
Abstract
Gene transfer involves the use of an engineered biologic vehicle known as a vector to introduce a gene encoding a protein of interest into a particular tissue. In diseases with known defects at a genetic level, gene transfer offers a potential means of restoring a normal molecular environment via vector-mediated entry (transduction) and expression of genes encoding potentially therapeutic proteins selectively in diseased tissues. The technology of gene transfer therefore underlies the concept of gene therapy and falls under the umbrella of the current genomics revolution. Particularly since 1995, numerous attempts have been made to introduce genes into intracranial blood vessels to demonstrate and characterize viable transduction. More recently, in attempting to translate cerebrovascular gene transfer technology closer to the clinical arena, successful transductions of normal human cerebral arteries ex vivo and diseased animal cerebral arteries in vivo have been reported using vasomodulatory vectors. Considering the emerging importance of gene-based strategies for the treatment of the spectrum of human disease, the goals of the present report are to overview the fundamentals of gene transfer and review experimental studies germane to the clinical translation of a technology that can facilitate genetic modification of cerebral blood vessels.
It is clear that potential key benefits of a gene therapy approach include correction of a disease at its molecular origin and use of the host's own transcriptional and translational apparatus as an integral part of the therapeutic process. However, to use this technology successfully in humans, the following salient elements of an optimal gene therapy paradigm (Friedmann and Roblin, 1972) must be addressed (Table 1):
The disease in question must have known molecular pathogenesis to construct a specific and effective vector. The vector should be engineered to contain at least one gene of interest (encoding at least one potentially therapeutic protein) relevant to the disease in question. A genetic regulatory element, such as a promoter, that is tissue-specific or responsive to local concentrations of an exogenously administered drug can be incorporated into a vector to confer added control over the site and degree of recombinant protein expression. There must be comprehensive preclinical work involving the vector (including substantial data from relevant in vivo experiments in animals and, where possible, ex vivo experiments using human tissues) supporting a protocol designed for use of that vector in humans. A highly purified and biosafe version of the vector (i.e., a clinical grade vector) should be available for use in humans Patients in whom the vector is to be introduced must be fully informed of and give consent to the procedure and should be aware of risks known to be associated with this technology (Brenner, 2000; Verma, 2003). There should be an appropriate means of delivering the vector into a patient and a system in place for safely and effectively tracking its uptake and distribution. Objective means of detecting and quantifying clinical outcome must be available to accurately ascertain the impact of the technology on the patient and the course of the disease. The entire process needs to be subject to rigorous peer review at both an intramural level (via an Institutional Review Board and Biosafety Committee) and an extramural level (via government funding bodies such as the National Institutes of Health; NIH). Federal regulatory bodies equivalent to the NIH Recombinant Advisory Committee (RAC), and the Food and Drug Administration (FDA) in the United States should oversee the approval and subsequent follow-up for all human gene therapy protocols.
Key elements of an optimal gene therapy paradigm

In the context of the above criteria, despite numerous excellent and pioneering laboratory studies involving gene transfer to intracranial blood vessels (see reviews by Chen et al., 1998; Gunnett and Heistad, 2002; Heistad and Faraci, 1996; Khurana and Katusic, 2001; Weihl et al., 1999), to date there has been no published clinical trial with the goal of targeting and genetically modifying the cerebral vasculature. There is, however, an array of cerebrovascular disease entities that may be amenable to such a trial. Potential disease candidates include intracranial atherosclerosis, hypertensive arteriopathy, ischemic stroke, inflammatory or immune-mediated vasculitis, Moyamoya disease and Takayasu's arteritis (including a gene therapy-based proangiogenic adjunct to cerebral revascularization procedures), cerebral vasospasm following aneurysmal subarachnoid hemorrhage (SAH), venous hypertension, arteriovenous malformations, and neoplasms in which antiangiogenic gene transfer may be beneficial (Anderson and Meyer, 2000; Chen et al., 1998; Freedman and Isner, 2001; Gunnett and Heistad, 2002; Heistad and Faraci, 1996; Khurana and Besser, 1997; Khurana and Katusic, 2001; Meyer et al., 1987; Regli et al., 1996; Ueki et al., 1994). The goals of the present report are, therefore, to overview the fundamentals of cerebrovascular gene transfer technology and, in particular, review those studies germane to clinical translation of this technology as it gradually makes its passage toward a clinical trial.
PRINCIPLES OF CEREBROVASCULAR GENE TRANSFER
At the outset, it should be noted that we define cerebrovascular gene transfer as the process whereby genetic information is primarily introduced into cellular components of cerebral blood vessels and not the process whereby cerebral blood vessels are used merely as conduits for the transfer of genetic information across the blood brain barrier and into cells of the central nervous system (CNS). The goals of gene transfer for the treatment of intracranial vascular disease are to safely, specifically, and efficiently introduce genetic material into the wall of a cerebral blood vessel to produce a substance that favorably modulates vessel growth, function, or both (Chen et al., 1998; Gunnett and Heistad, 2002; Heistad and Faraci, 1996; Khurana and Katusic, 2001). Key issues to consider in any cerebrovascular gene transfer paradigm include the type of vector to be used, the gene (or genes) to transfer, and the means of achieving targeted vector delivery. These can be illustrated in the example of post-SAH cerebral vasospasm, a prototypic disease model for cerebrovascular gene transfer experiments in animals (see below).
Cerebral vasospasm, which can complicate rupture of an intracranial aneurysm, involves transient narrowing of the cerebral arteries in response to the pathologic presence of blood in the subarachnoid space (Khurana and Besser, 1997; Weir, 1995) (Fig. 1). This narrowing may be severe enough to cause symptoms and signs of cerebral ischemia and accounts for approximately 20% of the morbidity and mortality after aneurysmal rupture. The pathogenesis of post-SAH vasospasm is complex and remains incompletely understood (Dietrich and Dacey, 2000; Khurana and Besser, 1997), but there is extensive evidence that it is (1) triggered by the formation and release of oxyhemoglobin from erythrocytes degraded in the subarachnoid blood clot, and (2) maintained, at least in part, by impaired local bioavailability and action of a key vasodilatory molecule, nitric oxide (NO) (Fig. 2). In this context, it has been the hypothesis of several cerebrovascular gene transfer studies (Chen et al., 1997a,1997b; Khurana et al., 2000b, 2002; Onoue et al., 1998; Stoodley et al., 2000) that a vector such as a recombinant adenovirus encoding the gene for endothelial nitric oxide synthase (eNOS) can be delivered to cerebral arteries to augment and hopefully restore local production of NO in the setting of vasospasm (Fig. 3).
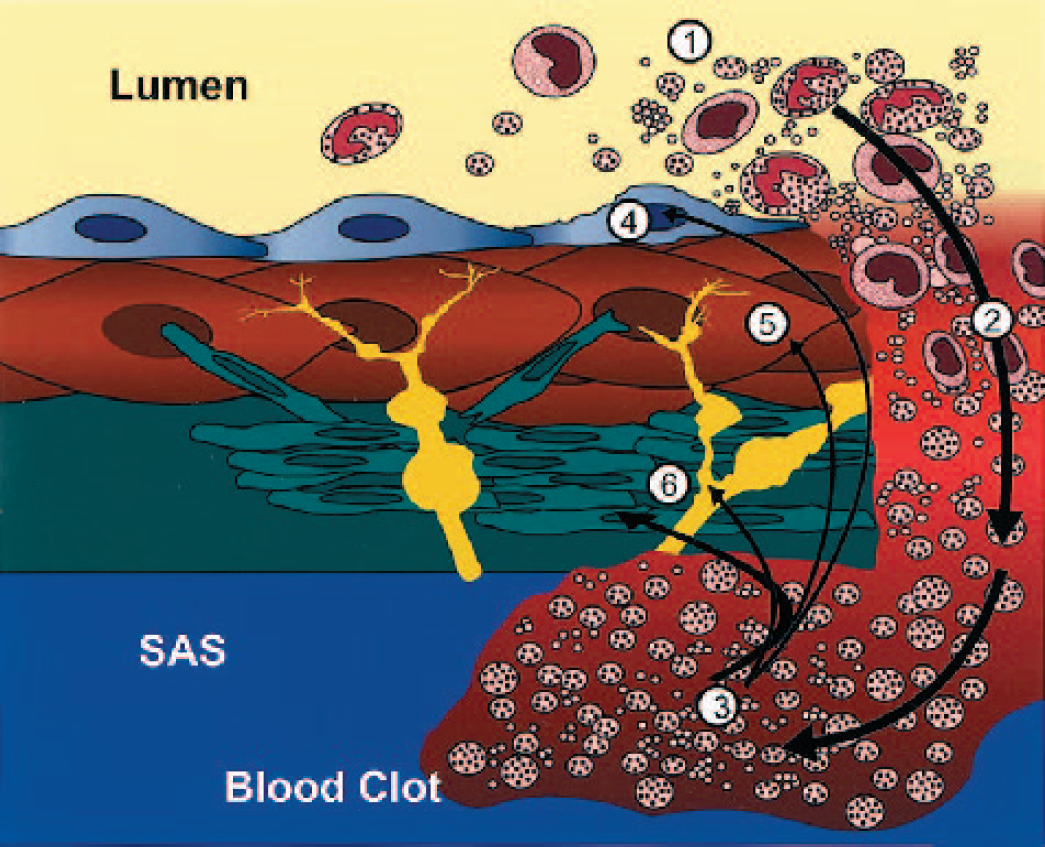
Pathogenesis of cerebral vasospasm after aneurysmal subarachnoid hemorrhage. After intracranial aneurysm rupture, blood products egress from the arterial lumen (1) through the ruptured aneurysm wall (2) and into the subarachnoid space (SAS). Here, a blood clot (thrombus) forms (3), which is intrinsically vasoactive. Degeneration of blood products in the clot leads to the formation of oxyhemoglobin, which triggers a cascade of pathologic events in the vascular endothelium (4), smooth muscle (5), and adventitia (6), leading to sustained contraction of the arterial wall and secondary brain ischemia. (Yellow) adventitial neurons; (green) fibroblasts.
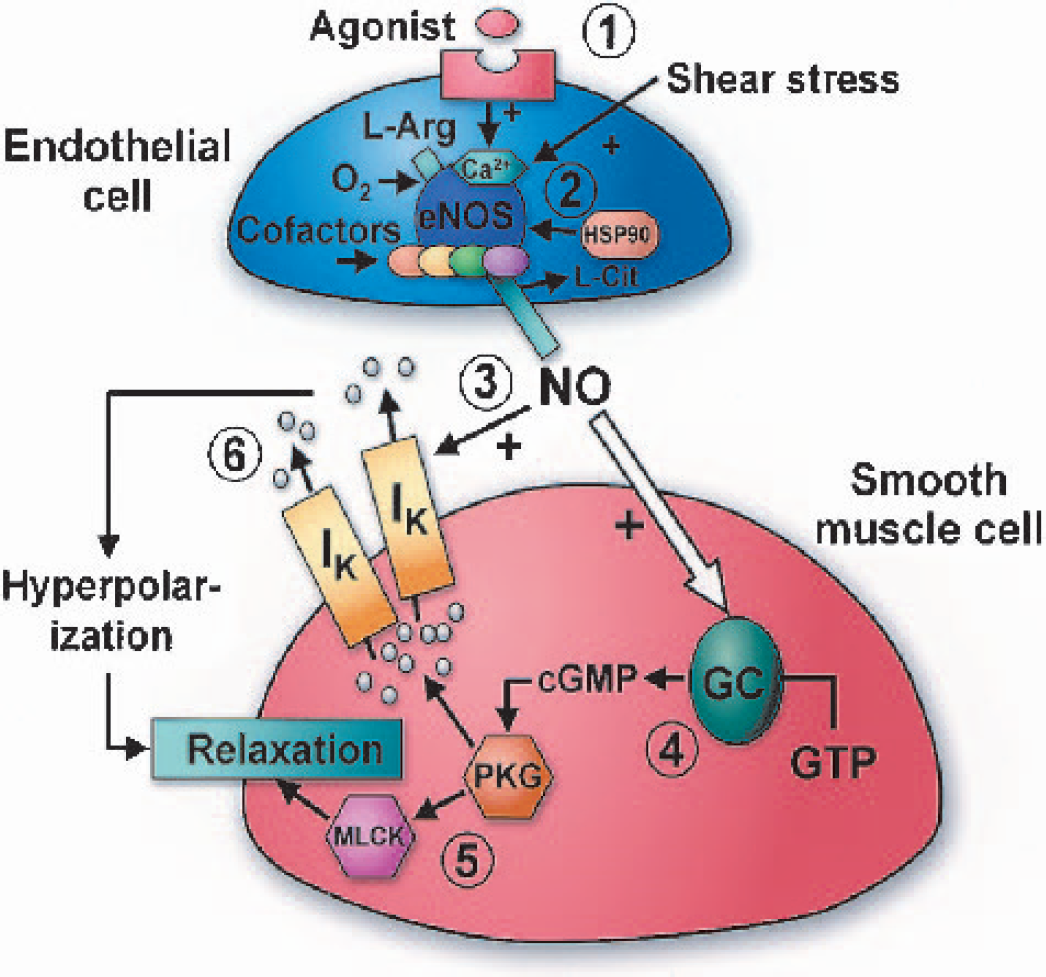
Nitric oxide biosynthesis and action in blood vessels. Biosynthesis of the key endogenous vasodilator NO is principally performed by the calcium-dependent endothelial isoform of eNOS. This is triggered by the binding of agonists or by shear stress (1) and facilitated by a variety of cofactors and the molecular chaperone HSP90. The amino acid L-Arg is converted by eNOS into NO (2), with L-Cit as a byproduct. NO diffuses into adjacent smooth muscle cells (3) where it activates its effector enzyme, GC. GC (4) converts GTP into the second messenger cGMP, which activates PKG (5), leading to modulation of myosin light chain kinase and smooth muscle relaxation. PKG also modulates the activity of potassium channels (IK; 6), thereby increasing cell membrane hyperpolarization and causing relaxation. As shown, NO can also modulate potassium channels in a direct, cGMP-independent manner (Bolotina et al., 1994). NO, nitric oxide; eNOS, nitric oxide synthase; HSP90, heat-shock protein 90; L-Arg, L-arginine; L-Cit, L-citrulline; GC, guanylate cyclase; GTP, guanosine triphospate; cGMP, cyclic guanosine monophosphate; PKG, protein kinase G.
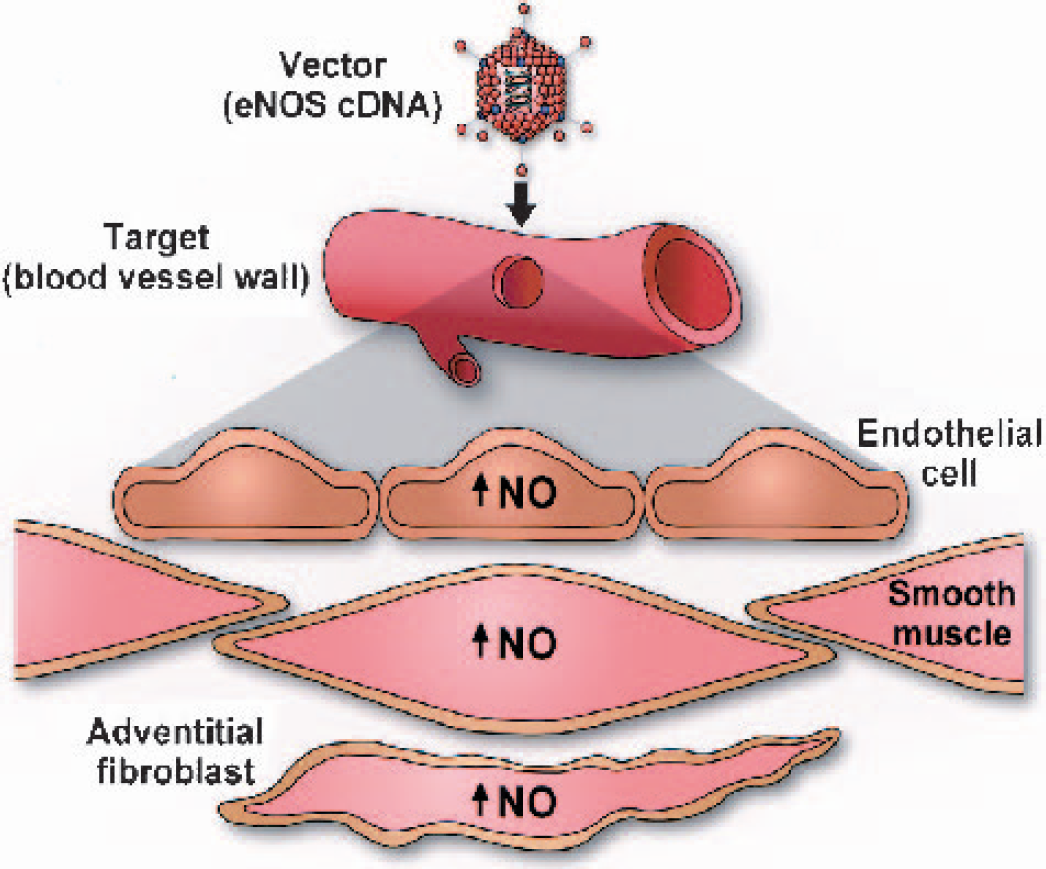
Blood vessel transduction. An engineered biologic vehicle known as a vector (e.g., a replication-incompetent adenovirus or naked DNA in the form of a plasmid) containing a gene of interest (e.g., cDNA for eNOS, the endothelial isoform of nitric oxide synthase) is introduced into a target tissue (e.g., blood vessel wall). The vector gains entry into cells of the target tissue (i.e., a process known as transduction) where it produces a protein (in this case, recombinant eNOS) encoded by the gene of interest. Using this approach, owing to relative overexpression of eNOS, NO biosynthesis is effectively augmented in the blood vessel wall. In vascular diseases such as atherosclerosis, diabetes mellitus, vasospasm, and hypertension, all of which involve impaired NO-mediated signaling, such an approach may be potentially therapeutic. NO, nitric oxide.
Vectors
The choice of biologic vehicle to deliver a recombinant gene into an artery can be broadly divided into viral versus nonviral versus hybrid vectors (Dyer and Herrling, 2000; Heistad and Faraci, 1996; Richter et al., 2000). Viral vectors may be RNA viruses (retroviruses), such as mouse Moloney leukemia virus (MoMLV), or lentiviruses, including human (HIV), bovine, and simian immunodeficiency viruses. DNA viral vectors, on the other hand, include strains linked to the common cold pathogen (adenovirus), herpes simplex virus, or parvoviruses such as adeno-associated virus (AAV). Nonviral vectors include naked DNA (plasmids) and DNA-containing cationic lipid particles (liposomes). Hybrids such as plasmid-liposome (Ono et al., 1998) and Sendai virus-liposome (Von der Leyen et al., 1995) conjugates have also been developed and used in vascular gene transfer. Each of these classes of vectors has a characteristic profile related to genomic capacity (relevant to the size of genetic information of interest, i.e., the “expression cassette”, that can be inserted into the vector), DNA integration (i.e., propensity to incorporate into the host or target cell's genome), efficiency of transduction, cell avidity (i.e., ability to infect dividing, nondividing cells, or both), induced inflammatory response, and length of expression (i.e., how long the vector-transduced cell can produce the recombinant product).
In general, viral vectors demonstrate appreciably greater gene transduction efficiency compared with nonviral vectors (Dyer and Herrling, 2000; Heistad and Faraci, 1996). DNA integration after entry into the nucleus is a feature of RNA viruses and the DNA-containing AAV but not adenovirus, which remains epichromosomal. Although the benefit of DNA integration is that it results in relatively long-term recombinant gene expression, there is an appreciably higher risk of insertional mutagenesis. This type of risk is real and has recently received much attention regarding the development of a leukemic illness in some infants with subacute combined immunodeficiency (SCID) who were injected with retrovirus-altered autologous hemopoietic stem cells as part of approved human gene therapy protocols (Verma, 2003). With regard to the type of cell infected by viruses, MoMLV exclusively targets dividing cells (thereby limiting tissue selection and in vivo applications), whereas lentiviruses can transduce some but not all types of dividing and nondividing cells. On the other hand, adenoviruses and AAV have the broadest known cell avidity. The major disadvantages of adenoviruses are their propensity to induce a brisk inflammatory response in vivo, their immunogenicity upon reexposure, and relatively short-term transgene expression (Newman et al., 1995; Thomas et al., 2000; Vassalli et al., 1999; Wen et al., 2000; Wood et al., 1996). The advent of newer generation “gutted” adenoviruses with minimal native viral genome is a move toward less immunogenic and cytotoxic vectors (Brenner, 2000; Dyer and Herrling, 2000; Richter et al., 2000). Hybrid vectors, such as virus-liposome conjugates, may yield certain advantages in terms of improved efficiency of gene transfer compared with the use of liposomes alone and reduced inflammatory response compared with viruses alone (Dyer and Herrling, 2000; Von der Leyen et al., 1995).
To date, the adenovirus (Fig. 4), particularly serotype 5, remains the predominant vector used in cerebrovascular gene transfer studies (Table 2), most likely because of its broader cell avidity, greater efficiency of transduction, and ability to be generated in relatively high titers (Chen et al., 1998). As elaborated below, a considerable amount of information has been acquired regarding this vector and its applications. Briefly, for use in gene transfer, the adenoviral genome is combined with a gene of interest whose expression (i.e., transcription followed by translation into a particular protein) is driven by a promoter, frequently a cell-nonspecific one derived from cytomegalovirus (CMV). The adenovirus is rendered replication-incompetent through the deletion of certain replication-associated genetic sequences (e.g., early regions E1 and E3) (Chen et al., 1997b; Heistad and Faraci, 1996). Entry of the modified virus into target cells (Fig. 5) typically involves attachment of the viral fiber knob to the host-cell plasmalemma facilitated by the coxsackie virus-adenovirus receptor (CAR) (Bergelson et al., 1997) and is followed by viral pentone-base and host αv-integrin mediated internalization (Wickham et al., 1993). Once it has entered the cell, the adenovirus retains an epichromosomal (nonintegrated) position in the nucleus and uses the biosynthetic machinery of the host to generate the recombinant protein of interest. In experimental models, expression of such proteins is detectable morphologically, biochemically, and functionally. It is important to note that most cerebrovascular gene transfer studies involving adenoviruses have used early generation vectors, which contain considerably more viral genome than the more recent adeno-associated and “gutted” vectors, thereby accounting for increased immunogenicity and cytotoxicity, particularly from the biosynthesis of peptides derived from nondeleted late-region adenoviral genome sequences (Blau and Springer, 1995; Heistad and Faraci, 1996; Khurana et al., 2002; Newman et al., 1995; Richter et al., 2000; Thomas et al., 2000; Vassalli et al., 1999; Wen et al., 2000; Wood et al., 1996).
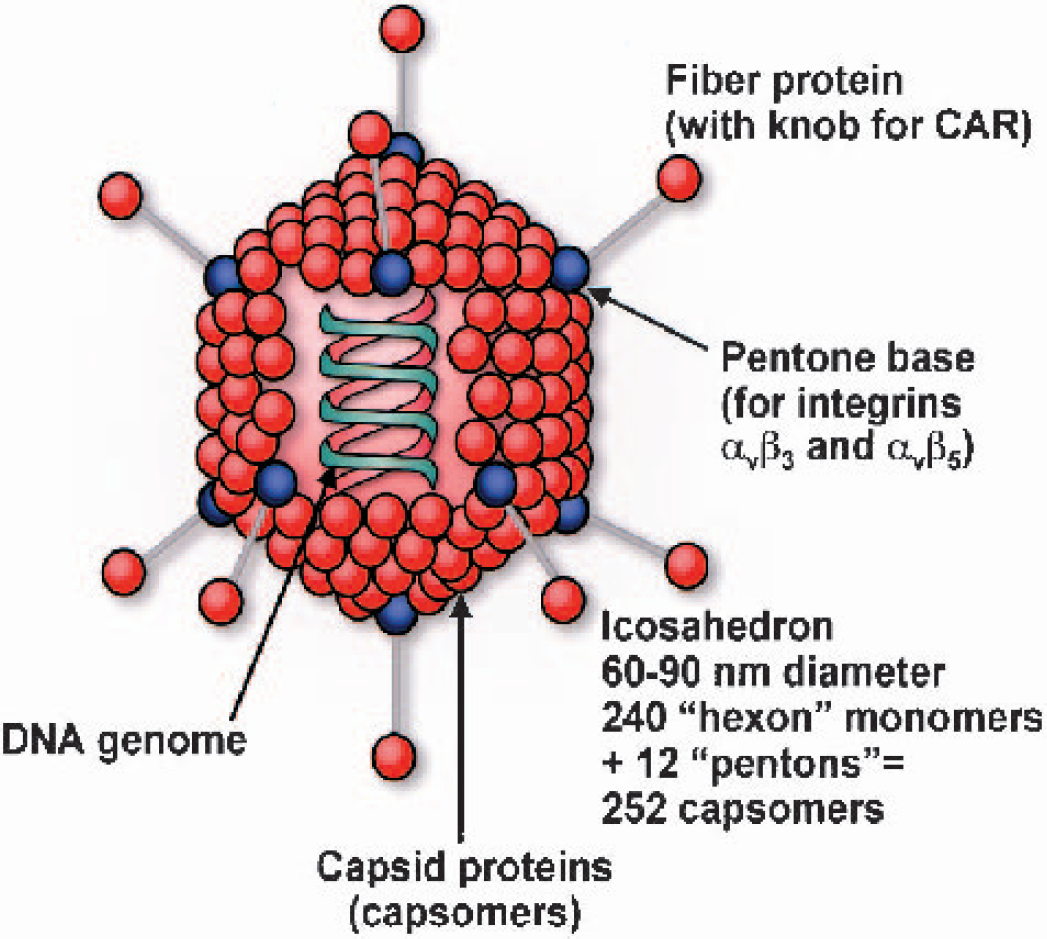
Adenoviral virion. The icosahedral adenoviral virion is comprised of a DNA genome enclosed in a protein shell (capsid). Attachment and cell entry is mediated by the fiber protein (whose knob interacts with the host cell coxsackie virus-adenovirus receptor) and pentone base (which interacts with host αv integrins).
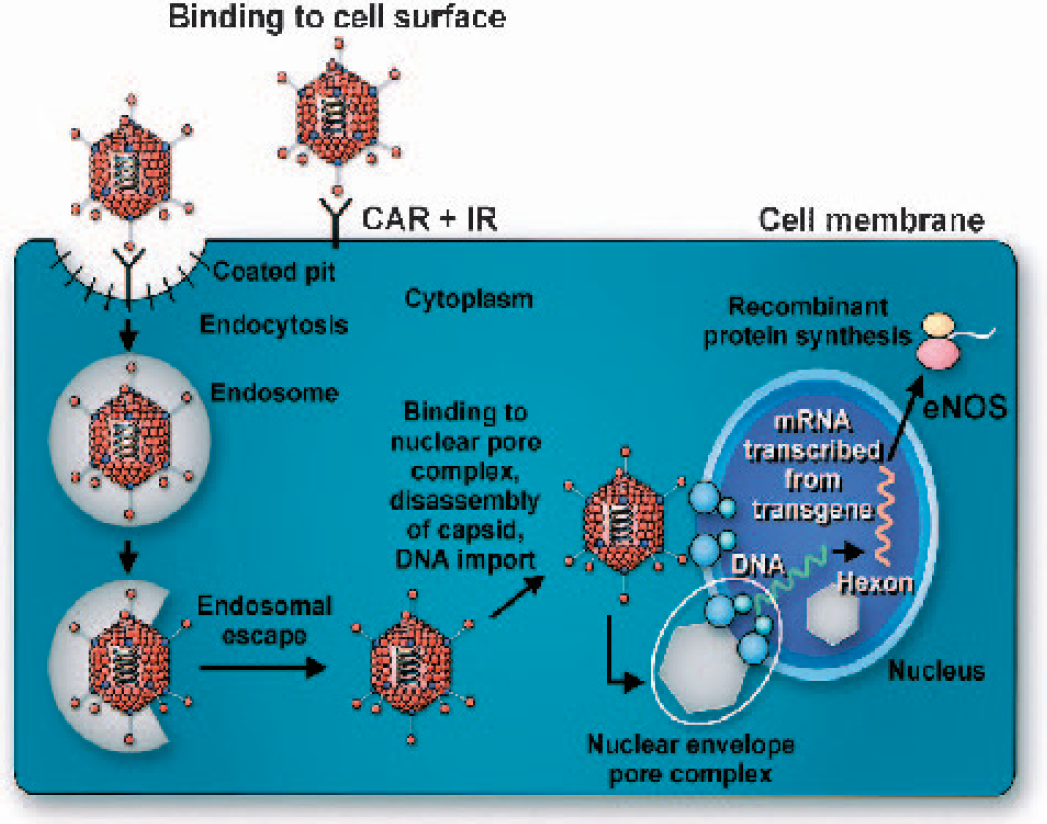
Adenoviral transduction. Recombinant adenovirus enters cells via CAR- and IR-mediated binding. Adenoviral particles are endocytosed, escape from endosomes, and enter into the nucleus via the nuclear envelope pore complex. Genetic material in the adenovirus is not incorporated into the host cell genome, but rather assumes an epichromosomal location, where it can still use the host cell's transcriptional and translational machinery to synthesize recombinant protein, in this case the endothelial isoform of eNOS. CAR, coxsackie virus-adenovirus receptor; IR, integrin receptor; eNOS, nitric oxide synthase.
Gene transfer studies in the cerebral vasculature
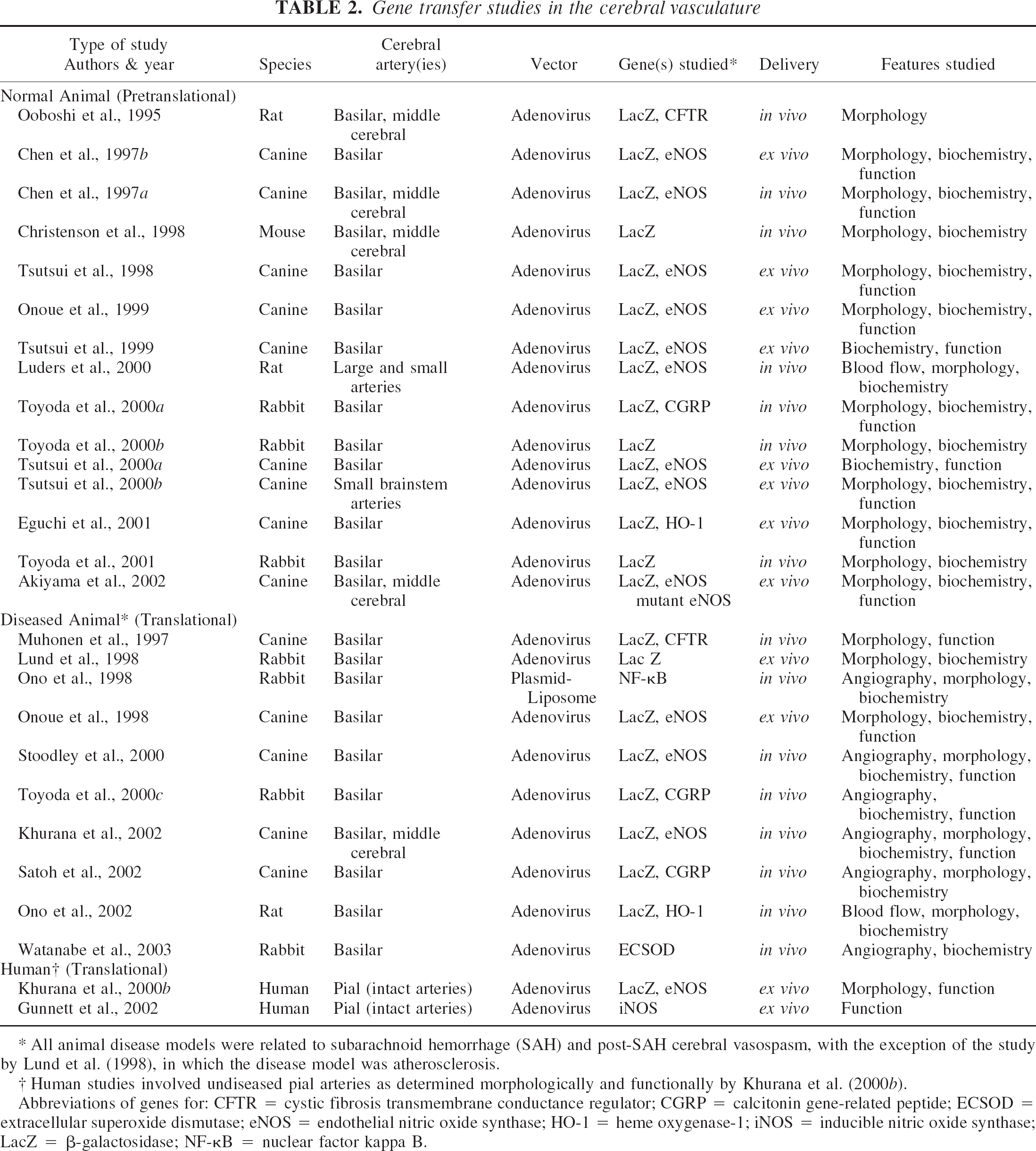
All animal disease models were related to subarachnoid hemorrhage (SAH) and post-SAH cerebral vasospasm, with the exception of the study by Lund et al. (1998), in which the disease model was atherosclerosis.
Human studies involved undiseased pial arteries as determined morphologically and functionally by Khurana et al. (2000b).
Abbreviations of genes for: CFTR = cystic fibrosis transmembrane conductance regulator; CGRP = calcitonin gene-related peptide; ECSOD = extracellular superoxide dismutase; eNOS = endothelial nitric oxide synthase; HO-1 = heme oxygenase-1; iNOS = inducible nitric oxide synthase; LacZ = β-galactosidase; NF-κB = nuclear factor kappa B.
Genes
Selection of an appropriate gene (part of a vector “expression cassette” encoding a potentially therapeutic protein of interest) is based primarily upon the level of understanding of disease pathogenesis. The development of bicistronic vectors capable of expressing more than one gene, the products of which may be amenable to tracking via imaging modalities such as positron emission tomography (PET), has been a major advance in the utility of gene transfer technology (Lin et al., 2002; Wu et al., 2002). Pertaining to the cardiovascular system, the pivotal role played by NO in cerebral vasomotor function and its implication in the pathogenesis of a wide variety of diseases, including atherosclerosis-thrombosis, diabetes mellitus, vasospasm, and hypertension, make this molecule a prime candidate for potentially therapeutic gene transfer (Channon et al., 1996; Chen et al., 1998; Heistad and Faraci, 1996; Hobbs et al., 1999; Khurana and Besser, 1997). This is certainly substantiated in the cerebrovascular gene transfer literature, in which almost one half of the studies to date have involved a vector encoding eNOS cDNA (Table 2). It should be noted that the choice of constitutive eNOS over inducible iNOS is principally based upon the propensity of iNOS to cause undesirable local oxidative stress via free radical generation; for further information pertaining to the choice of NOS isoforms in cardiovascular gene transfer, see the review by Chen et al. (1998).
Regarding the cerebral circulation, there are two other considerations in determining the most appropriate choice of gene to be inserted into a vector. First, NO may be only one of a number of important mediators underlying the pathogenesis of cerebrovascular disease; other candidates include calcitonin gene-related peptide (CGRP), endothelin-1 (ET-1), and altered enzymatic activities of cyclooxygenase, superoxide dismutase (SOD), and heme oxygenase-1 (HO-1) (Chen et al., 1997a; De Belder and Radomski, 1994; Dietrich and Dacey, 2000; Furchgott and Zawadzki, 1980; Khurana and Besser, 1997; Luders et al., 2000; Ono et al., 2002; Onoue et al., 1998, 1999; Toyoda et al., 2000c; Vanhoutte, 1998; Watanabe et al., 2003; Weir, 1995). Therefore, gene transfer using cDNAs encoding one or more of these proteins may need to be considered in addition to NOS gene transfer alone. Second, relative insufficiency of enzymatic substrates, cofactors, or molecular chaperones after NOS gene transfer may potentially limit the efficacy of this process if uncorrected by exogenous or endogenous means (Fleming and Busse, 1999; Garcia-Cardena et al., 1998; Khurana et al., 2000a; Moncada et al., 1991). Further in vivo and ex vivo gene transfer studies along these lines will aid in addressing these important issues, as will information gleaned from cell signaling assays, the Human Genome Project, and the use of microarray technology to precisely identify genes and their products differentially expressed in normal versus diseased cerebral arteries.
Delivery
For cardiovascular gene transfer in vivo, the route of delivery of a vector (Fig. 6) may be either through the vessel lumen, that is, endovascular, or via the outer wall, that is, periadventitial (Caplice, 2001; Chen et al., 1998; Heistad and Faraci, 1996; Khurana et al., 2002, 2003; Kullo et al., 1997; Laitinen et al., 2000). Endovascular transduction involves use of a catheter- or stent-based delivery device (Caplice, 2001; Laitinen et al., 2000). Appreciable limitations include the risk of vascular injury during navigation or deployment of the device, the need for temporary interruption of blood flow leading to tissue ischemia when using certain (nonperfusion) catheters, diffusion of vector distal to the target vessel segment particularly after flow restoration, and possible resistance to transduction from the blood-brain barrier. Periadventitial delivery, on the other hand, may be a sound alternative for several reasons. First, for a rapidly diffusible gas such as NO, this route of delivery may be as effective as, if not more effective than, intraluminal delivery in terms of access of NO to smooth muscle cells of the tunica media, which is somewhat akin to physiologic NO release from perivascular nitrergic nerve endings (Heistad and Faraci, 1996; Thomas et al., 1999). Second, from the periadventitial side, transduction is unhindered by interendothelial tight junctions of the blood-brain barrier and by the subendothelial elastic lamina. Third, choices of periadventitial delivery include injection or infusion of vector into the cerebrospinal fluid (CSF; i.e., intrathecal delivery), the efficacy of which has been repeatedly demonstrated in animal experiments (Chen et al., 1997a; Heistad and Faraci, 1996; Khurana et al., 2002; Ooboshi et al., 1995; Stoodley et al., 2000; Toyoda et al., 2000c) or direct application of vector upon carotid and cerebral arteries ex vivo or during open surgery (Fig. 7) (Khurana et al., 2003). Last, although adventitial fibroblasts do not express endogenous eNOS, they do express receptors that are coupled by calcium to the activation of recombinant enzyme (Onoue et al., 1999; Tsutsui et al., 1998, 2000a). This is fortunate given that these cells are primary targets for adenovirus entry and recombinant eNOS synthesis after periadventitial delivery, as demonstrated using immunoelectron microscopy in animal cerebrovascular gene transfer studies (Chen et al., 1998; Tsutsui et al., 1998) and, more recently, in a study involving intact human cerebral arteries (Khurana et al., 2000b).
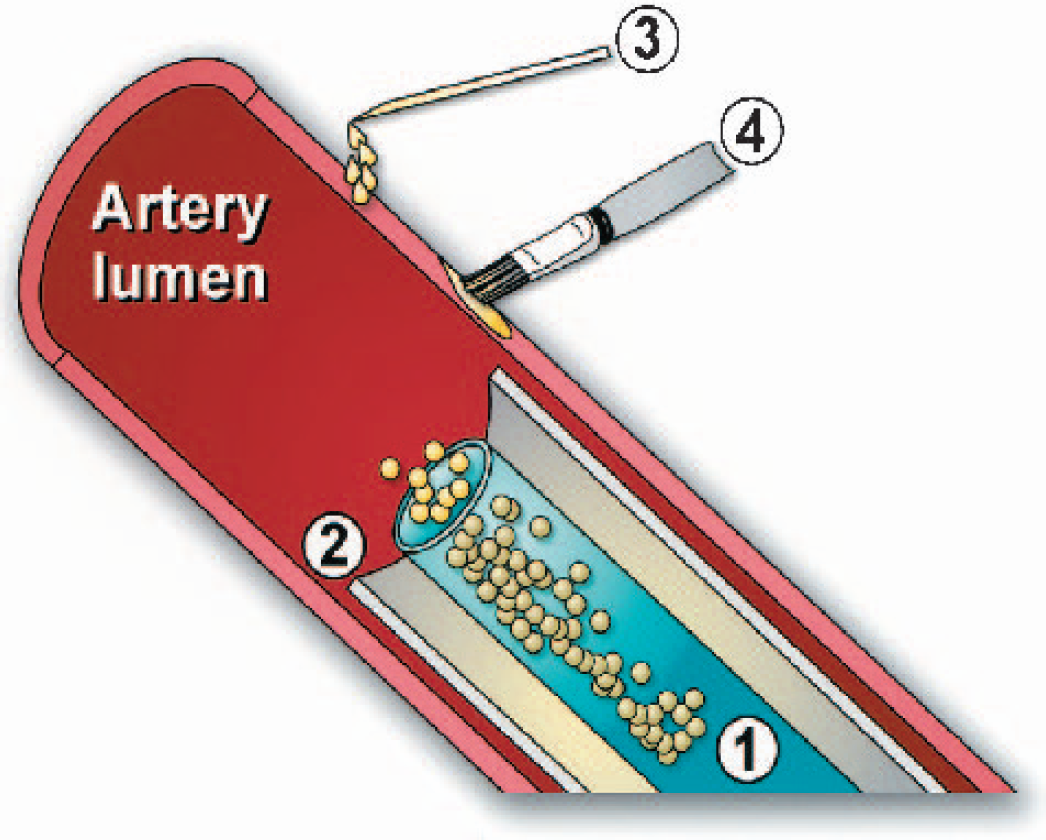
Methods of blood vessel transduction in vivo. Transduction of blood vessels in vivo can be carried out via deployment of an intravascular catheter (1), using a vector-impregnated stent (2), by injection of a vector into the adventitial surface of the artery (3), or by direct paintbrush-mediated adventitial delivery (4).
Paintbrush mechanical technique for cerebrovascular gene transfer.
Transduction
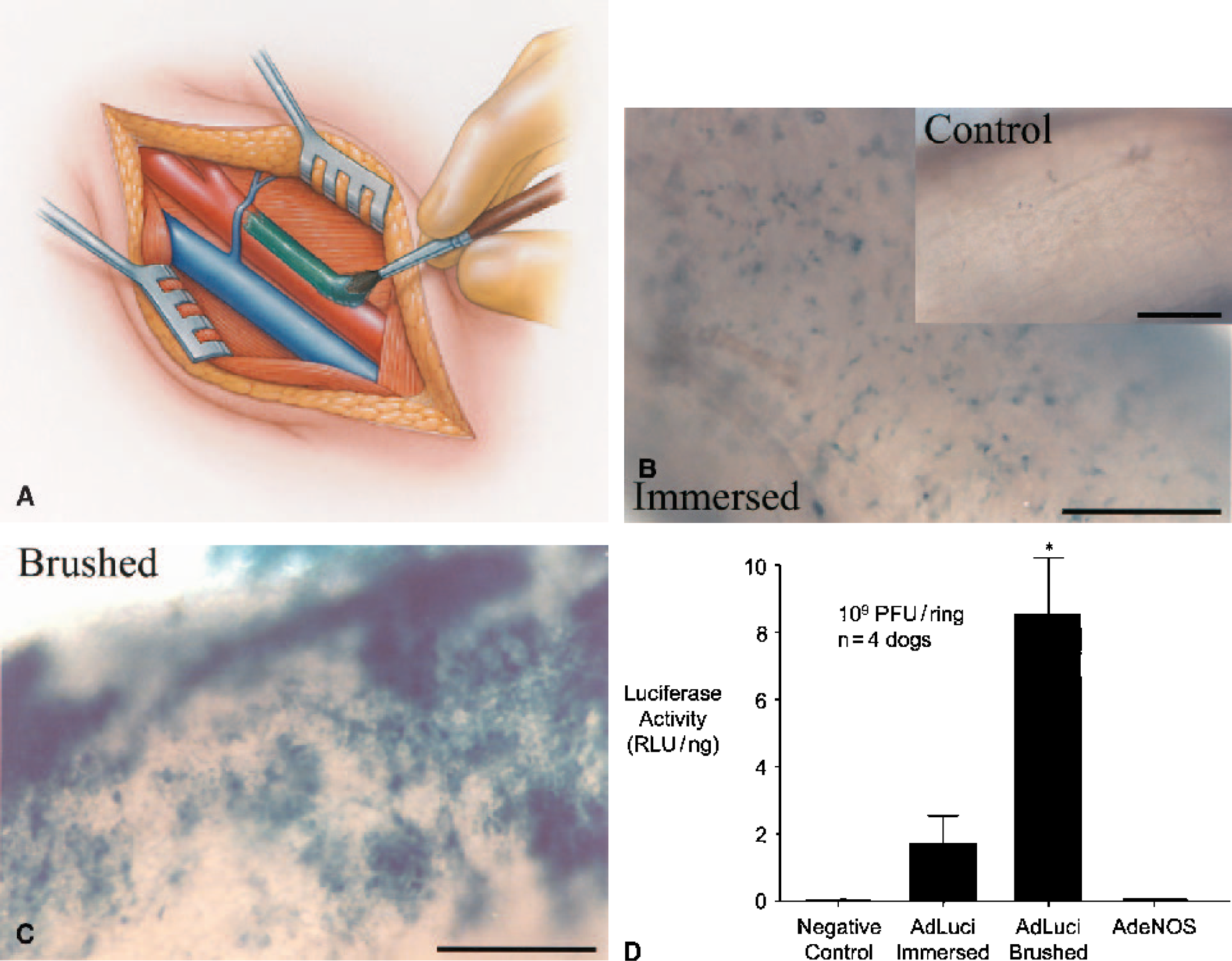
Targeting vectors to specific cells or tissues remains a major obstacle that needs to be overcome before clinical implementation of cerebrovascular gene transfer techniques is achieved. Heistad and colleagues (Heistad and Faraci, 1996; Ooboshi et al., 1995) first reported the use of a mechanical method, namely controlled animal head-tilt, to assist in localizing vectors injected into the CSF via the cisterna magna to arteries in the circle of Willis. Whereas this technique is indeed helpful in this regard, it remains relatively nonspecific and operator-dependent. With the aim of improving vector targeting to tissues, a variety of biochemical methods have also been described (e.g., see Toyoda et al., 2000a, 2001) but again lack in territorial specificity. A molecular targeting technique using a cell-specific promoter such as SM22α (selective for smooth muscle cells; cf. cell-nonspecific CMV-derived promoters) has been demonstrated to be effective in vitro (Kim et al., 1997) and may be useful in vivo via selectively targeting vascular versus neuronal or glial tissue. However, at present there is no way to reliably distinguish between smooth muscle cells in different cerebral arteries, and therefore the question of being able to target a vector to specific vascular territories remains unanswered using this approach. More recently, a direct mechanical method for accurate and efficient delivery of adenoviral vector to a variety of tissues has been described (Khurana et al., 2003) and holds promise for use in the cerebral circulation (Fig. 7) (Khurana and Katusic, 2001; Khurana and Katusic, unpublished observations, 2001).
TRANSLATIONAL PARADIGMS IN CEREBROVASCULAR GENE TRANSFER
After the pioneering study by Davidson et al. (1993) describing in vivo delivery of adenovirus into murine central nervous system, Ooboshi et al. (1995), using Sprague-Dawley rats, morphologically characterized in vivo adenovirus-mediated gene transfer to cerebral arteries. Subsequently, numerous cerebrovascular gene transfer studies have been carried out, and we were able to identify a total of 27 such studies published between 1995 and 2003, inclusively (Table 2). From this body of literature, the following is apparent:
Transduced, intact arteries from numerous species have been studied including, more recently, humans. Both large-diameter (basilar and middle cerebral) and small-diameter (pial and brainstem secondary) arteries have been studied following gene transfer. Nearly all studies involved adenoviral vectors. LacZ (encoding β-galactosidase) and eNOS cDNA were most frequently the genes of interest. Numerous ex vivo and in vivo studies have been carried out in animals, although to date only two cerebrovascular gene transfer studies have involved intact human arteries, and both of these studies were carried out ex vivo. The morphologic, biochemical, and functional features of cerebrovascular gene transfer have been extensively characterized.
It should be noted that in the present work, translational cerebrovascular gene transfer studies are defined as those involving the transfer and expression of potentially therapeutic recombinant genes in diseased intracranial animal vessels ex vivo or in vivo or using human arterial tissue. All other studies are referred to as pretranslational, that is, those involving gene transfer to normal animal intracranial blood vessels ex vivo or in vivo.
Animal pretranslational studies
A seminal study in cerebrovascular gene transfer was reported by Ooboshi et al. (1995) and involved morphologic characterization of transduction intracranially after CSF delivery of a recombinant adenovirus (AdLacZ) encoding the LacZ gene for bacterial β-galactosidase. In this rat in vivo study, the authors showed that after direct injection of AdLacZ into the cisterna magna (i.e., intracisternal delivery), recombinant protein could be detected by X-gal histochemical staining particularly in the perivascular leptomeninges and in the adventitia of cerebral blood vessels. Two years later, Chen et al. (1997b) showed that canine basilar artery rings transiently immersed ex vivo in a solution containing recombinant adenovirus encoding eNOS (AdeNOS) resulted in expression of recombinant eNOS in the endothelium and adventitia, enhanced endothelium-dependent relaxation to calcium ionophore A23187, reduced contractile responses to uridine triphosphate, and increased basal production of the NO second messenger, cyclic guanosine monophosphate (cGMP). These findings suggested engendering of a more favorable relaxation profile in AdeNOS transduced arteries. This work was shown to be reproducible in vivo by Chen et al. (1997a) after intracisternal delivery of AdeNOS in dogs. In this study, using electron microscopy with immunogold labeling of recombinant eNOS, the authors additionally showed that the principal cell in which recombinant protein synthesis occurred after intracisternal vector delivery was the adventitial fibroblast.
Together, the aforementioned three studies comprise the seminal body of pretranslational literature in cerebrovascular gene transfer, representing a proof of principle for the usefulness of this technology in cerebral arteries as demonstrated by morphologic, biochemical, and functional methods (Chen et al., 1997a,1997b; Ooboshi et al., 1995). Twelve subsequent pretranslational studies (Table 2) have repeated and added to these findings ex vivo and in vivo in mouse, rat, rabbit, and canine experimental models, as well as in large and small intracranial arteries. In each study, recombinant adenovirus was the vector of choice. Of the recombinant genes expressed, all studies used LacZ typically in the role of a reporter or control gene encoding a protein whose expression could be detected histochemically but which did not otherwise alter vessel structure or function. Most studies also used eNOS as a functional gene encoding a protein that could potentially modify vasomotor function. These studies consistently found that expression of recombinant eNOS in cerebral arteries augmented their ability to relax in response to a variety of NO-mediated agonists. This suggested a potential therapeutic role for eNOS gene transfer in the setting of a cerebrovascular disease processes in which impaired NO-mediated signaling leading to reduced vasodilatation was implicated. Other potentially therapeutic genes characterized in the pretranslational literature include those encoding (1) the perivascular nerve-derived vasodilatory polypeptide CGRP (Toyoda et al., 2000a), (2) the heme product-metabolizing enzyme HO-1 (Eguchi et al., 2001), which is likely to be of benefit in the setting of SAH for enhanced clot lysis, and (3) S1179D ‘super’ eNOS (Akiyama et al., 2002), an enzyme encoded by a mutated version of the eNOS gene resulting in enhanced agonist-independent production of NO (Fulton et al., 1999).
Animal translational studies
Of the ten animal translational studies to date, nine have involved experimental SAH models, and one (Lund et al., 1998) has involved an animal model for atherosclerosis (Table 2). Nine of the ten studies used a recombinant adenovirus as the vector of choice, whereas one (Ono et al., 1998) used a plasmid-liposome hybrid vector. Rat, rabbit, and canine models were studied, and the functional genes selected for investigation included eNOS, CGRP, HO-1, and SOD, in addition to nuclear factor-kappa B (NF-κB) decoy DNA (Table 2). Like their pretranslational counterparts, these animal translational studies have involved comprehensive morphologic, biochemical, and functional methodologies, in addition to cerebral angiography used by some investigators (e.g., Khurana et al., 2002; Ono et al., 1998; Stoodley et al., 2000; Toyoda et al., 2000c) to determine the presence of radiologic attenuation of cerebral vasospasm after eNOS, CGRP, or decoy NF-κB gene transfer. Together, the findings of these studies were critically important in establishing the feasibility of using gene transfer technology for the putative treatment of cerebrovascular disease. Note that the variety of genes used in this body of literature supports the notion that disease entities can be treated using different molecular therapies. For example, in post-SAH cerebral vasospasm, whose pathogenesis is complex (Dietrich and Dacey, 2000; Khurana and Besser, 1997), (1) targeting the deleterious presence of heme products can be carried out by augmenting local biosynthesis of the heme-metabolizing enzyme HO-1 (Ono et al., 2002), (2) attenuating the vascular inflammatory changes associated with vasospasm can be achieved by inhibiting a proinflammatory transcription factor such as NF-κB (Ono et al., 1998), (3) countering the tendency towards sustained vasoconstriction can be done by augmenting local production of eNOS and therefore NO (Khurana et al., 2002; Stoodley et al., 2000) or of CGRP (Toyoda et al., 2000c), and (4) reducing the high oxidative stress associated with vasospasm can be achieved by increasing local production of a free radical scavenger such as SOD (Watanabe et al., 2003).
In the context of the translational animal cerebrovascular gene transfer literature, certain studies (Khurana et al., 2002; Ono et al., 2002; Toyoda et al., 2000c; Watanabe et al., 2003) can be used to illustrate the progress in this field. First, the central hypothesis of these in vivo studies was that intracisternal delivery of an adenoviral vector would lead to overexpression of a recombinant protein of interest in cerebral arteries that could confer some degree of protection against the development of experimental post-SAH vasospasm. Second, the authors of these studies repeatedly performed the same experimental procedures in animals using a functional adenoviral vector (encoding a potentially therapeutic gene of interest), a control adenoviral vector at the same titer (encoding a nontherapeutic “reporter” gene), and, additionally in some studies (Khurana et al., 2002; Ono et al., 2002), a vector-free vehicle (negative control). Third, each of these studies used multiple experimental techniques to test the hypothesis, including two or more of the following methodologies: morphologic (e.g., histochemistry, immunohistochemistry), biochemical (e.g., Western analysis, CSF NO chemiluminescence, SOD or CGRP CSF assays), functional (e.g., isometric force recording, direct transcranial arterial videoscopy), and radiologic (e.g., cerebral angiography, laser Doppler flowmetry). Fourth, the potentially therapeutic gene of interest in each of these studies was different, with the candidates being CGRP (Toyoda et al., 2000c), eNOS (Khurana et al., 2002), HO-1 (Ono et al., 2002), and SOD (Watanabe et al., 2003). Fifth, all studies achieved a statistically significant degree of vasomotor or radiologic benefit against post-SAH vasospasm in both small (Ono et al., 2002; Toyoda et al., 2000c; Watanabe et al., 2003) and large (Khurana et al., 2002) animal models known to mimic the human disease counterpart. Last, putative benefits and limitations of these cerebrovascular gene therapy models were also addressed by the authors with an aim of optimizing progress in future translational studies (see below).
Human translational studies
It is clear that the ultimate objective of using human cerebral blood vessels in gene transfer studies is to demonstrate the usefulness of this technology in the treatment of human cerebrovascular disease. To date, there have been only two gene transfer studies involving cerebral blood vessels in humans, and both were carried out ex vivo (Gunnett et al., 2002; Khurana et al., 2000b) (Table 2). Both studies used pial arteries isolated rapidly from the cortical surfaces of temporal lobectomy specimens that would otherwise have been discarded as surgical waste. The study by Khurana et al. (2000b) commenced with a systematic characterization of the morphologic (using histology, histochemistry, and both regular and immunogold electron microscopy) and functional (using isometric force recording and an array of contracting and relaxing compounds) features of human pial arteries before gene transfer and then repeated morphologic and functional studies 24 hours after gene transfer using AdLacZ and AdeNOS. The study by Gunnett et al. (2002) characterized the functional effects of gene transfer in human pial arteries after exposure to adenovirus encoding iNOS. Both studies demonstrated successful transduction of human pial arteries after transient ex vivo immersion in solution containing adenoviral vector. The study by Khurana et al. (2000b) showed that the main cellular target for adenoviral uptake and recombinant protein synthesis was the adventitial fibroblast and that eNOS gene transfer resulted in a markedly improved vasorelaxation profile to the endothelium- and NO-dependent agonist bradykinin in these vessels. The study by Gunnett et al. (2002) demonstrated impaired vasorelaxation to bradykinin and the endothelium-independent NO donor nitroprusside following iNOS gene transfer, thereby confirming the notion that iNOS, whose production is induced in inflammatory pathologic states, is associated with vasomotor dysfunction. This phenomenon has been attributed to enhanced free radical formation associated with the sustained high-level NO production caused by iNOS, compared with eNOS (Chen et al., 1998; Kibbe et al., 1999; Szabo et al., 1995).
Toward a cerebrovascular gene therapy trial
Pertaining to the question as to which disease to first target in a clinical trial, it seems likely that the disease entity most widely studied using this technology in animal models will be the first to undergo investigation in the setting of a clinical trial. In this context, post-SAH cerebral vasospasm has been the focus of nearly all of the animal translational studies to date. This is because (1) there is a readily reproducible experimental SAH model in animals, which results in angiographically demonstrable vasospasm mimicking human disease, (2) intracisternal delivery of vector can be performed relatively easily without marked morbidity or mortality in animals, (3) results of animal studies involving gene transfer in the setting of experimental SAH have been very encouraging, and (4) the pathobiology of vasospasm and our capacity to treat it at a molecular level are fundamentally linked to our level of understanding of the essence of vasomotor reactivity itself. For these reasons, post-SAH cerebral vasospasm may represent a feasible disease for the first clinical cerebrovascular gene therapy trial. Note that although spasm is readily demonstrable in large arteries in the vicinity of the ruptured aneurysm (i.e., typically where subarachnoid blood is most dense), it is not established whether spasm in humans occurs diffusely in small arteries, that is, those beyond the scope of angiographic detection and remote from the site of aneurysm rupture. Despite this, it seems plausible that intracisternal gene delivery of a suitable vector in the future will allow its widespread diffusion throughout the subarachnoid space, perhaps facilitated by introduction of a thrombolytic agent after surgical or endovascular treatment of the aneurysm to prevent its rerupture. Alternatively, endovascular delivery of a suitable vector may be efficacious using a catheter or biostent-based delivery system, as long as gene transfer can be optimized to occur endoluminally without interference with endothelial function or cerebral perfusion during the processes of delivery and transduction themselves. Finally, another cerebrovascular disease target to consider for such a trial in the foreseeable future may be cerebral ischemia from thromboembolic occlusive disease or from obliterative arteriopathies such as Moyamoya disease or Takayasu's arteritis. There is certainly a large body of literature pertaining to CNS gene transfer and therapy for cerebral ischemia in which affected brain regions can be approached via endovascular or periadventitial (e.g., intracisternal) delivery of a suitable vector (see reviews by Gunnett and Heistad, 2001; Papadopoulos et al., 2000; Yenari et al., 2001). In such paradigms, the principal gene therapy targets are cells of the CNS.
Given the tremendous progress that has been made in cerebrovascular gene transfer since 1995, the question arises as to what further needs to be done before the first human cerebrovascular gene therapy clinical trial? Considering the findings of the 27 studies to date (Table 2), it is clear that there has been a substantial amount of preclinical work performed toward fulfillment of this objective, including recent demonstrations of the therapeutic efficacy of this technology in animal models closely mimicking human disease (Khurana et al., 2002; Ono et al., 2002; Satoh et al., 2002; Toyoda et al., 2000c; Watanabe et al., 2003) and its applicability to the human cerebral vasculature (Gunnett et al., 2002; Khurana et al., 2000b). Having stated this, however, it is clear that much still remains to be done for the successful progression of this technology to actual clinical trial. In general, for this to occur, advancements are required in our understanding of human genomics and proteomics (as they pertain to identification of the precise molecular pathogenesis of a disease) and vector biology (particularly reduction of immunogenicity and enhancement of inherent transduction efficiency and target-specificity), and more effective and not necessarily more complex delivery systems also need to be developed and tested (Khurana et al., 2003). Specific to cerebrovascular gene transfer studies, to date, none has demonstrated therapeutic efficacy using a vector of clinical grade status, that is, one that is highly purified, minimally immunogenic, and regarded as safe enough for clinical use. To this end, refinement and testing of new “gutless” adenoviral vectors, AAV, and hybrids in such translational models may be helpful (Balicki and Buetler, 2002; Dyer and Herrling, 2000; Kay et al., 2001; Vorburger and Hunt, 2002). Further, such studies must not only address the issue of safety using such vectors, they must also endeavor to fully address the issues of gene transfer efficiency, tissue-target specificity, and therapeutic efficacy. With regard to vector targeting in cerebrovascular gene transfer paradigms, appropriate toxicology studies must be performed, including measurement of an array of biologic parameters, close monitoring of neurologic state, and experiments designed to detect the presence of vector and recombinant product in both local and remote tissue sites (e.g., via PET scanning using a bicistronic vector or reverse transcription-polymerase chain reaction). We feel that these studies are indeed possible and, if performed patiently and thoroughly, will undoubtedly facilitate the safe and effective use of this important technology for the treatment of cerebrovascular disease.
Footnotes
Acknowledgment
The authors thank Ms. Christine Nerland and Mr. John Hagen, Section of Illustration and Design, Mayo Clinic, for their expert assistance with the artwork used in this paper.