
Abstract
Deoxyribonucleic acid fragmentation at nucleosomal junctions is a hallmark of neuronal apoptosis in ischemic brain injury, for which the mechanism is not fully understood. Using the in vitro cell-free apoptosis assay, the authors found that caspase-3–dependent deoxyribonuclease activity caused internucleosomal DNA fragmentation in brain-cell extracts in a rat model of transient focal ischemia. This in vitro deoxyribonuclease activity was completely inhibited by purified inhibitor of caspase-activated deoxyribonuclease protein, the specific endogenous inhibitor of caspase-activated deoxyribonuclease, or by caspase-activated deoxyribonuclease immunodepletion. The induction of the deoxyribonuclease activity was correlated with caspase-3 activation and caspase-3–mediated degradation of inhibitor of caspase-activated deoxyribonuclease. Furthermore, inhibiting caspase-3–like protease activity prevented the endogenous induction of internucleosomal DNA fragmentation in the ischemic brain. These results suggest that caspase-3–dependent caspase-activated deoxyribonuclease activity plays an important role in mediating DNA fragmentation after focal ischemia.
Induction of DNA fragmentation at the nucleosomal junctions (internucleosomal DNA fragmentation) is a characteristic manifestation of apoptosis and a highly reproducible marker for neuronal cell death in cerebral ischemia (Linnik et al., 1993; MacManus et al., 1995; Chen et al., 1997; Fujimura et al., 1999), for which the molecular mechanism is not fully understood. Based on the observations that internucleosomal DNA fragmentation and other apoptotic nuclear changes resulting from ischemic injury can be blocked by inhibiting caspase-3like protease activity, it has been suggested that caspase-3 may be the key executioner of nuclear degradation in ischemic neurons (Graham and Chen, 2001). A potential mechanism by which caspase-3 contributes to internucleosomal DNA fragmentation in ischemic neurons may be the activation of caspase-activated deoxyribonuclease (CAD), a gene product that is highly conserved across species (Enari et al., 1998; Mukae et al., 1998; Chen et al., 2000; Mukae et al., 2000). Caspase-activated deoxyribonuclease normally exists in the cell as a nonactive heterodimeric complex with its natural inhibitor, ICAD. During apoptosis, inhibitor of caspase-activated deoxyribonuclease (ICAD) is cleaved by caspase-3 and released from the ICAD/CAD complex, leading to spontaneous activation of CAD and subsequent DNA fragmentation (Sakahira et al., 1998). This caspase-3–mediated mechanism of cellular destruction has been extensively studied in nonneuronal cells, but it may also be relevant to neuronal apoptosis resulting from neurologic disorders (Chen et al., 2000).
In this investigation, the potential role of caspase-3–mediated activation of CAD in the induction of internucleosomal DNA fragmentation was studied in a rat model of focal cerebral ischemia and reperfusion. We detected caspase-3–mediated proteolytic cleavage of ICAD and measured ICAD-inhibitable deoxyribonuclease activity in cell extracts from ischemic brains.
MATERIALS AND METHODS
Animal model of ischemia and in vivo drug administration
All experiments were performed on adult male Sprague-Dawley rats (weight, 275–300 g). Focal ischemia was induced in isoflurane-anesthetized rats for 30 minutes using the intraluminal vascular occlusion of the middle cerebral artery, as described previously (Cao et al., 2001). Blood pressure, blood gases, and blood glucose concentration were maintained in the normal range throughout the experiments. Temporalis muscle and rectal temperatures were maintained in the range of 36.5 to 37.5°C. Sham operations were performed in additional animals using the same anesthesia and surgical procedures but without insertion of the intraluminal suture; these brains served as nonischemic controls.
In selective experiments, rats were subjected to intracerebral ventricular infusion of the caspase-3/7 inhibitor z-DEVD.fmk (N-benzyloxycarbonyl- a sp(ome)- g lu(ome)- v al- a sp-(ome)-fluoromethylketone) using a procedure described by Chen et al. (1998). Each animal received a bonus infusion of 4.5 μg z-DEVD.fmk (in 5 μL diluted dimethyl sulfoxide in mock cerebrospinal fluid), or the same volume of vehicle for a 10-minute period at the indicated time points after middle cerebral artery occlusion.
Western blot analysis and immunohistochemistry
Animals were anesthetized using 8% chloral hydrate and decapitated 1, 3, 6, 12, 24, or 72 hours after 30 minutes of ischemia or 24 hours after sham operation (n = 3 per experimental condition). The caudate-putamen, which is highly reproducibly affected by middle cerebral artery occlusion in this model, was dissected for extraction of cellular proteins and immunoblotting using standard methodologies (Chen et al., 1998). The following antibodies were used for Western blot analysis: affinity-purified rabbit polyclonal antibody against the C-terminal sequence of CAD (Chemicon, Temecula, CA, U.S.A.), affinity-purified rabbit polyclonal antibody against rat ICAD, which recognized both intact and the larger cleavage form of ICAD (Chen et al., 2000); and rabbit polyclonal antibody recognizing the active form (p17) of caspase-3 (Chen et al., 1998).
Immunofluorescent staining for activated caspase-3 was performed using frozen coronal sections prepared from brains 12 and 24 hours after ischemia or sham operation (n = 3 per condition). The procedures for immunofluorescence (caspase-3) and double-label immunofluorescence [caspase-3 and neuron-specific nuclear protein (NeuN) or caspase-3 and TUNEL (terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling)] were the same as previously described (Chen et al., 1998; Cao et al., 2001).
Measurement of caspase-3–like activity
The animals were killed 1, 3, 6, 12, 24, or 72 hours after ischemia or sham surgery (n = 4 per group). Brains were quickly removed and tissues were dissected from the caudate of the two hemispheres separately. The procedures for protein extraction and subsequent measurement of caspase-3–like protease activity in cell extracts were performed as described previously (Cao et al., 2001). The substrate for the measurement of caspase-3like activity was Ac-DEVD-AFC (Medical and Biological Laboratories, Watertown, MA).
Detection of CAD activity in cell extracts
This assay measures the deoxyribonuclease activity in cellular extracts under apoptotic conditions (Liu et al., 1997; Chen et al., 2000). To determine whether CAD activity was induced in the brain after ischemia, 100 μg protein was extracted from the caudate of brains 6, 24, or 72 hours after 30 minutes of ischemia or after sham operation (n = 3 per condition) and incubated with genomic DNA isolated from normal brain cells (5 × 105 per reaction) overnight at 32°C in the reaction buffer supplemented with an adenosine triphosphate-regeneration system (Chen et al., 2000). The reaction was terminated by adding the buffer containing 50 mmol/L Tris-HCl (pH 8.0), 10 mmol/L edetic acid, protease K (0.5 mg/mL), and ribonuclease (5 μg/mL), and incubated at 50°C for 1 hour. The DNA was extracted with two changes of chloroform-isoamyl alcohol (volume-to-volume ratio, 24:1) and precipitated overnight with cold ethanol. The pellet was resuspended in Tris/EDTA buffer, and DNA samples (5 μg) were incubated in 50 μL terminal deoxynucleotidyl transferase buffer containing 15 μCi α-32 P-dideoxyadenosine triphosphate and 30 U terminal deoxynucleotidyl transferase for 2 hours at 27°C. The reaction was stopped by the addition of 2 μL 0.5M edetic acid. The DNA was precipitated, electrophoresed on an agarose gel, and autoradiographed onto X-Omat RP-5 X-Ray film (Eastman Kodak, Rochester, NY).
Two methods were used to determine the specific CAD activity using the above assays. In the first, purified caspase-resistant rat ICAD recombinant protein (0.5 μg/mL) was added at the beginning of reactions to test whether the deoxyribonuclease activity present in the nuclear extracts could be inhibited. The production, purification, and characterization of this recombinant protein have been described previously (Chen et al., 2000). In the second, nuclear extracts were immunoprecipitated using the anti-CAD antibody to deplete the endogenous CAD protein before the activity assays.
Data analysis
All data are presented as mean ± SD. Comparisons between experimental groups were made using analysis of variance and post hoc Fisher t-tests. A level of P < 0.05 was considered statistically significant.
RESULTS
Evidence of CAD activation after ischemia
Representative Western blots showing the proteolytic degradation of ICAD in the ischemic caudate-putamen is presented in Fig. 1A. Degradation of ICAD from its intact form (35 kDa) to the larger fragment (16.5 kDa) was significantly increased 6 to 72 hours after 30 minutes of ischemia (P < 0.05); this degradation was absent in the contralateral hemisphere or in sham-operated control brains. Furthermore, intracerebral ventricular infusion of z-DEVD.fmk completely blocked the degradation of ICAD in the brain 24 and 72 hours after ischemia (Fig. 1B).
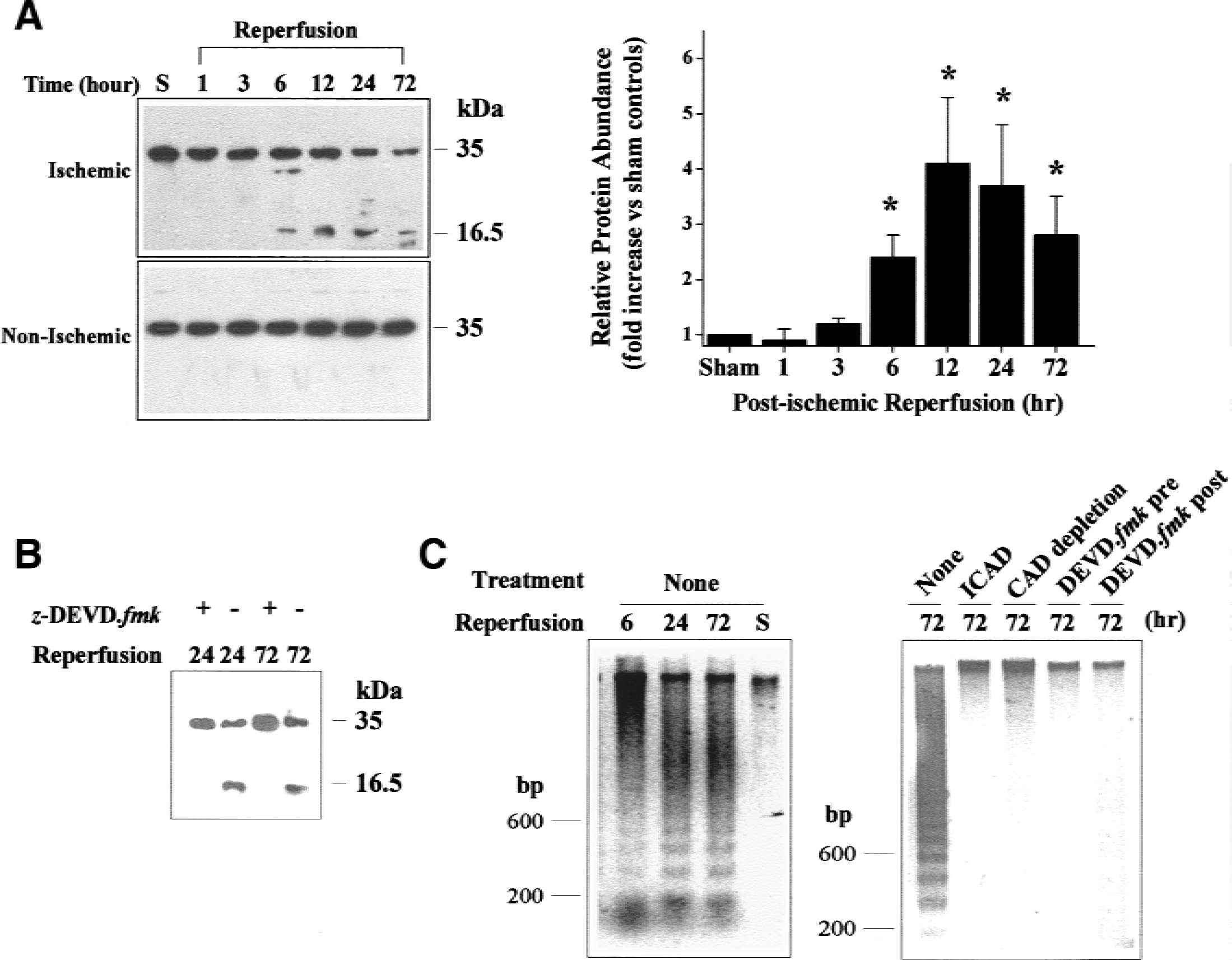
Caspase-activated deoxyribonuclease (CAD) activation after focal ischemia and reperfusion.
The DNA fragmentation-inducing activity (deoxyribonuclease activity) was clearly detectable in caudate-putamen cell extracts obtained 6, 24, or 72 hours after 30 minutes of ischemia (Fig. 1C), but not in sham-operated control brains. This deoxyribonuclease activity was completely blocked by adding the caspase-resistant ICAD recombinant protein to the reaction mixture or by immunodepletion of CAD protein in the cell extracts before the reaction. Moreover, the deoxyribonuclease activity was not detectable in extracts prepared from brains that received z-DEVD.fmk infusion 30 minutes before or 3 hours after ischemia.
Caspase-3 activation and endogenous DNA fragmentation after ischemia
Gel electrophoresis was performed to determine whether endogenous induction of internucleosomal DNA fragmentation in ischemic caudate-putamen was the consequence of caspase-3 activation. As shown in Fig. 2a, internucleosomal DNA fragmentation was detected in the caudate-putamen at 12, 24 or 72 h after ischemia. z-DEVD.fmk, when administrated at 3 or 6 h, but not at 12 h, after ischemia, prevented the induction of DNA fragmentation after an ischemic insult (Fig. 2b).
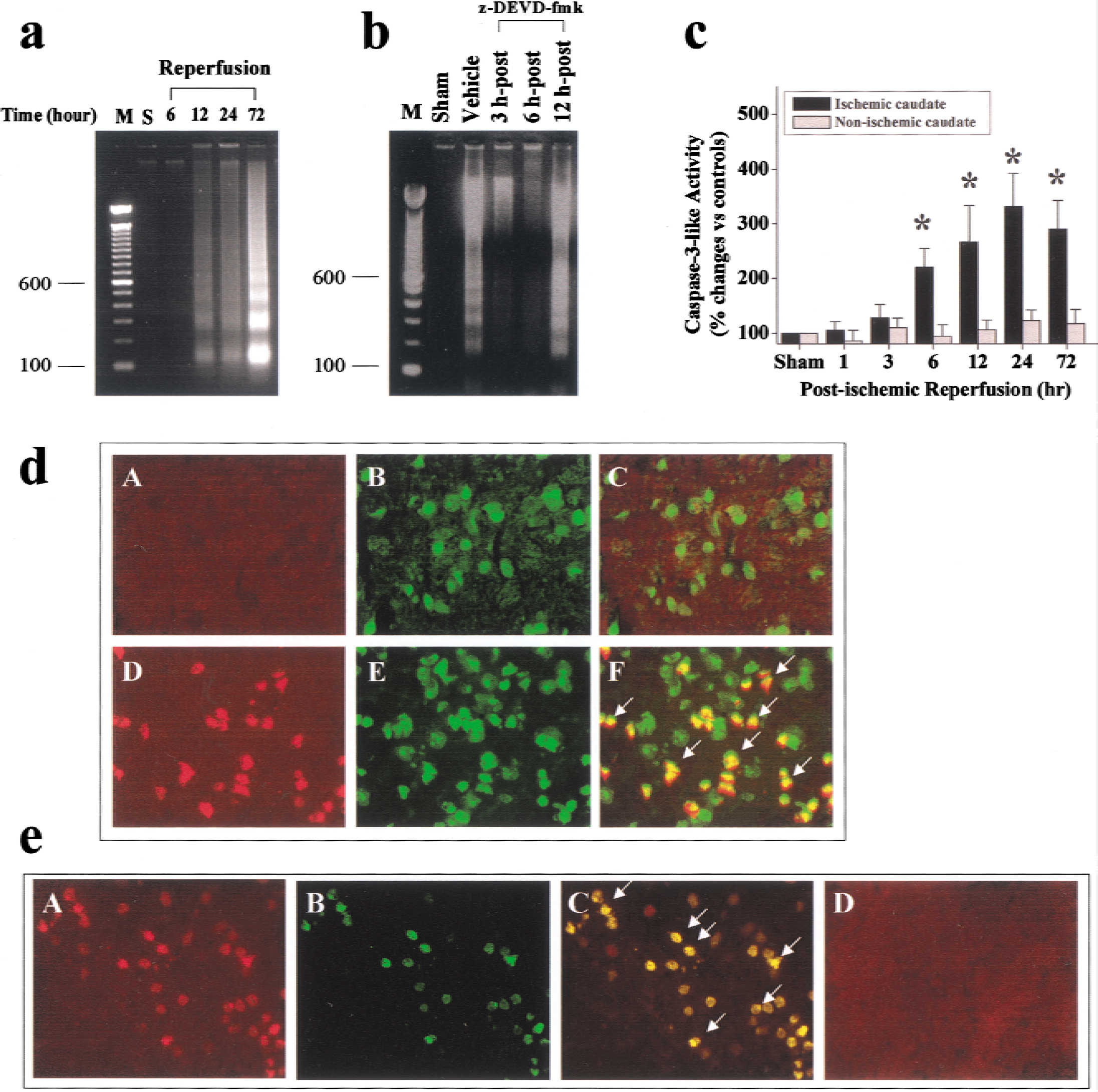
Caspase-3–mediated induction of endogenous DNA fragmentation after ischemia.
Measuring caspase-3like protease activity and performing immunohistochemistry confirmed the activation of caspase-3 in the ischemic caudate-putamen. Caspase-3like activity was significantly increased in the caudate 6 to 72 hours after ischemia (Fig. 2c); this increase was absent in the nonischemic hemisphere or sham-operated control brains.
Immunofluorescent staining showed the activation of caspase-3 in the ischemic caudate-putamen at the cellular level (Fig. 2d). Increased caspase-3 immunofluorescence in ischemic brains was detected primarily in neurons (NeuN positive). A colocalization of caspase-3 immunoreactivity and DNA fragmentation (TUNEL positive) was also detected in the nucleus of many neurons 72 hours after ischemia (Fig. 2e).
DISCUSSION
The major finding of this study is that the caspase-3–dependent CAD activity was induced in the brain after focal ischemia and reperfusion. The induced CAD activity was spatially correlated with (caudate-putamen) and preceded by the endogenous induction of internucleosomal DNA fragmentation after ischemia (6 vs. 12–24 hours). Moreover, inhibition of caspase-3like activity in the brain using a tetrapeptide inhibitor diminished CAD activation and prevented endogenous DNA fragmentation after ischemia. Taken together, these results suggest that the activated CAD may be responsible for the induction of internuleosomal DNA fragmentation after ischemia. These findings add to the currently growing body of evidence emphasizing the role of caspase-3–mediated mechanisms in the final execution of neuronal apoptosis in various forms of central nervous system injuries (Yakovlev et al., 1997; Namura et al., 1998; Springer et al., 1999; Zhang et al., 1999; Citron et al., 2000; Clark et al., 2000).
Three lines of evidence derived from this study support the specificity of the CAD activity detected in cell extracts using the cell-free apoptosis assay. First, there was clear evidence of caspase-3–mediated proteolytic degradation of ICAD in the ischemic samples, and the temporal profile of caspase-3 activation coincided with that of ICAD degradation and induction of the CAD activity. Second, the DNA fragmentation–inducing activity in the cell extracts was completely blocked by the purified ICAD recombinant protein. Third, immunodepletion of CAD in the cell extracts abolished the DNA fragmentation-inducing activity. However, this assay was unable to identify the phenotypes of cells that expressed CAD activity. Based on the results of double-label caspase-3 immunofluorescent staining, which showed that the majority of caspase-3–activated cells (> 90%) were neurons (NeuN positive), it is likely that caspase-3–dependent CAD activity was induced in neurons after ischemia. However, this speculation needs to be confirmed in future studies.
In summary, this study provides the first direct evidence that a caspase-3–dependent and ICAD-inhibitable deoxyribonuclease activity is induced in the brain after focal ischemia and reperfusion. This observation suggests a novel mechanism by which internucleosomal DNA fragmentation is induced in the ischemic brain. Because ICAD is the specific endogenous inhibitor for CAD activity, future studies are warranted to determine whether enhancement of cellular ICAD activity can prevent ischemia-induced DNA degradation as an additional therapeutic strategy for the management of experimental stroke.
Footnotes
Acknowledgments:
The authors thank Carol Culver for editorial assistance and Pat Strickler for secretarial support.