
Abstract
Rupture of an intracranial aneurysm (subarachnoid hemorrhage) is a potentially devastating condition frequently complicated by delayed cerebral ischemia from sustained contraction of intracranial arteries (cerebral vasospasm). There is mounting evidence linking the formation of intracranial aneurysms and the pathogenesis of post-subarachnoid hemorrhage vasospasm to aberrant bioavailability and action of the vasodilator molecule nitric oxide generated by isoforms of nitric oxide synthase. In humans, the gene encoding the endothelial isoform of nitric oxide synthase (eNOS) is known to be polymorphic, with certain polymorphisms associated with increased cardiovascular disease susceptibility. In this prospective clinical study involving 141 participants, we used gene microarray technology to demonstrate that the eNOS gene intron-4 27-base pair variable number tandem repeat polymorphism (eNOS 27 VNTR) predicts susceptibility to intracranial aneurysm rupture, while the eNOS gene promoter T-786C single nucleotide polymorphism (eNOS T-786C SNP) predicts susceptibility to post-subarachnoid hemorrhage vasospasm. We believe that genetic information such as this, which can be obtained expeditiously at the time of diagnosis, may be used as a helpful adjunct to other clinical information aimed at predicting and favorably modifying the clinical course of persons with intracranial aneurysms.
Keywords
Rupture of an intracranial aneurysm, referred to as aneurysmal subarachnoid hemorrhage (SAH), is a medical condition characterized by its distinctively sudden presentation, frequent lack of warning symptoms, high morbidity and mortality, and need for emergent surgical or endovascular treatment (Sundt and Whisnant, 1978). In patients surviving the original hemorrhage, major complications include aneurysm rebleeding and cerebral vasospasm, the latter a delayed sustained contractile response of intracranial arteries exposed to extravasated blood products, particularly oxyhemoglobin, that can lead to cerebral ischemia (Kassell and Torner, 1983; Macdonald and Weir, 1991; Weir and Macdonald, 1993; Khurana and Besser, 1997; Khurana et al., 1998). Cerebral vasospasm following aneurysmal SAH is a potentially life-threatening condition that occurs clinically in up to one in three patients with ruptured aneurysms (Weir and Macdonald, 1993; Khurana and Besser, 1997). To date, the only predictor for the occurrence of cerebral vasospasm following aneurysmal rupture is the amount of subarachnoid blood detected on the early post-hemorrhage head computerized tomography (CT) scan (Fisher et al., 1980). However, even in patients with similar extent of aneurysmal SAH radiologically (i.e., the same “Fisher CT grade”) at the time of rupture, the occurrence and neurological effects of vasospasm have been observed to vary markedly. Such variability in the clinical expression of cerebral vasospasm in patients with the same radiological grades of SAH raises the possibility that genes encoding key proteins implicated in cerebrovascular regulation may exhibit functionally significant aberrations that can account for interindividual differences in vasomotor integrity following aneurysmal rupture.
Of the array of molecular candidates potentially relevant to the pathogenesis of aneurysmal SAH and vasospasm, we deliberately chose the gene encoding the endothelial isoform of nitric oxide synthase (eNOS) as the focus for this investigation for several reasons. First, NO derived from constitutive isoforms of NOS acts as a potent vasodilator and inhibitor of inflammation, smooth muscle cell proliferation and platelet aggregation (Moncada et al., 1991; Dalkara and Moskowitz, 1997). Second, there is biochemical, immunohistochemical, and functional evidence for impairment of NO signaling after experimental SAH in animals (Weir and Macdonald, 1993; Khurana and Besser, 1997; Khurana et al., 2002), while abnormal cerebrospinal fluid NO levels have been reported in humans following aneurysmal SAH (Sadamitsu et al., 2001). Third, in gene transfer paradigms, (recombinant) eNOS overexpression in animal (Khurana et al., 2002) and human (Khurana et al., 2000) intracranial arteries, even in the setting of experimental SAH, has been shown to be vasoprotective. Fourth, eNOS gene knockout in animals has been shown to predispose to hypertension, atherosclerosis, coronary artery disease, and aortic aneurysm formation (Kuhlencordt et al., 2001). Fifth, the inducible isoform of NOS (iNOS) and metabolically uncoupled eNOS can generate free radicals that adversely affect the structure and function of vessel walls (Moncada et al., 1991; Guzik et al., 2002). Last, several studies have shown that the gene encoding eNOS, located on chromosome 7q35-36 (Marsden et al., 1993), is polymorphic in humans and that the presence of certain eNOS polymorphisms may increase susceptibility to conditions including atherosclerosis, hypertension, myocardial infarction, coronary vasospasm and formation of abdominal aortic aneurysms (Wang et al., 1996; Hingorani et al., 1999; Nakayama et al., 1999; Hingorani, 2000; Kotani et al., 2000; Wang and Wang, 2000; Lembo et al., 2001). Such genetic variations, referred to as “functional polymorphisms” owing to their association with clinical phenotypes (Evans and Relling, 1999; Rusnak et al., 2001), include the eNOS gene intron-4 27-base pair variable-number-tandem-repeat (eNOS 27 VNTR; Wang et al., 1996; Wang and Wang, 2000), eNOS gene promoter single-nucleotide-polymorphism (eNOS T-786C SNP; Nakayama et al., 1999; Khurana et al., 2003), and eNOS gene exon-7 SNP (eNOS G894T SNP;Hingorani et al., 1999; Hingorani, 2000; Table 1).
Polymorphisms investigated in this study

Putative abnormal allele in bold italics.
4a, four 27-base pair tandem repeats; 4b, five 27-base pair tandem repeats; AMI, acute myocardial infarction; C, cytosine; CAD, coronary artery disease; eNOS, endothelial isoform of nitric oxide synthase; G, guanine; SNP, single nucleotide polymorphism; T, thymine; VNTR, variable number tandem repeat.
In the present study, we hypothesized that the aforementioned eNOS gene polymorphisms could predict susceptibility to aneurysmal SAH and post-SAH cerebral vasospasm, and that knowledge of this information could be used to improve clinical management strategies and outcomes for persons with intracranial aneurysms. In this context, the principal aims of our prospective clinical study were to: (1) compare the prevalence of these eNOS gene polymorphisms in a random sample of the population (control group) with their prevalence in SAH patients (cases); and (2) determine if there was an association between the presence of such polymorphisms and the presence and clinical severity of vasospasm in patients stratified by Fisher CT grade of SAH.
METHODS
Study participants
This prospective case-control study, approved by our Institutional Review Board, involved 141 human subjects each of whom gave informed consent for participation. Our control group consisted of 90 persons selected randomly from a countywide adult registry. Our case group was comprised of 51 persons admitted to our hospital diagnosed with aneurysmal SAH based on history and radiological findings, including both admission head CT scan and 4-vessel cerebral angiography. All SAH patients were followed clinically and radiologically [including serial transcranial Doppler ultrasonography (Manno, 1997) in each case] from the time of admission to the time of dismissal (mean 22±10 days). Forty-six (90%) of SAH patients were admitted to our hospital within 48 hours of their initial hemorrhage. All SAH patients were managed in our Neurological-Neurosurgical Intensive Care Unit according to established clinical protocols.
Grading systems
Radiological severity of SAH on admission CT scan was classified according to Fisher's grading system [i.e., grades 1 (no subarachnoid blood on CT) to 4 (predominantly intraventricular blood), where Fisher grade 3 represents the most diffuse SAH on CT;Fisher et al., 1980]. Clinical severity of SAH on admission neurological exam was classified according to the World Federation of Neurological Surgeons (WFNS) SAH clinical grading system [i.e., grades 1 (headache only, no clinical deficit) to 5 (moribund);Drake, 1988]. Outcome at dismissal was classified according to modified Rankin score (i.e., from 0 to 6) where scores of 0–2 indicates no to slight disability and 6 indicates death (Van Swieten et al., 1988). Presence and severity of cerebral vasospasm was determined clinically by serial comprehensive neurological examination and radiologically by serial transcranial Doppler ultrasonography and, when indicated, repeat cerebral angiography. Cerebral vasospasm was classified as being absent (no clinical or radiological abnormality), versus asymptomatically present (radiological abnormality only), versus symptomatically present (both clinical and radiological abnormalities). We defined symptomatic vasospasm to include the delayed occurrence of confusion, aphasia, obtundation, or new motor deficit in the absence of any other pharmacological, metabolic or structural cause, and with radiological confirmation.
Microarray and microfluidic chip-based genotyping
A single 20 mL sample of peripheral venous blood was obtained from all participants for subsequent DNA extraction and genetic analysis. Genomic DNA was extracted from peripheral blood lymphocytes using QIAamp® DNA Blood Minikit (Qiagen, Germantown, MD, USA). SNPs were genotyped using Nanochip™ active electronic arrays (Nanogen, San Diego, CA, USA) as described elsewhere (Sohni et al., 2001). Oligo 6.61 software was used to design polymerase chain reaction (PCR) primers (IDT, Coralville, IA, USA) based on GenBank sequences. PCR mixtures consisted of 25 μL AmpliTaq Gold Master Mix (Applied Biosystems, Foster City, CA, USA), 1 μM primers, 20 ng DNA template and water to 50 μL. All oligonucleotides were synthesized by IDT. Primer sequences were 5′-biotin-GCATGCACTCTGGCCTGAAGT-3′ (forward) and 5′-CAGGAAGCTGCCTTCCAGTGC-3′ (reverse) for eNOS T-786C SNP, and 5′-biotin-CTGGAGATGAAGGCAGGAGAC-3′ (forward) and 5′-CTCCATCCCACCCAGTCAATC (reverse) for eNOS G894T SNP. Thermal cycling conditions for each were 95°C for 10 minutes, 30 cycles of 94°C for 30 seconds, 58°C for 30 seconds, and 72°C for 45 seconds, and final extension at 72°C for 7 minutes. For eNOS T-786C SNP, reporter probes were 5′-Cy3-AGGGTCAGCCA-3′ and 5′-Cy5-GGGTCAGCCG-3′ with stabilizer oligonucleotide 5′-GCCAGGGAAGAGCTTGATGCCCTGGTGGGAGC-3′. For eNOS G894T SNP, reporter probes were 5′-Cy3-GTTCTGGGGGC-3′ and 5′-Cy5-AGTTCTGGGGGA-3′ with stabilizer oligonucleotide 5′-TCATCTGGGGCCTGCAGCAGCAGGGGCAGCA-3′. Known heterozygotes, verified by dye-terminator sequencing performed on ABI 377 DNA sequencers in both forward and reverse directions, were used as controls to normalize hybridization efficiency between dye-labeled reporters. PCR conditions and methods for analyzing the eNOS 27 VNTR polymorphism are detailed elsewhere (Sohni et al., 2003). PCR products were analyzed using DNA 500 LabChip® kit on Agilent 2100 Bioanalyzer (Agilent Technologies, Wilmington, DE, USA) following manufacturer's instructions. DNA fragment sizes were determined for each sample from the calibration curve in conjunction with markers and sizing ladder. Genotypes were designated based on fragment sizes obtained at the end of the run. It should be noted that in the present study, SNPs are designated by nucleotide substitution nomenclature, and that eNOS Glu298 Asp is the amino acid substitution-equivalent of eNOS G894T.
Data analysis
Where specified, data are expressed as mean ± standard deviation (SD) or as percentage of column totals. In comparing cases and controls (or, where specified, within SAH subgroups), P-values for gender, medical history variables, genotypes, and Fisher grades [i.e., grade 3 (diffuse) SAH versus grades 1,2, or 4 (nondiffuse) SAH] were determined using Chi-square tests, or Fisher's exact tests when sample sizes were limited. The P-value for age compared between cases and controls was calculated via a two-sample t-test. A multiple logistic regression analysis controlling for the variables age, gender and smoking status was used to evaluate the association of each eNOS genotype and SAH/control status. The prevalence of the eNOS 27 VNTR “4a” allele between cases and controls was also evaluated via an adjusted logistic regression model. Fisher's exact test was used to compare each genotype (as well as the prevalence of the eNOST-786C SNP “C” allele) between patients with no vasospasm, asymptomatic vasospasm, and symptomatic vasospasm. Where specified, odds ratio associations with 95% confidence interval (CI) for the 4a and C alleles were determined. P-values of <0.05 were regarded as statistically significant.
RESULTS
Clinical data
Compared with controls, cases were significantly younger and had a greater preponderance of females and smokers (Table 2). Only two (4%) of our 51 SAH patients, but no controls, had a family history of aneurysmal SAH. Among persons presenting with SAH, the single most frequent radiological grade of SAH on initial CT scan was Fisher grade 3, observed in 28 (55%) of 51 patients (Table 3). Cerebral vasospasm occurred radiologically in 28 (55%) of 51 patients, and was symptomatic in 12 (24%; Table 3). Following the initial hemorrhage, the mean time of vasospasm onset was 5.4 ± 3.2 days (range 1–12 days) and the mean duration was 12.9 ± 4.5 days (range 6–21 days; n = 28). Of the 28 patients presenting with Fisher grade 3 SAH, 21 (75%) developed cerebral vasospasm. This was significantly different to the remaining 23 patients presenting with Fisher grade 1, 2 or 4 SAH, of whom 7 (30%) developed vasospasm (P = 0.002; Table 4).
Comparison of cases with controls ∗
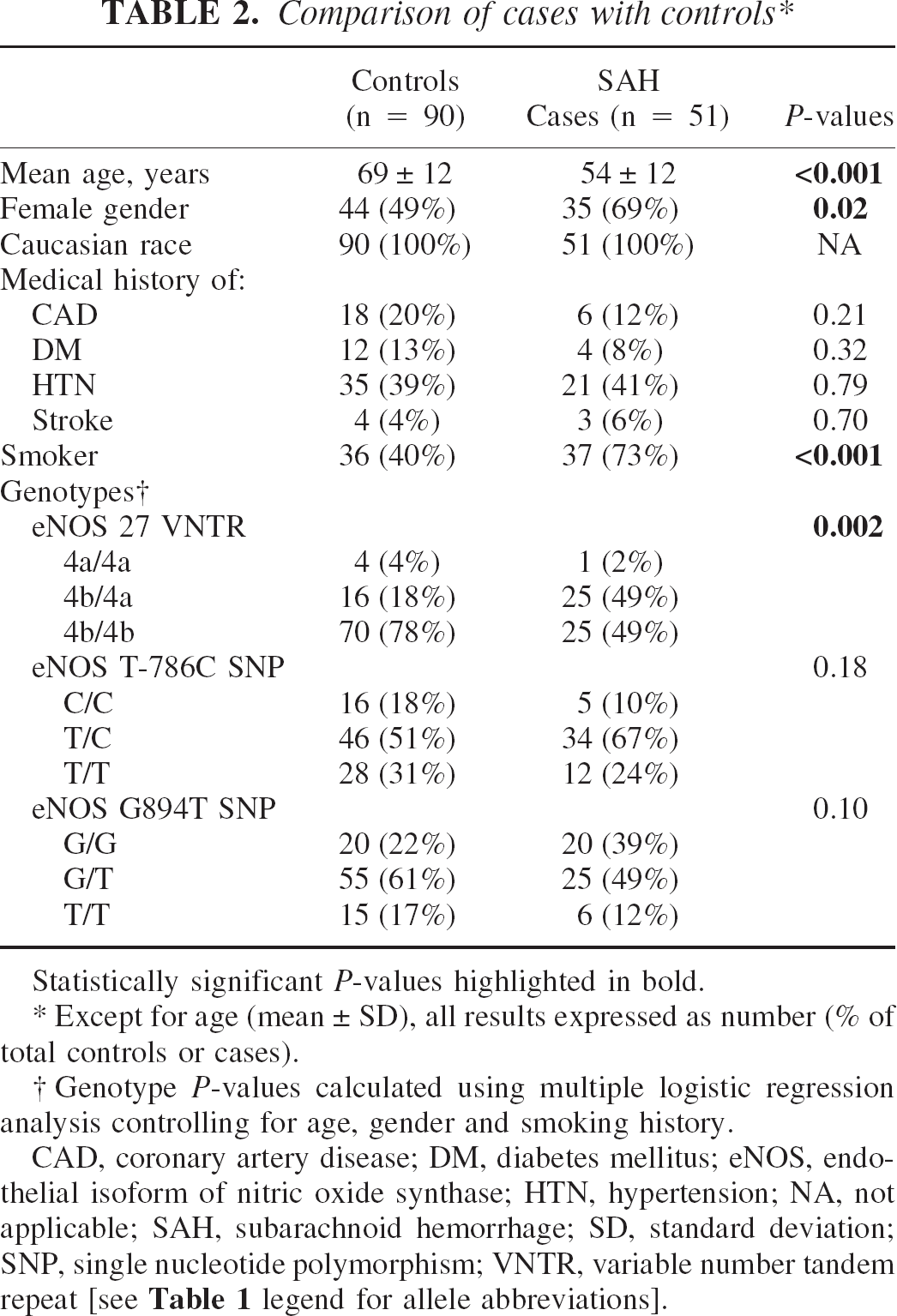
Statistically significant P-values highlighted in bold.
Except for age (mean ± SD), all results expressed as number (% of total controls or cases).
Genotype P-values calculated using multiple logistic regression analysis controlling for age, gender and smoking history.
CAD, coronary artery disease; DM, diabetes mellitus; eNOS, endothelial isoform of nitric oxide synthase; HTN, hypertension; NA, not applicable; SAH, subarachnoid hemorrhage; SD, standard deviation; SNP, single nucleotide polymorphism; VNTR, variable number tandem repeat [see Table 1 legend for allele abbreviations].
Clinical characteristics of SAH cases (n = 51) ∗

Expressed as number (% of cases).
See Methods for grading system details.
CT, Computerized tomography; SAH, subarachnoid hemorrhage; WFNS, World Federation of Neurological Surgeons.

Statistically significant P-values highlighted in bold.
CT = Computerized tomography.
See Methods for Fisher grading system details.
Genetic data
Aneurysmal SAH and the eNOS 27 VNTR:
Using multiple logistic regression analysis adjusting for age, gender and smoking history, there was a significant difference in the distribution of genotypes for the eNOS 27 VNTR polymorphism in SAH cases compared with controls (P = 0.002;Table 2). Heterozygosity for this polymorphism was almost 3 times as prevalent among cases compared with controls (P = 0.002;Table 2). The “4a” allele was found in 26 of 51 (51%) of cases, compared with 20 of 90 (22%) controls; this difference was significant (P = 0.007). In comparing SAH cases with controls, the odds ratio for being a case for those with at least one 4a allele was 3.95 (95% CI 1.45–10.65; P = 0.007) after controlling for differences between age, smoking and gender.
Post-SAH cerebral vasospasm and the eNOS T-786C SNP:
Among the 28 patients with Fisher grade 3 SAH, the eNOS T-786C SNP significantly differentiated between the presence and severity of cerebral vasospasm (P = 0.04;Table 5). Among these 28 patients, 21 experienced cerebral vasospasm and 18 (86%) of these were heterozygous for this SNP. The “C” allele occurred in 4 of 7 (57%) patients with no vasospasm, 8 of 10 (80%) of patients with asymptomatic vasospasm, and in all 11 (100%) patients with symptomatic vasospasm; this trend was significant (P = 0.046). Among the 28 patients with Fisher grade 3 SAH, the odds ratio for any spasm for those with at least one C allele was 7.1 (95% CI 0.88–57.5; P = 0.065).
Genotype versus presence and clinical severity of cerebral vasospasm among SAH cases with Fisher grade 3 SAH (n = 28) ∗
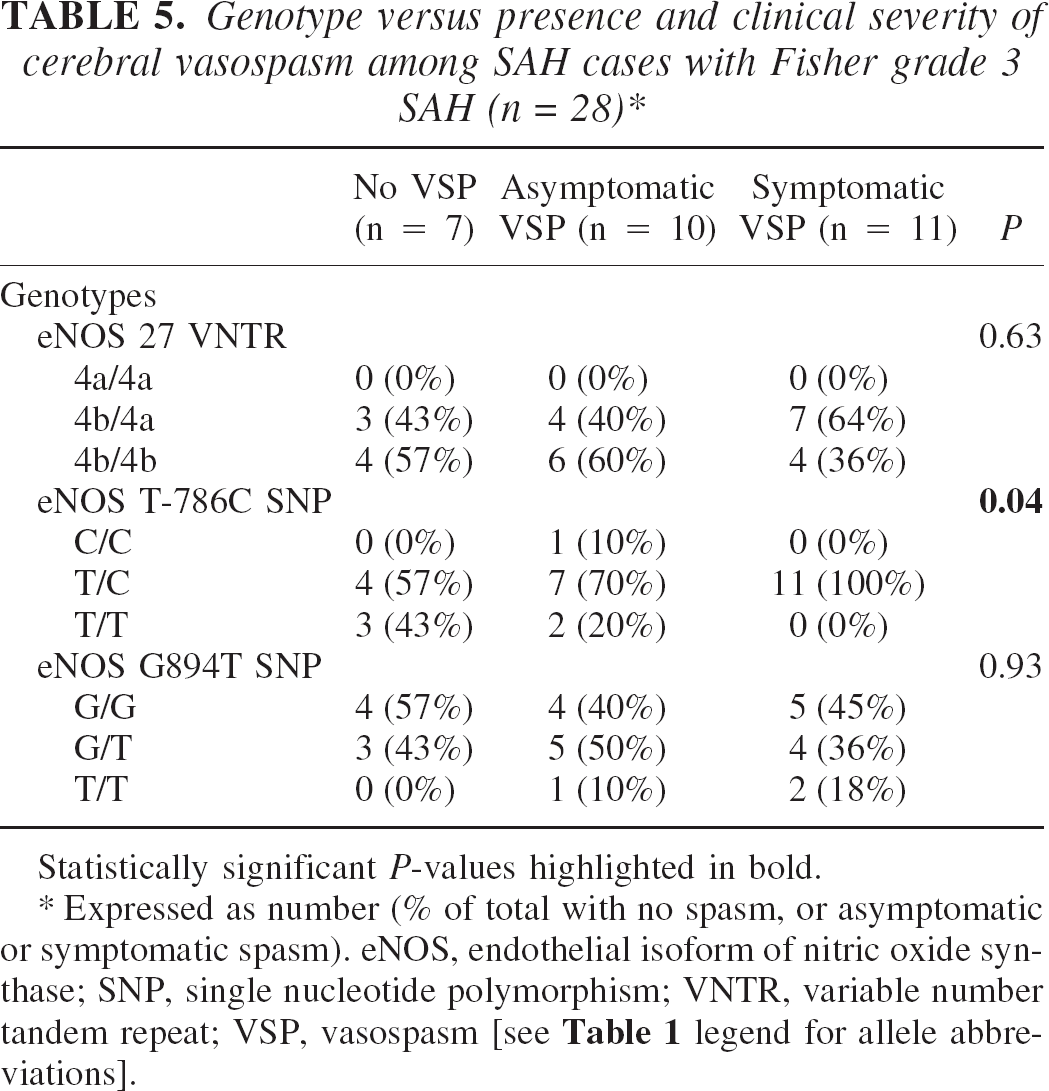
Statistically significant P-values highlighted in bold.
Expressed as number (% of total with no spasm, or asymptomatic or symptomatic spasm). eNOS, endothelial isoform of nitric oxide synthase; SNP, single nucleotide polymorphism; VNTR, variable number tandem repeat; VSP, vasospasm [see Table 1 legend for allele abbreviations].
DISCUSSION
Key findings
Despite the fact that in approximately 5–10% of persons with intracranial aneurysms genetic susceptibility to the formation of such lesions is indicated by the presence of a heritable connective tissue disorder and/or positive family history (Schievink, 1997), to our knowledge, the present study demonstrates for the first time genetic susceptibility to aneurysmal rupture (SAH) and post-SAH vasospasm. The unique findings of our study are that: (1) the eNOS 27 VNTR (in particular, the “4a” allele) is significantly associated with the occurrence of aneurysmal SAH; and (2) the eNOS T-786C SNP (in particular, the “C” allele) is significantly associated with the occurrence of post-SAH vasospasm. Our study lends further support to the notion that polymorphism of the eNOS gene may contribute in part to predetermining disease susceptibility throughout the cardiovascular system.
Aneurysmal SAH and the eNOS 27 VNTR:
Our finding with regard to the eNOS 27 VNTR and increased susceptibility to intracranial aneurysm rupture supports the work of Wang et al. (1996) and Kotani et al. (2000) who found that the relatively rare 4a allele of this polymorphism was associated with increased susceptibility to cardiovascular disease, specifically, coronary artery disease and aortic aneurysm rupture, respectively. Although the rare prevalence of the 4a/4a homozygote in participants of our study precluded statistical comparisons of this genotype between cases and controls, we found that heterozygosity (involving carriage of the putative abnormal 4a allele) occurred almost three times as frequently in SAH cases compared with controls. With regard to the prevalence of the 4a allele itself, we found this allele to be significantly overrepresented among our ruptured intracranial aneurysm cases compared with controls (i.e., 51% versus 22%, respectively). This is consistent with the study of this eNOS polymorphism by Kotani et al. (2000) who reported a significantly higher prevalence of the 4a allele among individuals susceptible to abdominal aortic aneurysm formation. Further support for a putative role for the 4a allele in aneurysmal disease pathogenesis comes from the present study, in which we found an odds ratio of approximately 4 for being an aneurysmal SAH case for those persons with at least one 4a allele.
Post-SAH cerebral vasospasm and the eNOS T-786C SNP:
With regard to determining susceptibility towards development of post-SAH vasospasm, we studied eNOS genotypes of the 28 patients who had Fisher grade 3 SAH, our largest radiological subgroup. As Fisher grade 3 hemorrhage has previously been shown to be associated with increased propensity towards developing vasospasm (Fisher et al., 1980), by studying the genotypes of individuals in this subgroup, we were able to standardize to the best of our ability for the amount of subarachnoid blood present. By comparing the incidence of vasospasm among patients presenting with Fisher grade 3 SAH (i.e., diffuse subarachnoid blood on CT) with the incidence among patients presenting with Fisher grades 1, 2 and 4 SAH (i.e., no subarachnoid blood, minimal subarachnoid blood, and predominantly intraventricular blood on CT, respectively), we confirmed that Fisher grade 3 SAH itself was significantly associated with the development of cerebral vasospasm (Fisher et al., 1980). However, the unreliability of this grade (i.e., this extent of subarachnoid blood) alone as being a predictor of vasospasm occurrence was suggested by our finding that 7 of 28 (25%) Fisher grade 3 patients did not develop vasospasm, and 7 of 23 (30%) patients with radiologically lesser amounts of subarachnoid blood (i.e., Fisher grades 1, 2, or 4) did develop vasospasm. This suggested to us that the amount of subarachnoid blood on a CT scan was unlikely to be a sole predictor for the occurrence of post-SAH vasospasm. In this context, we found that among our 28 Fisher grade 3 patients, 21 developed vasospasm and, of these, the vast majority (18 of 21; 86%) were heterozygous for the eNOS T-786C SNP. The only C/C homozygote among the 28 patients with Fisher grade 3 hemorrhage also developed cerebral vasospasm, albeit asymptomatically. These findings corroborate those reported in the seminal study by Nakayama et al. (1999) who found that the T-786C SNP was associated with increased susceptibility to coronary vasospasm. In their study, Nakayama et al. (1999) reported that the C allele was significantly overrepresented among cases with coronary vasospasm compared with controls (the rare C/C genotype was exclusively present in cases while the C-containing heterozygous genotype was four times more frequent in cases). Interestingly, among the 28 patients with Fisher grade 3 SAH in our study, the prevalence of the C allele showed a significant trend in terms of its association with the occurrence and severity of cerebral vasospasm: it was found in 57% of patients no vasospasm, 80% of patients with asymptomatic vasospasm, and 100% of patients with symptomatic vasospasm. Although we found an odds ratio of approximately 7 for the presence of spasm for those with at least one C allele, this approached but did not reach statistical significance, possibly related to the fewer number of patients in this analysis subgroup.
Pathogenic mechanisms
While our findings suggest that the eNOS 27 VNTR and T-786C gene polymorphisms are themselves functionally involved in increased risk of aneurysm rupture and post-SAH vasospasm, respectively, we recognize that these polymorphisms may be markers in linkage disequilibrium with other yet undefined genetic aberrations predisposing towards these disease entities (Evans and Relling, 1999; Rubin and Tall, 2000). This may explain our observation that not all persons with the putative variants (i.e., containing “4a” and “C” alleles) experienced SAH or post-SAH vasospasm. However, despite this possibility, it is important to note that polymorphic NOS dysfunction may result from the transcription and translation of a putatively abnormal allele in either the homozygous or heterozygous form (Nakayama et al., 1999). The precise molecular effects of these polymorphisms have not been elucidated, although there is biochemical evidence for decreased eNOS gene promoter activation associated with the T-786C SNP (Nakayama et al., 1999) and reduced eNOS protein expression and enzymatic activity associated with eNOS 27 VNTR and T-786C polymorphisms (Song et al., 2003). Further, it is possible that polymorphic eNOS may contribute towards aneurysm pathobiology and cerebral vasospasm through increased local oxidative stress leading to vessel wall damage (Khurana and Besser, 1997; Fukuda et al., 2000; Johanning et al., 2001; Kuhlencordt et al., 2001; Guzik et al., 2002; Johanning et al., 2002), predilection towards development of atherogenic intimal hyperplasia (Hingorani, 2000; Kuhlencordt et al., 2001) and systemic hypertension (Wang and Wang, 2000; Kuhlencordt et al., 2001), the presence of aberrant vascular smooth muscle proliferation (Moncada et al., 1991; Khurana and Besser, 1997), and increased platelet aggregation and pro-inflammatory monocyte adhesion (Moncada et al., 1991; Dumont et al., 2003), all of which are associated with NO signaling dysfunction. Such mechanisms may account for the impaired vasorelaxation and heightened vascular wall inflammation characteristic of post-SAH vasospasm (Macdonald and Weir, 1991; Weir and Macdonald, 1993; Khurana and Besser, 1997; Khurana et al., 2002; Dumont et al., 2003). Last, our observations that the eNOS 27 VNTR polymorphism is associated with increased risk of rupture and that the eNOS T-786C SNP is associated with increased risk of vasospasm after rupture suggest that there are two divergent mechanisms of action, as one would not expect these results if only decreased production of NO by the eNOS variants occurred.
Limitations and clinical implications of this study
We recognize certain limitations of our study. First, while our control group consisted of a random adult population sample from our county, it may have included a small number of subjects (likely n ≤ 4) with undiagnosed unruptured intracranial aneurysms whose population prevalence is estimated to be 1–5% (Inagawa et al., 1990; Schievink et al., 1997). It was felt that inclusion of this small, putative unruptured aneurysm cohort in the control group would avoid the selection bias of using only subjects who had undergone intracranial vascular imaging, i.e., those who might be more likely to have systemic cardiovascular disease, itself associated with eNOS genetic aberration (Hingorani, 2000; Wang and Wang, 2000). Second, although the clinical characteristics of our SAH group reflect those observed in other series of ruptured intracranial aneurysms (Sundt and Whisnant, 1978; Testa et al., 1985; Forget et al., 2001), our case and control groups differed significantly, but not unexpectedly (Schievink, 1997), in terms of age, gender and smoking status. Despite detecting no difference in the incidence of hypertension between the groups, ideally our control group would have been better matched if it had the same incidence of smoking, a strong SAH risk factor (Schievink, 1997). Therefore, in evaluating the distribution of eNOS genotypes between cases and controls, we adjusted for age, gender and smoking status using multiple logistic regression analysis, as has been carried out in other related studies (Nakayama et al., 1999). Last, another limitation of our work was that it involved only 51 aneurysmal SAH patients, a relatively small number. We intend to address this issue in an expanded cooperative study.
Despite the aforementioned limitations, we are encouraged by our observation of significant trends that bear striking similarities to findings in other cardiovascular disease cohorts (Wang et al. 1996; Wang and Wang, 2000), including abdominal aortic aneurysm (Kotani et al., 2000) and coronary vasospasm (Nakayama et al., 1999; Luscher and Noll, 1999). In identifying an association between eNOS gene polymorphism and susceptibility towards aneurysmal SAH and post-SAH vasospasm, the clinical implications of the present study are mainly twofold. First, in patients with unruptured intracranial aneurysms, knowledge of eNOS 27-VNTR genotype can be used as a helpful adjunct to knowledge of structural features of the aneurysm such as its size, location, and morphology when determining risk for rupture. In this context, it is conceivable that with more precise genetic and anatomical information about an unruptured aneurysm, more aggressive investigation and earlier treatment of the lesion can be initiated, i.e., prior to its potentially devastating rupture. Second, in patients with ruptured intracranial aneurysms, knowledge of eNOS T-786C genotype can potentially be used as a prejunct to clinical and radiological stigmata of vasospasm, through the identification of a subset of individuals more likely to develop vasospasm. We feel that more aggressively monitoring and prophylactically treating such individuals before vasospasm occurs will likely abrogate the neurological impairment associated with vasospasm, and reduce the substantial morbidity and mortality associated with aneurysmal disease.
Footnotes
Acknowledgments
We thank Drs. Jonathan Friedman, Ed Manno, Ronald Petersen, David Piepgras, Brian Weinshenker and Richard Weinshilboum for their insight and helpful comments regarding this study. We also thank Mr. Neal Jorgensen for assistance with statistical analyses, and Ms. Mary Soper and Ms. Constance Hoeft for assistance with preparation of the manuscript.