover, the antiapoptotic gene bcl-2 appears to protect against ischemia-induced neuronal cell death after either global or focal ischemia (Shimazaki et al., 1994; Chen et al., 1995). However, several reports have shown that protein synthesis such as HSP72 was suppressed after ischemia (Nowak, 1991). We do not know yet whether the rather robust increase in caspase-3 mRNA expression is accompanied by a similar increase in translated protein. Currently, we cannot confirm an increase in caspase-3 protein because of the lack of specific antisera necessary for such studies.
We also observed a transient increase in caspase-3 mRNA in CA3 neurons 24 hours after ischemia. However, these neurons do not show delayed cell death after transient global ischemia such as that employed in our study. This seemingly paradoxical observation may be explained by the recent finding that the antiapoptotic proto-oncogene bcl-2 also is induced in hippocampal CA3 neurons and granule neurons of the dentate gyrus (but not in CA1 pyramidal neurons) after ischemia (Chen et al., 1996). Thus, the transient elevation of caspase-3 mRNA expression in CA3 neurons may be balanced by the inhibitory effect of induced bcl2 expression. Alternatively, the transient and relatively minor elevation of caspase-3 mRNA may not be sufficient to induce cell death of CA3 neurons. Recent studies also have shown up-regulation of bcl2-related proteins, such as Bax and induced Bax transcripts in CA1 neurons after transient global ischemia (Krajewski et al., 1995; Chen et al., 1996). The increased expression of Bax may play a role in abrogating the ability of bcl-2 to suppress apoptosis by dimerizing with bcl-2. These observations are consistent with the fact that programmed cell death for any given population of cells occurs by a mechanism orchestrated by several proteins that may either promote or inhibit apoptosis, but that ultimately require cysteine proteases (Gagliardini et al., 1994; Milligan et al., 1995; Enari et al., 1995; Los et al., 1995). The possible involvement of cysteine protease(s) in ischemia-induced CA1 cell death also is supported by the observation that inhibition of proteolysis protects hippocampal neurons from ischemia-induced CA1 cell death (Lee et al., 1991). Finally, although caspase-1 mRNA expression has been shown recently to be increased in the hippocampus after transient global ischemia, the increased caspase-1 immunoreactivity was in this case localized to microglia, suggesting an indirect role of caspase-1 in neuronal damage after ischemia (Bhat et al., 1996).
Our observation of a differential expression of caspase-3 and caspase-2 mRNAs in response to transient global ischemia suggests that ischemia can selectively induce certain cell death proteases, presumably as a part of the mechanism underlying the apoptotic cascade. The transcriptional activation of caspase-3 mRNA in CA1 pyramidal neurons, which are committed to undergo apoptosis after ischemic injury, is consistent with our recent findings on cultured cerebellar granule neurons, in which an early and prolonged up-regulation of caspase-3 mRNA by apoptotic insults (e.g., reduced extracellular [K+]) is followed by apoptotic cell death (Ni et al., 1997a). Moreover, down-regulation of the overexpressed caspase-3 mRNA by exposure to neurotrophic factors, such as brain-derived neutrotrophic factor, or depolarization with [K+] will protect these neurons if such treatments are applied before the commitment point for apoptosis induced by K+ withdrawal (Ni et al., 1997a). Significantly, neuronal injury/death in the rat brain after ischemia (Hara et al., 1997) and in cultured cerebellar granule neurons by reduced extracellular K+/serum (Armstrong et al., 1997) are greatly attenuated by inhibitors of caspase-3 and caspase-1, suggesting a role for caspase-3 in neuronal apoptosis in vivo and in vitro.
It appears from our studies that the caspase-3 gene is readily inducible and may serve as a biological sensor to govern cell fate after metabolic or toxic insults. However, it seems unlikely that any single protease is solely responsible for ischemia-induced apoptosis of neurons in the mammalian central nervous system (Martin and Green, 1995). We cannot rule out the involvement of other members of caspase family in CA1 hippocampal cell death after ischemia. Although caspase-2 is unlikely to be involved directly in ischemia-induced CA1 pyramidal cell death, a possible role for caspase-2 in ischemia-induced apoptosis of other populations of neurons (e.g., in the cerebral cortex) is under investigation. Nonetheless, the existing data suggest that the development of brain-penetrable inhibitors of caspase-3 may be a useful therapeutic approach to preventing the delayed neurodegeneration that occurs after global or focal ischemic brain injury.
Footnotes
Acknowledgment
The authors thank Dr. S. Kumar for kindly providing caspase-2 cDNA.