
Abstract
In a prospective clinical investigation on neurochemical intensive care monitoring, the authors' aim was to elucidate the temporal profile of nitric oxide metabolite concentrations—that is, nitrite and nitrate (NOx)—and compounds related to energy-metabolism in the cerebral interstitium of patients after aneurysmal subarachnoid hemorrhage (SAH). During aneurysm surgery, microdialysis probes were implanted in cerebral white matter of the vascular territory most likely affected by vasospasm. Temporal profiles of NOx were analyzed in a subset of 10 patients (7 female, 3 male, mean age = 47 ± 14 years). Microdialysis was performed for 152 ± 63 hours. Extracellular metabolites (glucose, lactate, pyruvate, glutamate) were recovered from the extracellular fluid of the cerebral parenchyma. NOx was measured using a fluorometric assay. After early surgery, SAH patients revealed characteristic decreases of NOx from initial values of 46.2 ± 34.8 μmol/L to 23.5 ± 9.0 μmol/L on day 7 after SAH (P < 0.05). Decreases in NOx were seen regardless of development of delayed ischemia (DIND). Overall NOx correlated intraindividually with glucose, lactate, and glutamate (r = 0.58, P < 0.05;r = 0.32, P < 0.05;r = 0.28, P < 0.05; respectively). After SAH, cerebral extracellular concentrations of NO metabolites decrease over time and are associated with concomitant alterations in energy-or damage-related compounds. This could be related to reduced NO availability, potentially leading to an imbalance of vasodilatory and vasoconstrictive factors. On the basis of the current findings, however, subsequent development of DIND cannot be explained by a lack of vasodilatory NO alone.
Subarachnoid hemorrhage (SAH) caused by ruptured aneurysms is responsible for approximately one quarter of all cerebrovascular deaths. Reported fatality rates range from 25% and 50% (Fogelholm et al., 1993; Hop et al., 1997). In the postoperative phase after aneurysm occlusion, cerebral vasospasm (VSP) is the major complication of SAH, potentially leading to infarction and thus long-term morbidity and mortality. Despite extensive research, the pathomechanisms of SAH-induced VSP remain unclear.
Vasospasm is thought to result from an imbalance of vasoconstriction and vasorelaxation (Dietrich and Dacey, 2000). Endothelial cells of blood vessels generate factors that can modulate underlying smooth muscle tone, inducing vasorelaxation (endothelium-derived relaxing factor [EDRF] = nitric oxide [NO];Furchgott and Zawadzki, 1980; Ignarro et al., 1987; Palmer et al., 1987), or vasoconstriction (endothelium-derived contracting factors, EDCFs, including endothelins; (Yanagisawa et al., 1988), or both. Removal of endothelium or pathologic situations inducing endothelial dysfunction (atherosclerosis, diabetes, hypertension, or subarachnoid hemorrhage) cause increases of vascular contractility elicited by vasoconstrictive agents (noradrenaline, serotonin, EDCFs, and so on). These findings suggest that NO is a physiologic inhibitory modulator of vascular smooth muscle tone. Alterations of this pathway may initiate or facilitate the development of VSP.
During the last decade there has been increasing awareness that NO may play an essential role in the development of VSP. However, both reduced and excessive NO-metabolism have been suggested as important factors in the pathophysiology of VSP (Medele et al., 1996; Pluta et al., 1996; Widenka et al., 1999). Pioneering studies of experimental VSP in primates have implied that a lack of NO because of scavenging by liberated hemoglobin might be the cause for VSP (Edwards et al., 1992; Pluta et al., 1996, 1998). Recently, experimental studies of VSP reversed by NO replacement have triggered substantial interest (Ito et al., 2000; Pluta et al., 1997; Wolf et al., 1998) and have led to the first clinical applications (Thomas and Rosenwasser, 1999).
In vivo measurement of NO is rather complicated because of its short half-life and high reactivity with free oxygen, oxygen radicals, redox metals, sulphydryls, disulfides, and oxygenated hemoglobin (Stamler et al., 1992). Stable endproducts of NO—nitrate and nitrite (NO2−, NO3−)—however, can be assessed by photometry (Giovannoni et al., 1997; Schulz et al., 1999), fluorometry (Lei et al., 1999; Misko et al., 1993), and high performance liquid chromatography (HPLC) (El Menyawi et al., 1998; Everett et al., 1995; Kaku et al., 1994; Zecca et al., 1998).
In patients with SAH, total nitrate and nitrite (NOx) has been measured in cerebrospinal fluid (CSF) (Suzuki et al., 1997, 1999). However, it has been reported that VSP extends up to the distal parenchymal branches of cerebral arteries where CSF-NOx might be an insufficient estimator of the local conditions (Ohkuma et al., 1997). Microdialysis (MD) provides an elegant way to study the microenvironment of the brain. Established as “bedside” cerebral MD, it was proposed a safe procedure to monitor brain chemistry in the operating room and postoperatively in the neurointensive care unit (Nilsson et al., 1999; Schulz et al., 2000; Unterberg et al., 2001). Neurometabolic monitoring by cerebral MD of patients with SAH allows investigation of the extracellular fluid (ECF) for stable, low molecular weight substances such as energy-related parameters glucose, lactate, pyruvate, and glutamate as parameters of cell damage and excitotoxicity. In addition, NOx can be analyzed in microdialysates of patients with SAH undergoing either acute or elective surgical obliteration of aneurysms.
The aim of the current study included the following: (1) to define cerebral NOx concentrations of patients with SAH in contrast with patients who underwent elective surgery for cerebral aneurysms, (2) to evaluate the time course of NOx in the advent of symptomatic vasospasm in contrast with asymptomatic courses, and (3) to clarify the correlation of NOx with local neurometabolism.
MATERIALS AND METHODS
Patients
In 10 aneurysm patients with (n = 7) or without (n = 3) recent SAH, cerebral metabolism was monitored postoperatively by bedside MD. Extracellular concentrations of NOx were determined off-line.
On admission, SAH was confirmed by computerized tomography (CT) scanning and was graded by distribution and pattern of hemorrhage according to Fisher et al. (1980) and by clinical presentation according to the World Federation of Neurological Surgeons Scale (Drake, 1988), respectively. The presence of an aneurysm, its size, and location were assessed using four-vessel digital subtraction angiography.
Patients with recent SAH underwent surgery within 72 hours. Postoperative care monitoring included continuous measurement of arterial blood pressures and central venous pressures. Cerebral metabolism was monitored by bedside microdialysis (vide infra). Blood flow velocities were determined at least once daily by transcranial Doppler sonography.
Patients with SAH received intravenous nimodipine (2 mg h−1) and dexamethasone (24 mg d−1, tapered dosage). Only patients presenting with delayed ischemic neurological deficits (DIND) were treated with “triple-H” (hypertension, hypervolemia, and hemodilution) therapy. Insidious onset of confusion, appearance of a focal neurologic deficit, or both, were judged as symptomatic vasospasm. Cranial CT scans were obtained to rule out acute ischemic events, hemorrhage, or development of posthemorrhagic hydrocephalus.
Ethics
Intensive care monitoring of patients with SAH using the MD technique was approved by the local medical ethics comittee. Written informed consent was obtained from the patient or the closest relative.
Microdialysis
A MD catheter (CMA70; CMA, Sweden; 10-mm membrane) was inserted immediately after clipping of the aneurysm. Catheters were introduced 25 to 35 mm into the brain parenchyma of the vascular territory most likely to be affected by VSP, for example, the temporal lobe in patients with a middle cerebral (MCA) or an internal carotid artery (ICA) aneurysm or into the frontal lobe in patients with anterior communicating artery aneurysms. Care was taken to avoid insertion of the catheter into lesioned brain tissue surrounding intracerebral hemorrhage. Microdialysis catheters were externalized through a burr hole of the bone flaps, tunneled underneath the scalp, and attached to the skin with sutures.
Immediately after surgery, a microperfusion pump (CMA106, CMA, Sweden) was connected to the inlet tubing. Catheters were perfused with sterile Ringer's solution (Na+ 147 mmol/L, K+ 4 mmol/L, Ca++ 2 mmol/L, Cl− 155 mmol/L) at a fixed flow rate of 0.3 μL min−1. At this flow rate, catheters of the specified type yield approximately 70% recovery of low molecular weight analytes as defined by the extrapolation-to-zero flow method (Hutchinson et al., 2000). Thus, true interstitial concentrations are underestimated.
On the outlet tube, perfusates were collected in microvials, exchanged hourly, and analyzed immediately for concentrations of glucose, lactate, pyruvate, and glutamate. Severe ischemia was assessed by calculating lactate/pyruvate ratios (LPR). All analyses were performed in a mobile photometric, enzyme-kinetic analyzer (CMA600, CMA, Sweden). Results were obtained directly at bedside and displayed as trend curves on a mounted screen. NOx was measured “off-line” after storage of MD samples at −80°C.
Determination of NOx
NOx levels (NO2− and NO3−) were measured with a sensitive method by using 2,3-diaminonaphtalene (DAN; Sigma, Deisenhofen, Germany) as described previously (Lei et al., 1999). Briefly, NO3− was reduced to NO2− by nitrate reductase (from aspergillus species, Sigma) in the presence of nicotinamide adenine dinucleotide phosphate, reduced form (NADPH; Sigma). NO2− was incubated with DAN resulting in the formation of 1-(H)-naphtotriazole, the fluorescence activity of which was detected by spectrofluorometry. Stock solutions were prepared with DAN (0.05 mg mL−1 in 0.62 mol/L HCl, kept in the dark), tris-buffer (100 mmol/L, pH 7.6), NADPH (100 μmol/L), nitrate reductase (1.4 U L−1), NaOH (2.8 mol/L), NO2−, and NO3− (both 10 mmol/L; Merck, Darmstadt, Germany). For reductase reaction nitrate reductase (20 μL), NADPH (40 μL) were mixed with tris-buffer (20 μL) and the reaction was initiated by adding an aliquot (10 μL) of the sample. After incubation for 10 minutes at room temperature, probes were diluted with 100 μL destilled water and DAN (20 μL) was added. The reaction was stopped after 10 minutes at room temperature with NaOH (10 μL). Fluorescence activity was detected at an excitation wavelength of 358 nm and an emission wavelength of 405 nm with the spectrofluorometer. To calculate NOx levels from fluorescent units, 5 samples of NO2− (5 to 200 μmol/L) were incubated as standards in each incubation procedure. To determine the rate of NO3− to NO2− conversion, 5 samples of aequimolar NO3− (5 to 200 μmol/L) also were incubated. Conversion rate was always greater than 95%.
Transcranial Doppler sonography
Blood flow velocities were assessed using a 2-MHz pulsed Doppler probe (TC2–64; EME, Tubingen, Germany) through the transtemporal window. Doppler signals were sampled at various depths in steps of 5 mm. Mean flow velocities in the ICA, MCA, and anterior cerebral artery were obtained by the same investigator (O.S., A.S.) at least once daily. Flow velocities greater than 120 cm s−1 were regarded pathologic (Aaslid et al., 1984).
Angiography
Cerebral angiography was performed routinely in all patients upon admittance. According to the time course of VSP, control angiography was performed on day 7 to 8 after bleeding or earlier in cases of neurologic deterioration.
For angiographic assessment of cerebral vasospasm, vessel diameters were measured in the narrowest parts of the A1, A2, M1, M2, C1/C2 segments. Correction for magnification errors was performed by comparison with extradural ICA segments (C4/C5) not exposed to subarachnoid blood.
Angiographic vasospasm was defined as at least moderate (that is, >30%) narrowing of the cerebral vessel lumen diameter.
Data analysis
Statistical analysis was performed using SPSS 10.0 (SPSS, Chicago, IL, U.S.A.). Data averaged among groups are reported as means and standard deviation. For statistical testing of NOx time courses, Wilcoxon's matched-pairs rank tests were used. Microdialysis parameters were correlated after normalization by an intraindividual z-transform. Correlations were tested using Pearson's r- statistics. P < 0.05 was considered significant.
RESULTS
Patient characteristics
In this study, 10 patients with aneurysms (3 male, 7 female, mean age = 47 ± 14 years, demographic data summarized in Table 1) were investigated by MD monitoring. In 7 patients, recent hemorrhage had been confirmed by CT. Probes were inserted directly after microsurgical clip ligation of the aneurysm. Microdialysis was initiated 35 ± 22 hours after SAH. Three patients with incidental aneurysms underwent elective surgery. They had not experienced clinical symptoms of SAH before operation. All patients were monitored for 152 ± 63 hours. A total of 308 NOx determinations covered approximately 1200 hours of monitoring.
Demographics, microdialysis, and clinical course (n = 10)
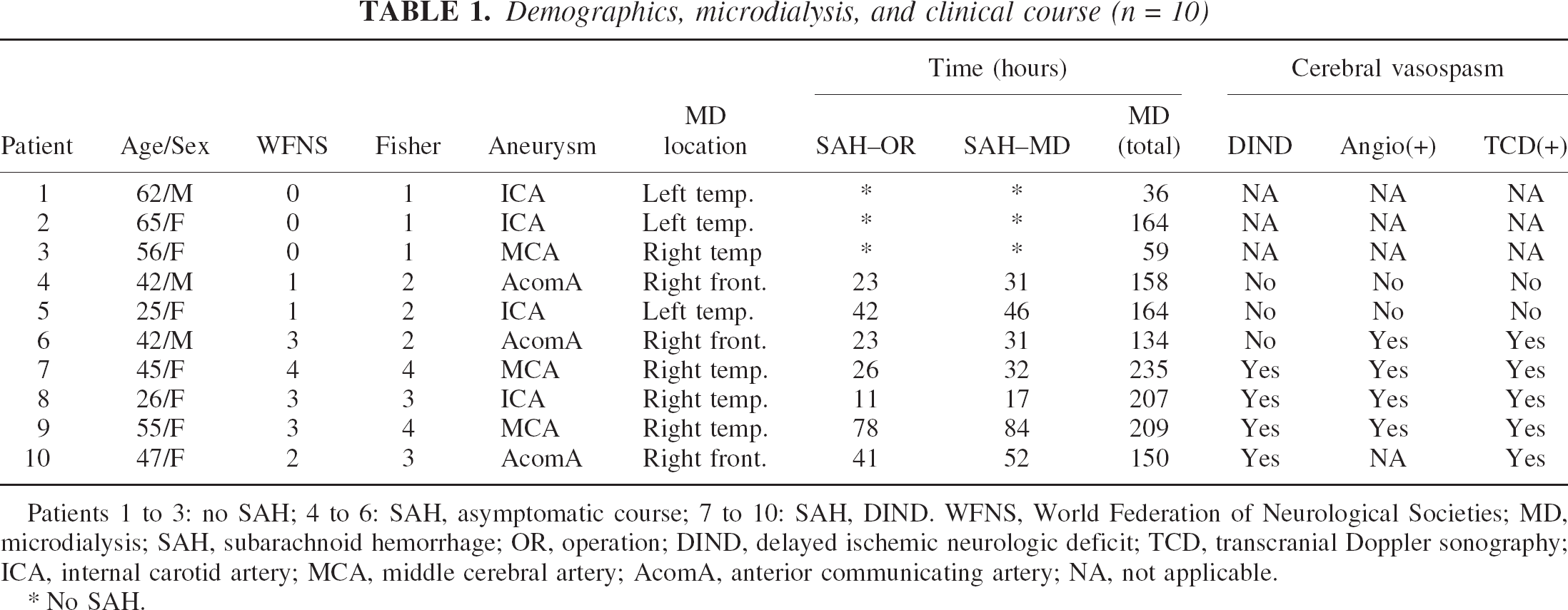
Patients 1 to 3: no SAH; 4 to 6: SAH, asymptomatic course; 7 to 10: SAH, DIND. WFNS, World Federation of Neurological Societies; MD, microdialysis; SAH, subarachnoid hemorrhage; OR, operation; DIND, delayed ischemic neurologic deficit; TCD, transcranial Doppler sonography; ICA, internal carotid artery; MCA, middle cerebral artery; AcomA, anterior communicating artery; NA, not applicable.
No SAH.
Patients fully recovered postoperatively. No occurrences of acute ischemic deficits were observed. There were no complications (hemorrhagic, infectious) related to MD.
In 3 patients with incidental aneurysms, MD values stabilized shortly after the procedure. None of the electively operated patients deteriorated secondarily.
Of the 7 SAH patients, 4 presented with DIND. This was confirmed by secondary deterioration of MD values (n = 4), significant angiographic vessel narrowing (n = 3), and elevated blood flow velocities (n = 3). In three SAH patients without DIND, significant angiographic vessel narrowing and elevated blood flow velocities were noticed once.
All of the patients with DIND received “triple-H” therapy aiming at mean arterial blood pressure values greater than 110 mm Hg. Permanent resolution of DIND was achieved in all patients except one, who retained a mild left-sided hemiparesis.
Temporal profile of NOx
Patients with SAH showed a specific temporal profile of NOx consisting of an initial peak followed by an exponential decay. Figures 1 and 2 represent the characteristic case of a patient who deteriorated neurologically on the fifth day after hemorrhage. Both angiographic and neurochemical studies confirmed this to be related to vasospasm and consecutive malperfusion, that is, DIND. However, NOx concentrations failed to display a course distinct from other postoperative patients who remained asymptomatic.
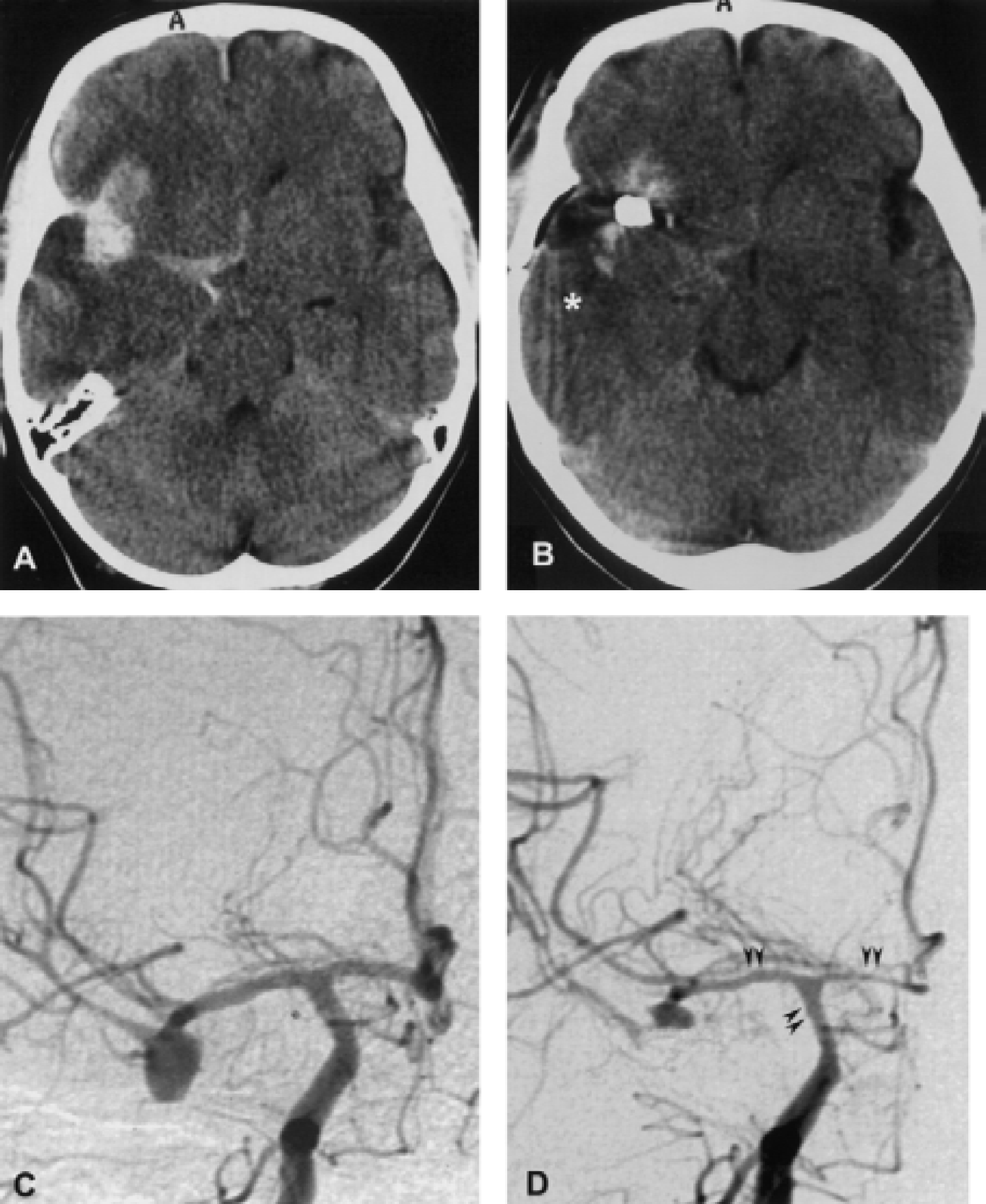
Patient 9, SAH Fisher grade 4, WFNS grade 3, DIND beginning on day 5 after SAH (see MD data in Fig. 2).
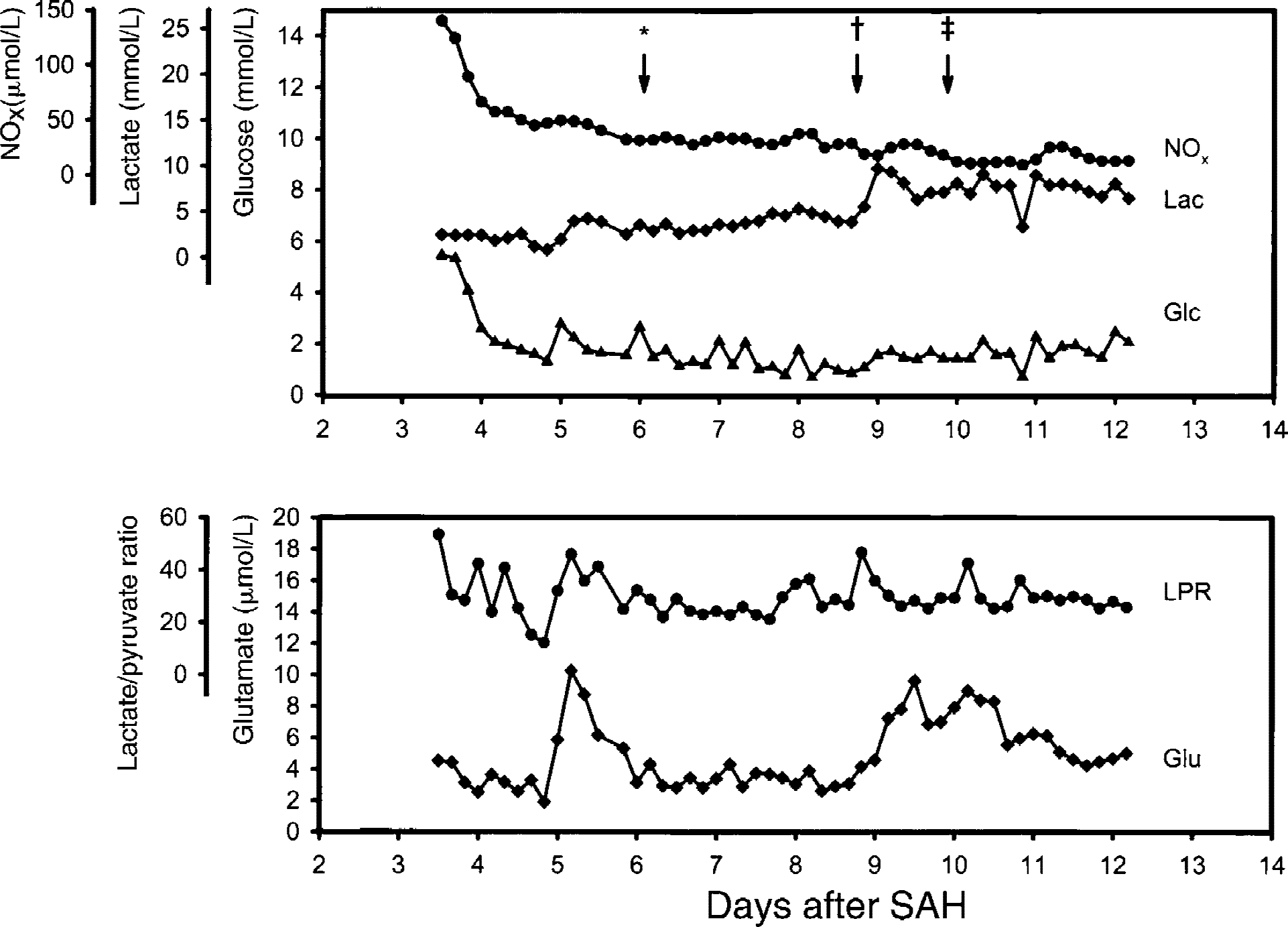
Microdialysis (MD) data of a 55-year-old female patient (patient 9) who developed DIND on day 5 posthemorrhage. Clinically presented drowsy, dysarthric with a left-sided brachio-facial hemiparesis (WFNS grade 3). CT disclosed a Fisher grade 4 SAH. Angiography revealed an aneurysm of the right MCA and an aneurysm of the right PICA. MCA aneurysm was clipped and a MD catheter was inserted into the right temporal lobe 84 hours post-SAH. After surgery, the patient improved clinically. On day 5 after SAH, hemiparesis worsened again adjoined by a temporary increase in ECF glutamate. Because of the unclipped PICA aneurysm, only modest triple-H therapy (mean arterial blood pressure 90 to 100 mm Hg) was initiated at first. This was carefully intensified with continued worsening of clinical symptoms. Control angiography on day 10 post-SAH showed moderate vasospasm of the right A1 and M1 segments. Blood flow velocities were <120 cm/s at all times. The patient recovered incompletely during triple-H therapy. MD was discontinued 12 days post-SAH. The PICA aneurysm was occluded endovascularly 7 months later. Glasgow outcome score at 6 months was moderate disability (4) because of persistent left-sided weakness. *Worsening of clinical symptoms (hemiparesis). †Continued worsening of clinical symptoms (hemiparesis and somnolence). ‡Control angiography.
In 3 patients without recent SAH, extracellular concentrations of NOx stabilized within 4 hours after insertion of the probe (Fig. 3A). After stabilization, amounts of total NOx ranged from 22.2 to 58.0 μmol/L. This range was used as visual control for comparison with SAH patients.
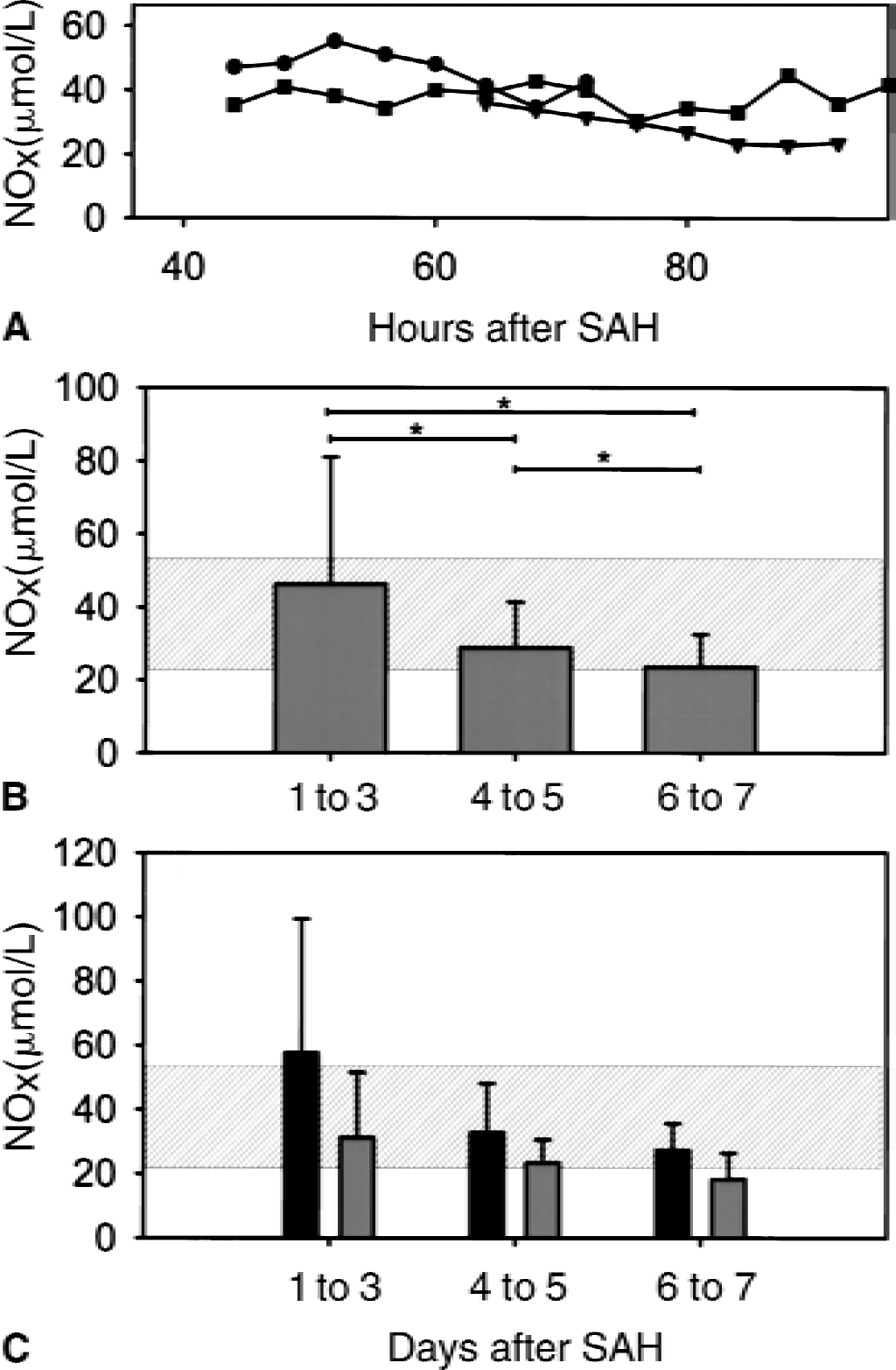
As summarized in Fig. 3B, values of NOx averaged 46.2 ± 34.8 μmol/L (n = 7) on days 1 to 3 in SAH patients. On days 4 to 5 and 6 to 7, NOx decreased significantly to 28.8 ± 12.6 μmol/L and 23.5 ± 9.0 μmol/L, respectively (n = 7, P < 0.05). Initial values varied intraindividually, thereby 3 of 7 patients presented with supranormal NOx concentrations, whereas NOx decreased to less than normal values in 6 of 7 patients.
Descriptive statistics of subgroups according to development of DIND revealed a trend for greater NOx concentrations in DIND patients at all times (Fig. 3C). NOx decreased to similar values in DIND and asymptomatic patients (27.3 ± 8.4 vs. 18.4 ± 8.2 μmol/L, not significant).
NOx and energy-related metabolites
Energy-related metabolite changes with DIND (n = 4) and overall correlations of metabolite concentrations with NOx were investigated (Table 2). In comparison, extracellular fluid concentrations were obtained from SAH patients without DIND on days 6 to 7 after hemorrhage (n = 3). With the development of DIND, ECF glucose concentrations decreased approximately 23% from 1.3 to 1.0 mmol/L. This was adjoined by an increase in lactate from 4.2 to 5.5 mmol/L (31%) and an increase in LPRs by 12%. Concentrations of NOx decreased on average by 21% from 42.7 μmol/L to 33.7 μmol/L.
Analyte changes with DIND and correlations with NOx

NOx, total nitrite and nitrate; LPR, lactate/pyruvate ratio.
Extracellular fluid concentrations in patients with subarachnoid hemorrhage (SAH) without delayed ischemic neurologic deficit (DIND) on days 6 to 7 after hemorrhage (n = 3). Alterations in patients with SAH with DIND (before vs. after, n = 4). Mean and standard deviations.
Correlation coefficients of intraindividually normalized data (z-transform), only correlation with NOx given.
Correlations of NOx with energy-related metabolites (glucose, lactate, LPR) were calculated from intraindividually z-transformed data pooled from all patients. Significant correlations with NOx were determined for glucose (r = 0.58, P < 0.05) and lactate (r = 0.32, P < 0.05). LPRs were not correlated with NOx (r = 0.06, not significant).
NOx and glutamate
In SAH patients without DIND, glutamate steadily declined with time averaging 1.4 ± 0.3 μmol/L on days 6 to 7 after hemorrhage (Table 2). Secondary increases in glutamate were observed in patients with DIND reflecting clinical deterioration (324% increase, 4.2 to 13.6 μmol/L) (Table 2). No alterations in NOx were seen at the same time.
Overall glutamate was positively correlated with NOx (r = 0.28, P < 0.05). In 1 patient (patient 8 in Table 1), however, glutamate steeply increased to the 50 to 100 μmol/L range on day 2 after SAH, and thus paralleled the advent of DIND. Meanwhile, NOx declined from initial maximum values of 70 to 90 μmol/L to average values of 14.7 ± 6.3 μmol/L on days 6 to 7.
DISCUSSION
In the current study, cerebral microdialysates of aneurysm patients with and without recent SAH have been analyzed for stable metabolites of NO. A sensitive fluorometric assay for aqueous samples was used. Samples from patients without SAH were analyzed to estimate the natural course of NOx. NOx decreased with time in patients after SAH. This was paralleled by decreases in ECF glucose and increases in ECF lactate, a pattern consistent with the assumption that lowered ECF-NOx leads to an imbalance of vasoconstrictive and vasodilatory factors, resulting in vasoconstriction and subsequent decreases in local CBF. After SAH, glutamate declined in parallel with NOx. However, glutamate increased secondarily with the advent of DIND, whereas no such alterations were observed in NOx. Concentrations and temporal profiles of NOx in patients with DIND did not differ significantly from those in patients not presenting with DIND.
Methodologic issues
As in the current study, using MD probes with a 10-mm membrane is methodologically favorable because of the relatively high (>70% recovery at 0.3 μL min−1 flow rate) yield of low molecular substrate (Hutchinson et al., 2000). This, however, technically imposes the need for placement in depths that correspond to cerebral white matter or cortico-medullary junction at best. Ischemic resistance of cerebral white matter is far better than that of cortex. However, previous studies have shown that increases in lactate and glutamate can be measured after SAH (Nilsson et al., 1999; Schulz et al., 2000). This is specifically the case in patients developing DIND (Unterberg et al., 2001). Possibly, MD might be more sensitive to neurometabolic alterations in vasospasm if used strictly cortical. This might be suggested considering the fact that in experimental stroke, glutamate appears in cerebral white matter after the cortical surge with a time lag (<2 hours) that is suggestive of slow diffusion (Shimada et al., 1993). With respect to NO, the authors hypothesize that as a gas it readily diffuses in cerebral tissue within seconds. Although this is limited by its biologic half-life of approximately 0.5 to 5 seconds (Wood and Garthwaite, 1994), stable end products of NO metabolism might diffuse further to reflect NO availability beyond cortex.
Oxidation of NO to NO3− by oxyhemoglobin is assumed to be the major elimination pathway (Perutz, 1996). However, stoichiometric relations of NO and NOx are highly dependent on oxygen saturation, availability of vacant hemes, and are thus not simple to estimate in vivo. In biologic systems, elimination pathways of NO can be oxidative or cooperative as well (Gow et al., 1999; Perutz, 1996). Hence, NO bioactivity and NOx can not be assumed to be directly proportional. The authors hypothesize that NOx measured in the current study reflects NO availability in cerebral tissue at the given time points after SAH.
Several methods can be used to measure NO2− and NO3− as stable end products of nitric oxide. Determination of NOx in aqueous samples can be achieved with the Griess reaction. Recently, assays using a highly sensitive, modified Griess reaction have been described (Giovannoni et al., 1997; Schulz et al., 1999).
Compared with methods based on the Griess reaction, the fluorometric method used in this study is approximately 50 to 100 times more sensitive making it ideal for small volume, ultrafiltrated samples with micromolar concentrations of NOx (Lei et al., 1999; Misko et al., 1993).
Several authors have reported the feasibility of HPLC-based methods to determine both NO2− and NO3− (El Menyawi et al., 1998; Everett et al., 1995; Kaku et al., 1994; Zecca et al., 1998). The major advantage of this technique is that analysis is unaffected by biogenic amines; thus, deproteinization of samples is not necessary. Because microdialysates have been ultrafiltrated over a 20-kD microporous membrane, the method used in this study can be assumed to be equally effective.
Experimentally, measurements of NOx have been proven effective using microdialysis (Ohta et al., 1994, 1996; Togashi et al., 1998). The main benefit of this method is the ability to measure NO in vivo in cerebral tissue. In this study, the authors reported the feasibility of NOx detection in extracellular fluid samples recovered from the human brain by microdialysis. The temporal profile of NOx in patients at risk for delayed ischemia because of vasospasm was described and correlated with measurements of metabolites reflecting energetic impairment and glutamate-mediated excitotoxicity.
Cerebral sources of NO
A time-dependent decrease in extracellular NOx was observed in patients with SAH in the current study. However, the origin of NO metabolites in cerebral tissue and CSF after aneurysmal SAH is not clear. Three isoforms of NO synthase of endothelial (eNOS), neuronal (nNOS), and various origin (inducible, iNOS) have been identified in the brain (Iadecola, 1997). There are several sources that have been demonstrated to contribute to the generation of NO. Vascular NO or its metabolites might diffuse directly into cerebral tissues or be carried further distal and diffuse secondarily. Nitric oxide also can be generated directly by nNOS from nitroxidergic neurons (Iadecola et al., 1993; Montecot et al., 1997; Nozaki et al., 1993). Glial production of NO can be inferred from the presence of eNOS in astrocytes (Wiencken and Casagrande, 1999). After SAH, iNOS has been identified primarily in a perivascular distribution (that is, in endothelial cells, in vascular smooth muscle cells, and in adventitial cells) (Medele et al., 1996; Widenka et al., 1999). Based on this distribution pattern of iNOS, a mechanism of peroxidative vessel damage has been attributed to NO. However, contradicting findings of low NOS after SAH have been reported (Pluta et al., 1996).
Nitric oxide metabolites in other human biologic fluid specimen
Concentrations of nitric oxide and its metabolites have been assessed in various biologic fluids, including arterial and venous plasma and CSF. In this study, brain MD was used as a method of surveillance for delayed ischemia in man. Systemic plasma levels of NO metabolites are significantly greater in arterial blood compared with paired peripheral venous blood samples (approximately 45.1 ± 17.7 vs. 22.5 ± 8.5 μmol/L) (Cicinelli et al., 1999). The values of cerebral extracellular NOx, as obtained in this study, are at same order (22 to 58 μmol/L). NOx concentrations of human CSF are lower (2.6 ± 0.4 to 13.0 ± 2.4 μmol/L), which might possibly reflect NO dilution or degradation by parenchymal, or adventitial enzymes, or both (Suzuki et al., 1997; Zecca et al., 1998).
Temporal profile of NO metabolites after SAH
Extracellular concentrations of NOx have been measured for several days after SAH in patients. A time-dependent decrease was observed in SAH patients independently of the operative procedure because NOx concentrations appeared stable in three patients undergoing surgery for incidental aneurysms.
It has been reported that cisternal CSF of patients with SAH contains elevated concentrations of NOx (Suzuki et al., 1999, 1997). Concentrations in CSF might well be different from those in other compartments (perivascular, vascular, brain parenchyma). With CSF samples it appears that the increase of NO metabolites with time depends on the degree of SAH (Suzuki et al., 1997) or severity of acute injury as it has been shown in severely head-injured patients (Clark et al., 1996). A recent MD study has also yielded increases of ECF NOx in the presence of infarctions (Staub et al., 2000). This may well explain the positive correlation of NOx and glutamate concentrations observed in this study.
In the perivascular or parenchymal environment after SAH the picture differs. In a primate model of cisternal hemorrhage, Pluta et al. (1998) provided in vivo evidence that the concentrations of oxyhemoglobin and deoxyhemoglobin increase in the subarachnoid perivascular space during the development of delayed cerebral vasospasm. In the SAH group, perivascular concentrations of oxyhemoglobin and deoxyhemoglobin peaked (about 100-fold higher than control) on day 7 when prevalence of VSP is known to be highest; hence, NO scavenging might account for the temporal profile of NOx observed in the current study.
Role of NO in the development of VSP
There is considerable evidence for the role of NO in the development of VSP. Recently, it has been shown that the availability of NO decreases in the acute phase of SAH, a pattern presumably causing acute vasoconstriction and ischemia (Schwartz et al., 2000; Sehba et al., 2000).
As for delayed vasoconstriction, the injection of hemoglobin or blood in the cisterna magna is known to elicit cerebral VSP subacutely (Edwards et al., 1992). Pluta et al. (1996) reported a decrease of total NOS in a primate model of SAH. However, a perivascular increase in iNOS staining has been reported in rats after SAH (Medele et al., 1996; Widenka et al., 1999). Decreased immunoreactive staining for eNOS was found in cerebral arteries induced by autologous blood injection (Hino et al., 1996). Meanwhile, increased compensatory eNOS reactivity occurred on the contralateral side.
Histomorphologically, constriction of intraparenchymal arterioles has been shown in a canine model of SAH (Ohkuma et al., 1997). This could be because of NO scavenging by products of hemolysis diffusing parenchymally. As with the current findings, this might be related to experimental findings of Dreier et al. (1998), in which a combination of decreased NO levels with either increased subarachnoid potassium levels or decreased glucose levels induce electrophysiologic spreading depression with an acute ischemic CBF response. The authors suggest that such disturbed coupling of metabolism and CBF may aggravate ischemia and speculate that similar alterations in the cellular microenvironment may possibly be related to the development of DIND and infarction after SAH (Dreier et al., 2000).
Role of NO in the treatment of VSP
Therapeutic intervention by NO replacement currently is being debated. It is known that NO antagonists raise the lower threshold of autoregulation in brain perfusion (Jones et al., 1999). Consecutively, in the absence of NO, even normal cerebral perfusion pressures might result in insufficient perfusion as seen in VSP. In rabbits, even low dose administration of nitroglycerin ameliorates angiographic vessel narrowing without affecting systemic blood pressures (Ito et al., 2000). In a primate model, NO replacement by continuous and acute administration of NO donor substances results in complete prevention or reversal of cerebral VSP (Pluta et al., 1997). NO donors are also capable of reversing VSP when applied intrathecally (Wolf et al., 1998). Encouraged by this, clinical studies have begun. A recent phase-1 clinical study demonstrated the safety of intraventricular nitroprus side to resolve cerebral vasospasm clinically (Thomas et al., 1999). In the past, beneficial effects of intraventricular nitroprusside on tissue oxygenation have been observed in a few cases of refractory VSP only, thus, a more systematic evaluation is underway (Thomas and Rosenwasser, 1999; Vajkoczy et al., 2000). Nevertheless, the current findings of time-dependent decreases of NO in cerebral tissue underscore the potential of these recently tested therapeutic measures in refractory VSP.
Conclusion
After SAH extracellular concentrations of NO, metabolites decrease with time and are associated with concomitant alterations in energy-or damage-related compounds. This might be related to a decrease in NO availability either by limited production or increased scavenging by products of hemolysis. However, subsequent development of DIND cannot be explained by a lack of vasodilatory NO alone.
Footnotes
Acknowledgments:
The authors thank Prof. U. Dirnagl (Dept. Experimental Neurology, Charité) for critical review, helpful comments, and discussions. The authors also acknowledge the contributions of the technical (S. Seidlitz, A. Stoessel) and nursing staff of the neurosurgical intensive care unit.