
Abstract
This study was designed to determine the effect of subarachnoid hemorrhage (SAH) on potassium (K+) channels involved in relaxations of cerebral arteries to nitrovasodilators. The effects of K+ channel inhibitors on relaxations to 3-morpholinosydnonimine (SIN-1) and sodium nitroprusside (SNP) were studied in rings of basilar arteries obtained from untreated dogs and dogs exposed to SAH. The levels of cyclic GMP were measured by radioimmunoassay. In rings without endothelium, concentration-dependent relaxations to SIN-1 (10−9 − 10−4 mol/L) and SNP (10−9 − 10−4 mol/L) were not affected by SAH, whereas increase in cyclic GMP production stimulated by SIN-1 (10−6 mol/L) was significantly suppressed after SAH. The relaxations to SIN-1 and SNP were reduced by charybdotoxin (CTX; 10−7 mol/L), a selective Ca2+-activated K+ channel inhibitor, in both normal and SAH arteries; however, the reduction of relaxations by CTX was significantly greater in SAH arteries. By contrast, the relaxations to these nitrovasodilators were not affected by glyburide (10−5 mol/L), an ATP-sensitive K+ channel inhibitor, in both normal and SAH arteries. These findings suggest that in cerebral arteries exposed to SAH, Ca2+-activated K+ channels may play a compensatory role in mediation of relaxations to nitric oxide. This may help to explain mechanisms of relaxations to nitrovasodilators in arteries with impaired production of cyclic GMP.
Chronic cerebral vasospasm is a major cause of morbidity and mortality in patients with subarachnoid hemorrhage (SAH; Heros et al., 1983; Kassell et al., 1985). Although precise mechanisms responsible for pathogenesis of vasospasm still remain to be defined, an impairment of endothelium-dependent relaxations may contribute to the narrowing of cerebral arteries (Nakagomi et al., 1987; Kanamaru et al., 1989). Nitric oxide, an important mediator of endothelium-dependent relaxations (Furchgott, 1983; Moncada et al., 1991), is believed to produce vascular relaxations via activation of the soluble guanylate cyclase and subsequent increase of cyclic GMP (Rapoport et al., 1983; Ignarro and Kadowitz, 1985). Several investigators have reported that expression of guanylate cyclase protein (Kasuya et al., 1995), and production of cyclic GMP (Kim et al., 1992; Pasqualin et al., 1992; Nakao et al., 1996) are decreased in cerebral arteries after SAH. These findings may explain the impairment of nitric oxide-mediated relaxations in spastic cerebral arteries. In contrast, it has been also documented that endothelium-independent relaxations to exogenous nitric oxide donors are maintained in arteries affected by experimental SAH (Kanamaru et al., 1989; Nakao et al., 1996), suggesting that certain compensatory mechanisms may preserve reactivity to nitrovasodilators despite impaired formation of cyclic GMP.
In recent years, electrophysiologic and pharmacologic studies showed an important role of potassium (K+) channels in the hyperpolarization and relaxations of smooth muscle cells (Brayden, 1990; Nelson and Quayle, 1995). It has been also suggested that nitric oxide liberated from nitrovasodilators can produce relaxations by activating K+ channels in cerebral arteries (Paterno et al., 1996; Onoue and Katusic, 1997). In cerebral arteries exposed to SAH, smooth muscle is depolarized because of a decreased membrane conductance to K+ (Waters and Harder, 1985; Harder et al., 1987); therefore, we decided to examine whether SAH may modify the role of K+ channels involved in cerebral arterial relaxations to nitrovasodilators. We characterized the effect of K+ channel inhibitors on relaxations to 3-morpholinosydnonimine (SIN-1) and sodium nitroprusside (SNP) in basilar arteries obtained from untreated dogs and dogs exposed to SAH (Varsos et al., 1983; Liszczak et al., 1983; Katusic et al., 1993).
MATERIALS AND METHODS
Animal model of subarachnoid hemorrhage
Mongrel dogs of either sex weighing 12 to 17 kg were used. Induction of SAH followed by cerebral vasospasm was conducted as described in our previous study (Katusic et al., 1993). Under general anesthesia with 15 mg/kg of intravenous thiopental, the cisterna magna was aseptically punctured with a No. 22 spinal needle, and 5 mL of CSF was removed. Five milliliters of autologous venous blood was injected through the spinal needle over 2 minutes. Subsequently, with the animal in a 30° head-down position for 15 minutes, the animal was allowed to recover. Two days later (day 2), the injection of venous blood into the cisterna magna was repeated with the same manner as the first injection. Because the identical procedures evoked reproducible cerebral vasospasm (Katusic et al., 1993), angiography was not performed in the present study. On day 7, the animals were killed, and isolated cerebral arteries were studied in organ chambers. Procedures and handling of the animals were reviewed and approved by the Institutional Animal Care and Use Committee of the Mayo Foundation.
In vitro studies
The brain was removed from dogs (untreated animals and those exposed to SAH) anesthetized with 30 mg/kg of intravenous pentobarbital and placed into cold modified Krebs-Ringer bicarbonate solution (control solution; millimolar composition: NaCl, 118.3; KCl, 4.7; CaCl2, 2.5; MgSO4, 1.2; KH2PO4 1.2; NaHCO3 25.0; calcium-ethylenediamine-tetraacetic acid, 0.026; and glucose, 11.1). Experiments were performed on rings (4-mm long) of basilar arteries. The arterial rings were placed in the control solution, and the endothelium was removed mechanically by gentle rubbing of the intimal surface with a stainless-steel wire (31-gauge diameter; Katusic et al., 1984). Each ring was connected to an isometric force-displacement transducer (Grass FT03; Grass Instrument Co., Quincy, MA) and suspended in an organ chamber filled with 25 mL of control solution (37°C, pH 7.4) aerated with 94% O2 −6% CO2. Isometric tension was recorded continuously. The rings were allowed to stabilize at a resting tension of 0.2 to 0.4 g for 1 hour. Each ring was then gradually stretched to the optimal point of its length-tension curve (approximately 3.0 g) as determined by the contraction to 10−5 mol/L of uridine 5′-triphosphate (UTP; Katusic et al., 1987). The successful removal of endothelium was verified by the absence of relaxation induced by 10−6 mol/L of bradykinin (Katusic et al., 1986).
Radioimmunoassay of cyclic GMP
A radioimmunoassay technique was used to determine the tissue content of cyclic GMP. Rings without endothelium were initially incubated in control solution bubbled with a 94% O2-6% CO2 gas mixture and maintained at 37°C. After 1 hour, the rings were incubated for additional 30 minutes in a solution containing 3-isobutyl-1-methylxanthine (10−3 mol/L) to inhibit the degradation of cyclic nucleotides by phosphodiesterases. A nitric oxide donor, SIN-1, was added after 20 minutes of incubation and was in contact with the rings for the remaining 10 minutes. After the incubation, the rings were immediately removed from the solution and frozen in liquid nitrogen. Cyclic GMP radioimmunoassay kits (Amersham, Arlington Heights, IL) were used to perform the measurements. Protein assay was conducted by DC Protein Assay Kit (Bio-Rad, Hercules, CA).
Drugs
The following pharmacologic agents were used: UTP (Sigma, St. Louis, MO), bradykinin (Sigma), SIN-1 (Molecular Probes, Eugene, OR), SNP (Sigma), 8-bromo-3′,5′-cGMP (8-bromo-cGMP; Sigma), charybdotoxin (CTX; Sigma), glyburide (Biomol Res. Lab. Inc., Natick, PA), 4-aminopyridine (Research Biochemicals International, Plymouth Meeting, MA), and papaverine hydrochloride (Sigma). Drugs were dissolved in distilled water so that volumes of less than 0.15 mL were added to the organ chambers. Stock solutions of glyburide were prepared in dimethyl sulfoxide (Sigma). Concentrations of all drugs are expressed as final molar (mol/L) concentration in the bathing solution. The rings were contracted with 10−5 mol/L UTP before the addition of vasodilator agents. Concentration-response curves were obtained in a cumulative fashion. Several rings prepared from the same artery were studied in parallel, and a concentration-response curve was established by each preparation. The relaxations were expressed as a percentage of maximal relaxations induced by papaverine (3 × 10−4 mol/L). The drugs used as K+ channel inhibitors were added 20 minutes before obtaining the concentration-response curve for each vasodilator agent. CTX (10−7 mol/L) and 4-aminopyridine (10−3 mol/L) caused contractions of quiescent basilar arteries obtained from untreated dogs and dogs exposed to SAH; however, the contractions were not significantly different between both groups (Table 1). In addition, because UTP produced only small contractions in the rings already contracted by K+ channel inhibitors, absolute values of tension did not differ significantly between control arteries and arteries treated with K+ channel inhibitors (see figure legends 1–9). In certain experiments, the median effective concentration was calculated for each ring by linear interpolation between the two concentrations evoking responses just above and below 50% of the maximum.
Concentration-response curves to 3-morpholinosydnonimine (SIN-1)
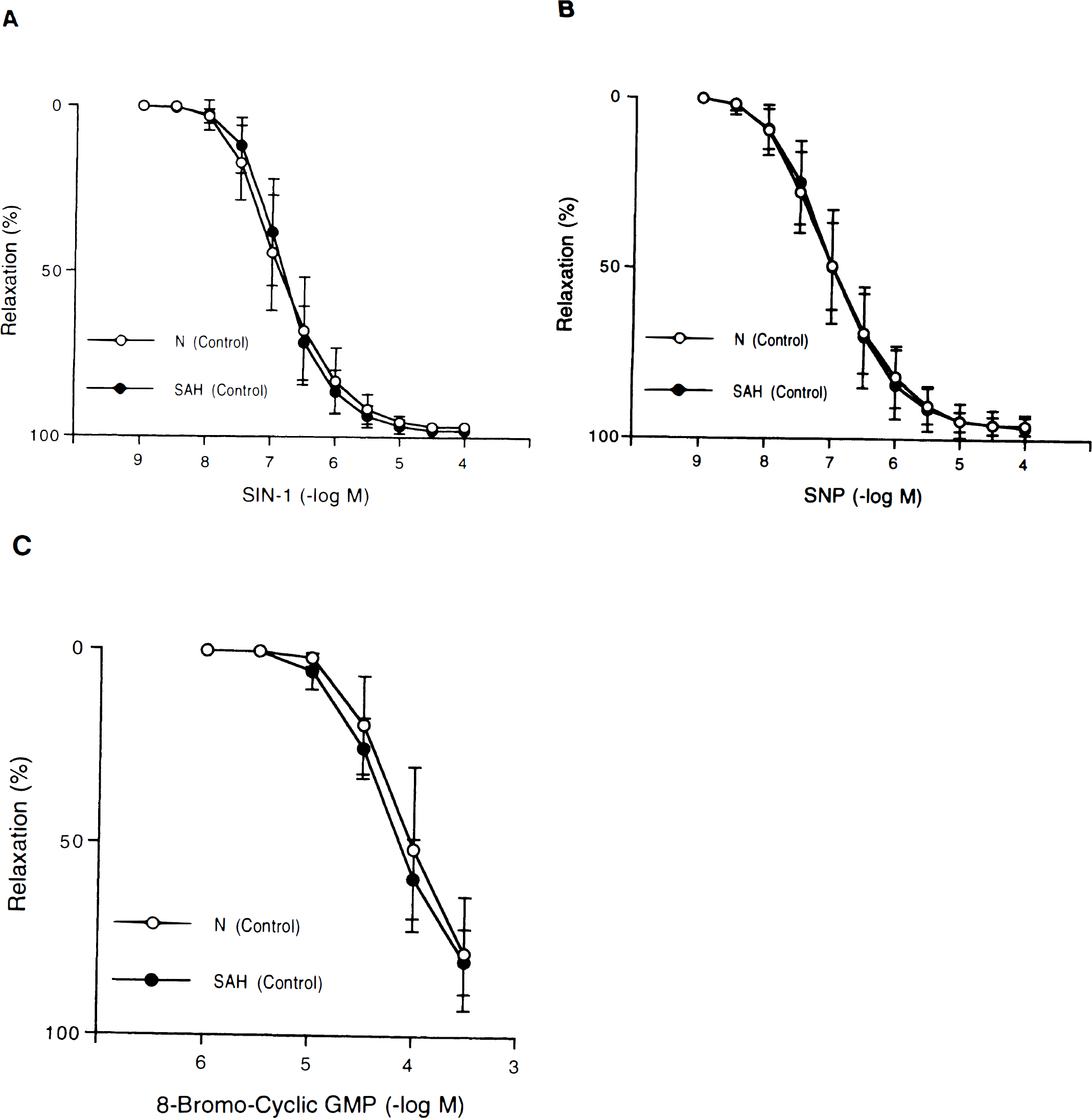
Concentration-response curves to 3-morpholinosydnonimine (SIN-1) in canine basilar artery rings without endothelium obtained from normal (N)
Concentration-response curves to sodium nitroprusside (SNP) in canine basilar artery rings without endothelium obtained from normal (N)
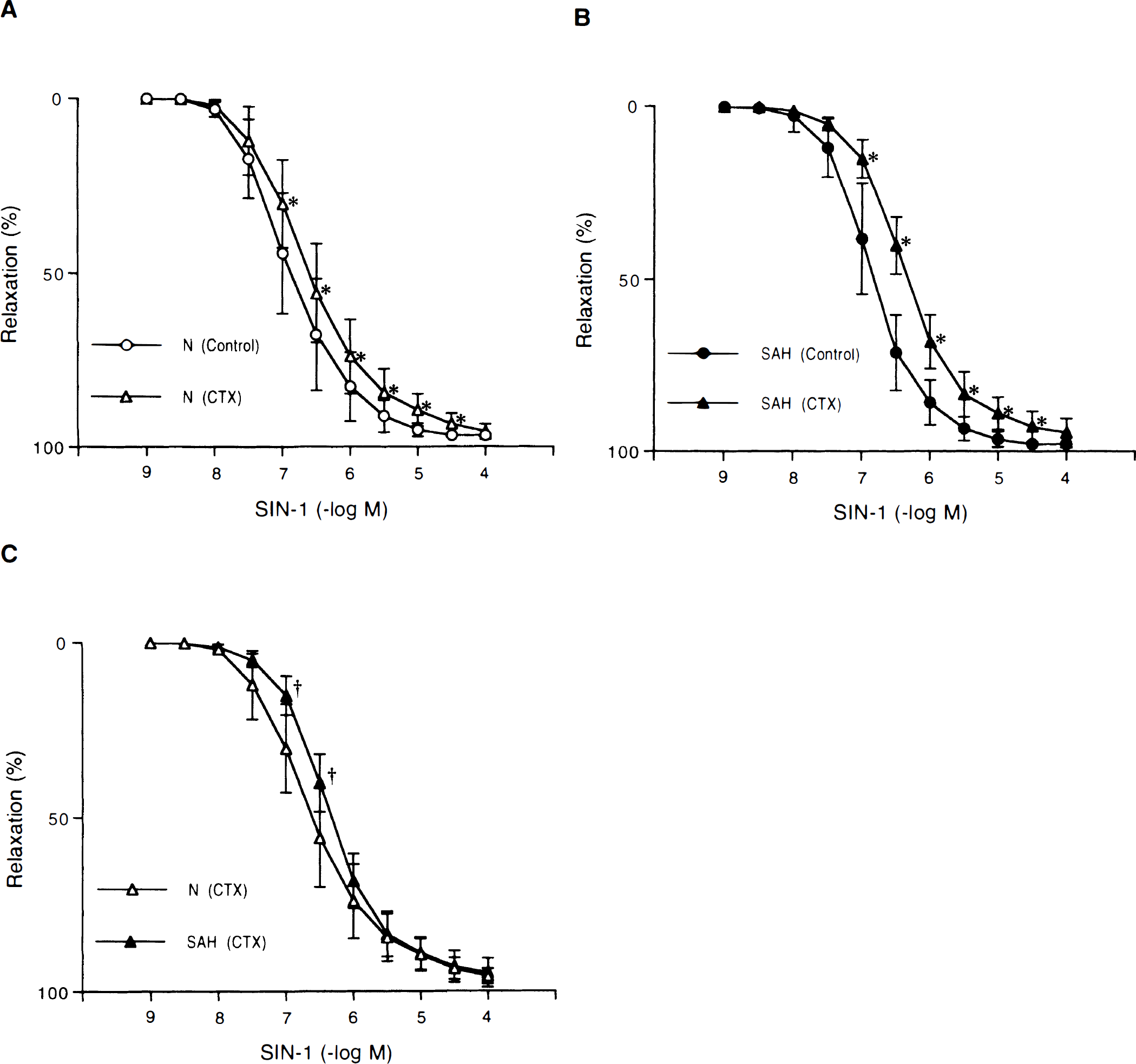
Concentration-response curves to 3-morpholinosydnonimine (SIN-1) in canine basilar artery rings without endothelium obtained from normal (N)
Concentration-response curves to sodium nitroprusside (SNP) in canine basilar artery rings without endothelium obtained from normal (N)
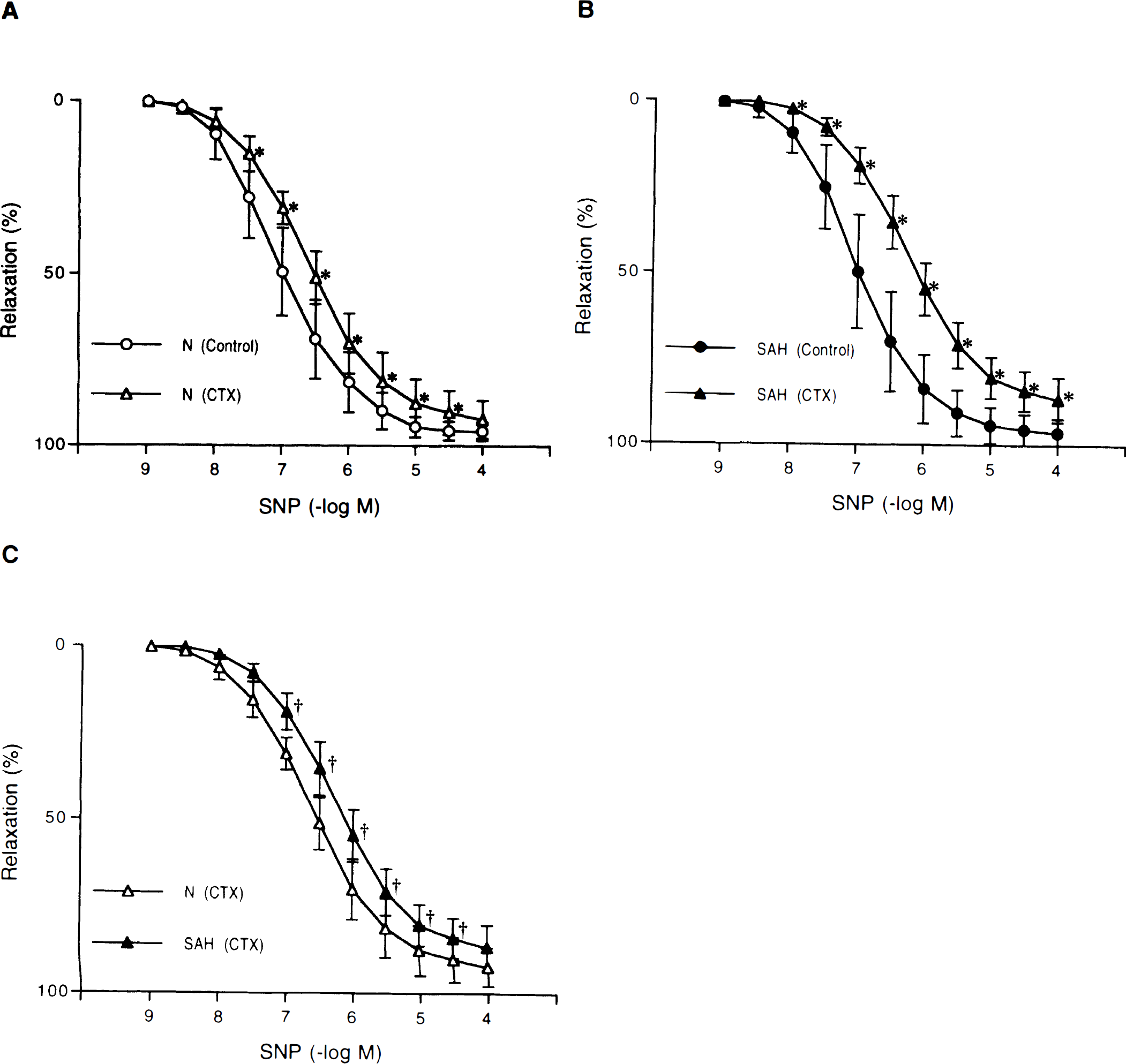
Concentration-response curves to 3-morpholinosydnonimine (SIN-1) in canine basilar artery rings without endothelium obtained from normal (N)
Concentration-response curves to sodium nitroprusside (SNP) in canine basilar artery rings without endothelium obtained from normal (N)
Concentration-response curves to 8-bromo-3′,5′-cyclic guanosine monophosphate (8-bromo-cyclic GMP) in canine basilar artery rings without endothelium obtained from normal (N)
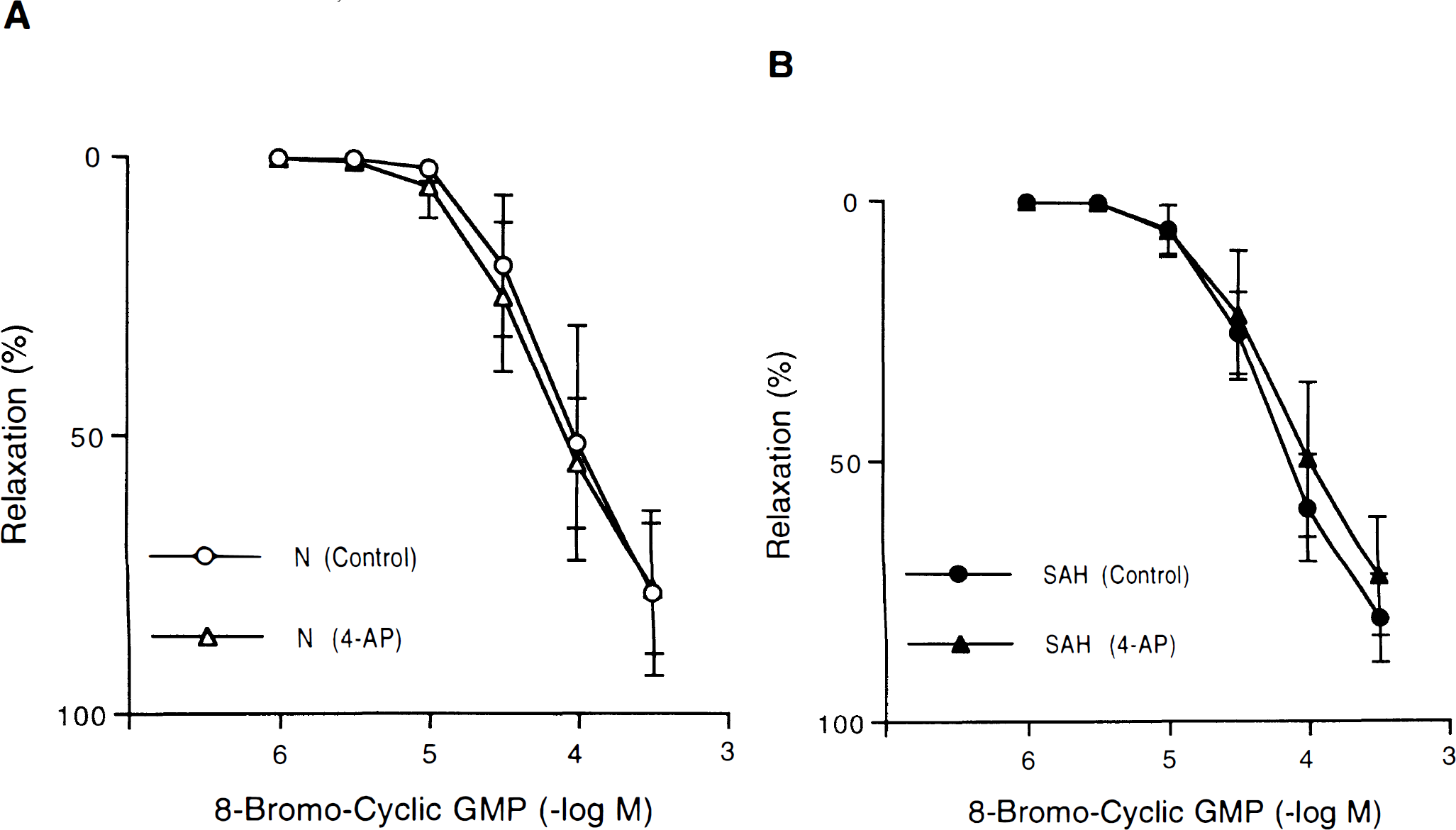
Concentration-response curves to 8-bromo-3′,5′-cyclic guanosine monophosphate (8-bromo-cyclic GMP) in canine basilar artery rings without endothelium obtained from normal (N)
Effect of charybdotoxin and 4-aminopyridine on the resting tension of canine basilar arteries without endothelium obtained from untreated or subarachnoid hemorrhage dogs

Values are expressed as mean ± SD. SAH, subarachnoid hemorrhage.
Statistical analysis
The results are expressed as means ± SD; n refers to the number of animals studied. Statistical evaluation of the data was performed by using analysis of variance, followed by Bonferroni/Dunn's post-hoc test. Statistical significance was accepted at the level of P < .05.
RESULTS
During contractions induced by UTP (10−5 mol/L), SIN-1 (10−9 − 10−4 mol/L), SNP (10−9 − 10−4 mol/L), and 8-bromo-cyclic GMP (10−6 − 3 × 10−4 mol/L) caused concentration-dependent relaxations in rings of canine basilar arteries without endothelium. The relaxations to these agonists were similar in rings obtained from untreated dogs and dogs exposed to SAH (Fig. 1).
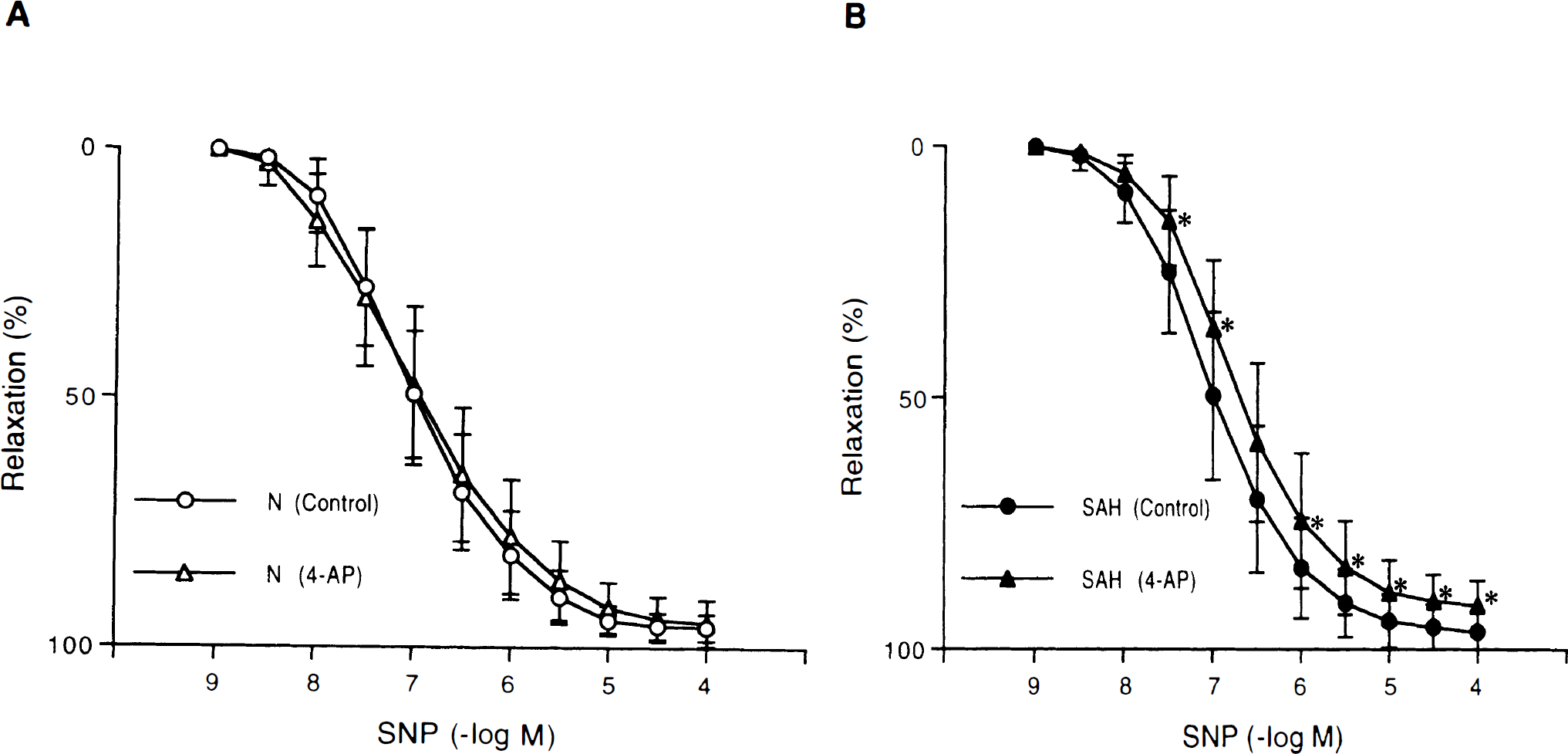
CTX (10−7 mol/L) reduced the relaxations to SIN-1 or SNP in rings from both untreated and SAH dogs (Figs. 2A, 2B, and 3A, 3B). The reduction of relaxations to SIN-1 or SNP by CTX was significantly greater in SAH arteries than in normal arteries (Figs. 2C and 3C).
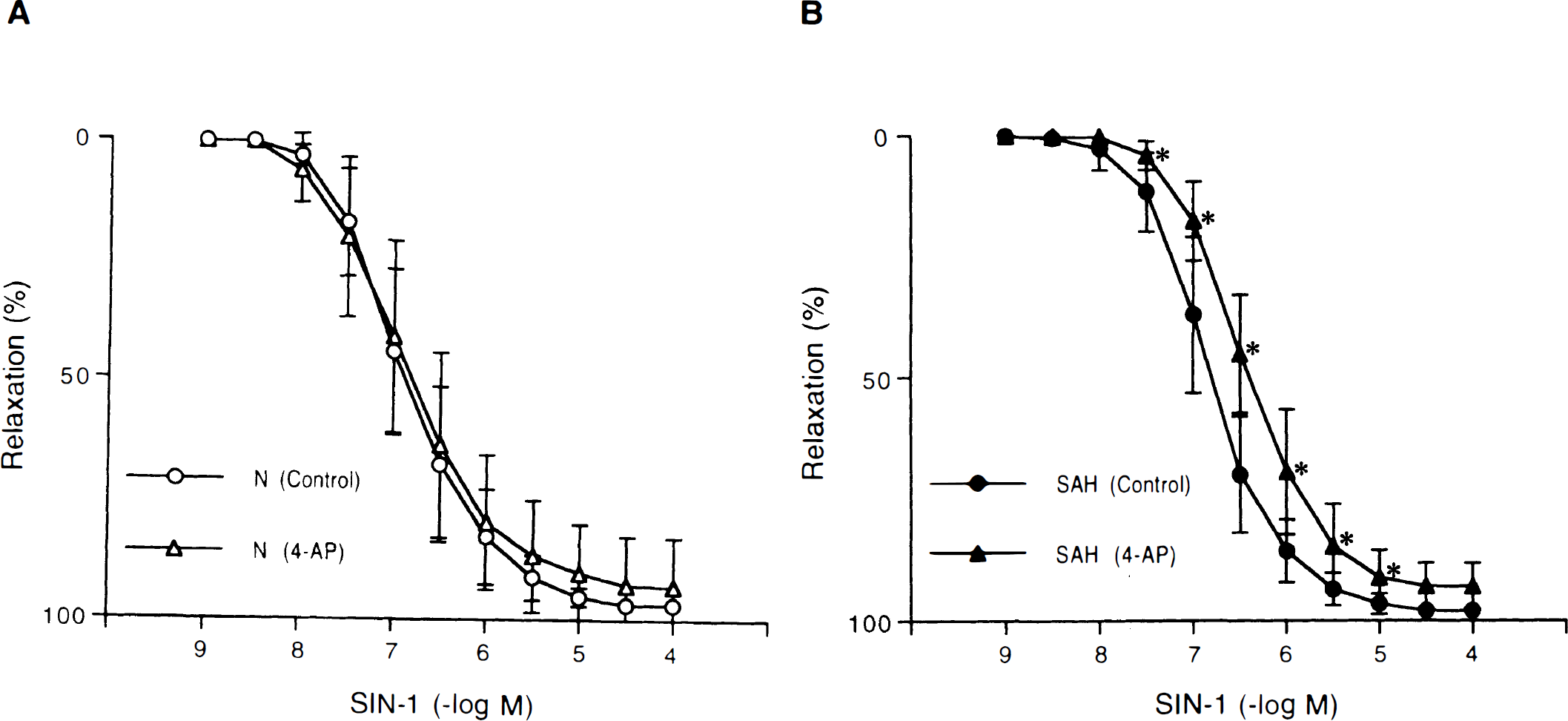
4-aminopyridine (10−3 mol/L) did not affect the relaxations to SIN-1 or SNP in normal arteries (Figs. 4A and 5A); however, it significantly reduced the relaxations to these nitric oxide donors in arteries obtained from SAH dogs (Figs. 4B and 5B).
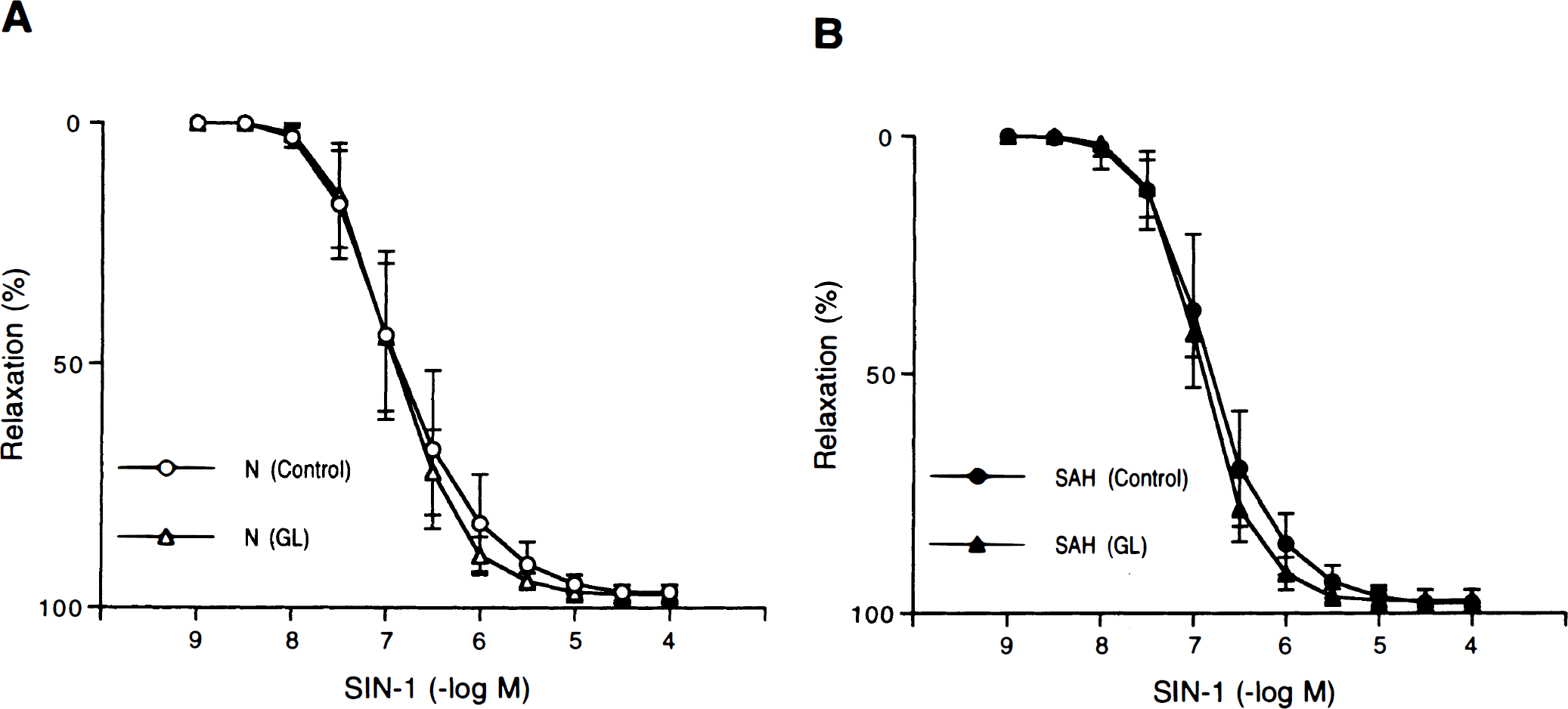
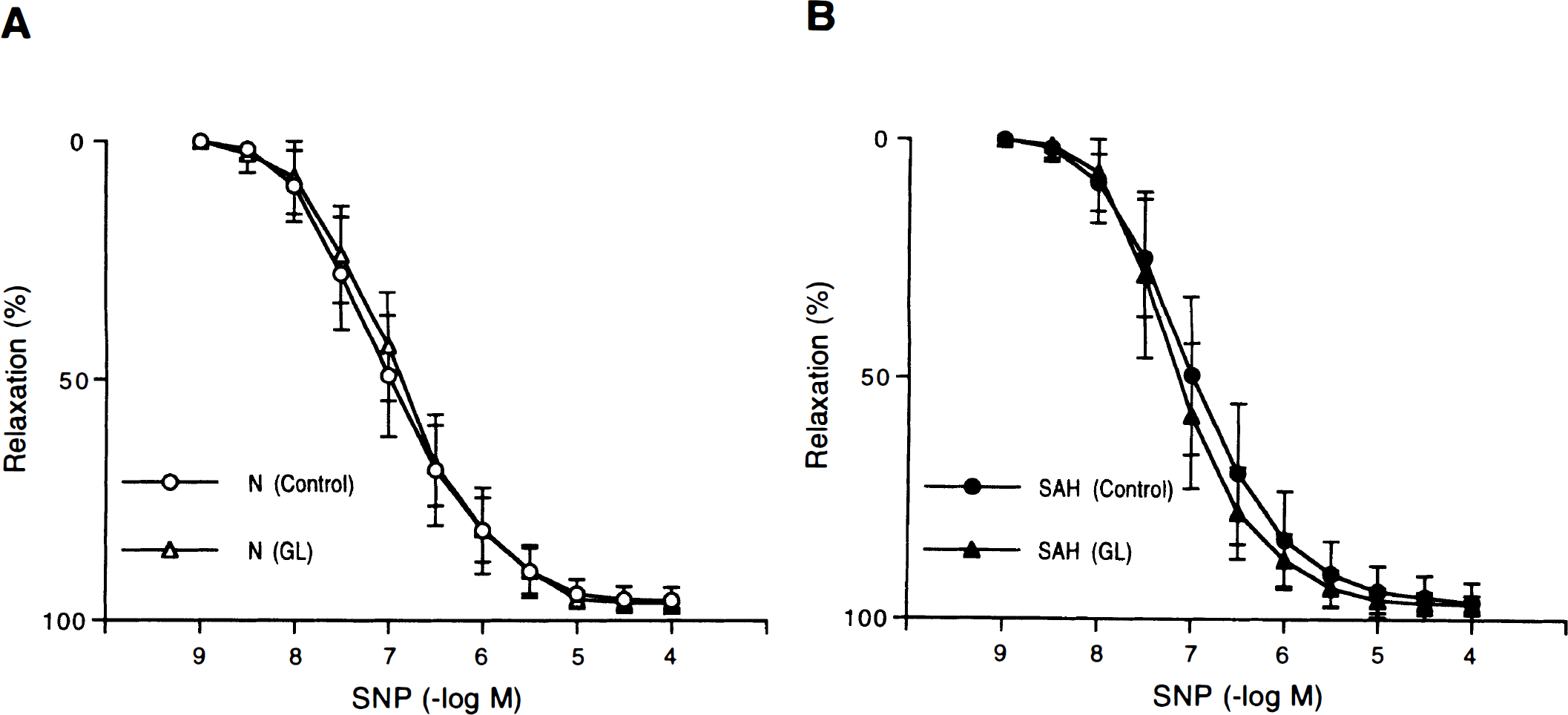
Glyburide (10−5 mol/L) did not affect the relaxations induced by SIN-1 or SNP in both normal and SAH arteries (Figs. 6 and 7).
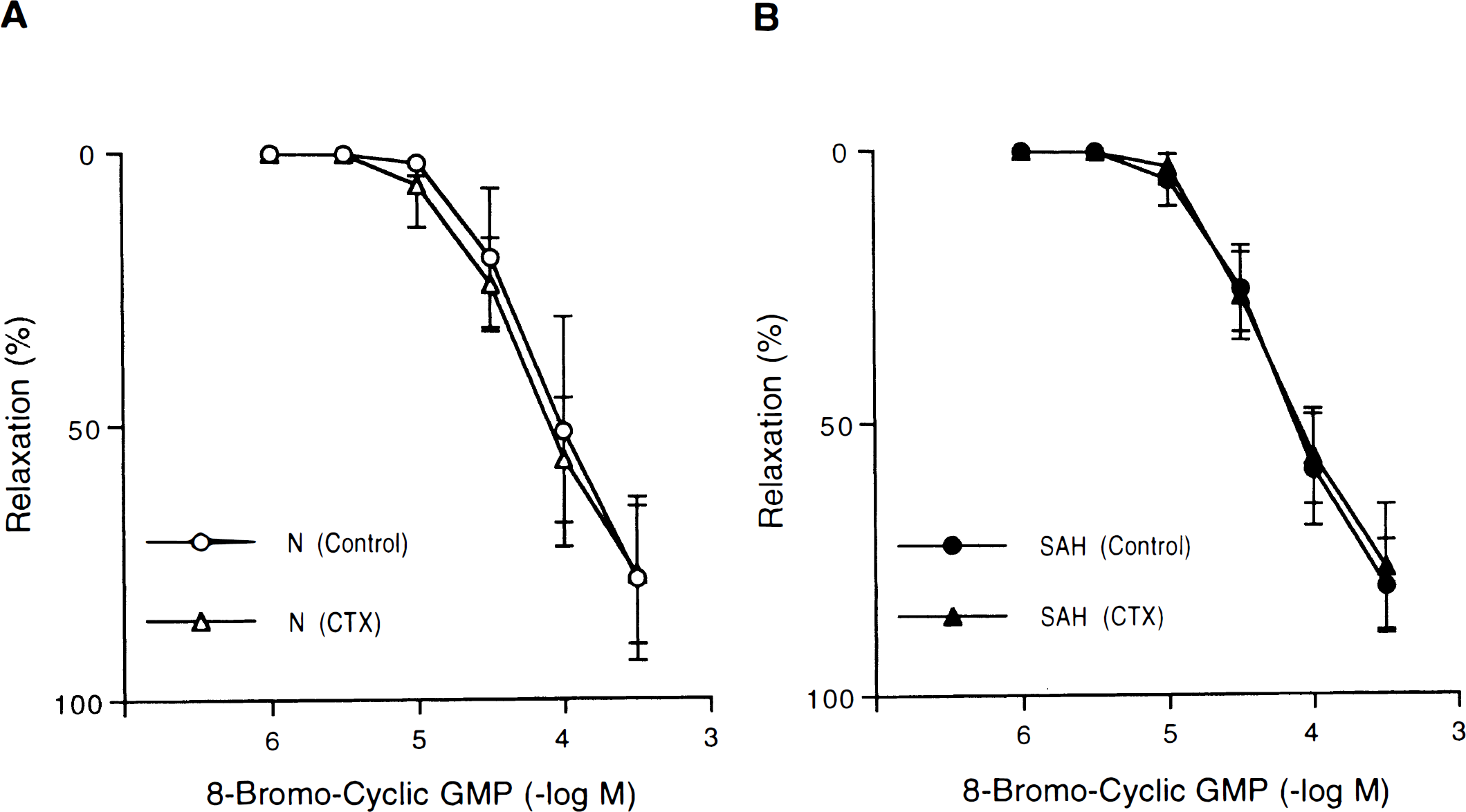
In contrast to nitric oxide donors, the relaxations to 8-bromo-cyclic GMP were not affected by CTX (10−7 mol/L) or 4-aminopyridine (10−3 mol/L) in both normal and SAH arteries (Figs. 8 and 9).
In normal canine basilar artery rings without endothelium, SIN-1 (10−7 − 10−6 mol/L) produced a concentration-dependent increase in levels of cyclic GMP (Fig. 10A). The basal levels of cyclic GMP did not differ between rings obtained from untreated dogs and dogs exposed to SAH; however, production of cyclic GMP stimulated by SIN-1 (10−6 mol/L) was significantly suppressed by SAH (Fig. 10B). Increase in cyclic GMP levels induced by 10−6 mol/L SIN-1 in SAH arteries was equivalent to increase induced by lower concentration (3 × 10−7 mol/L) of SIN-1 in arteries from untreated dogs.
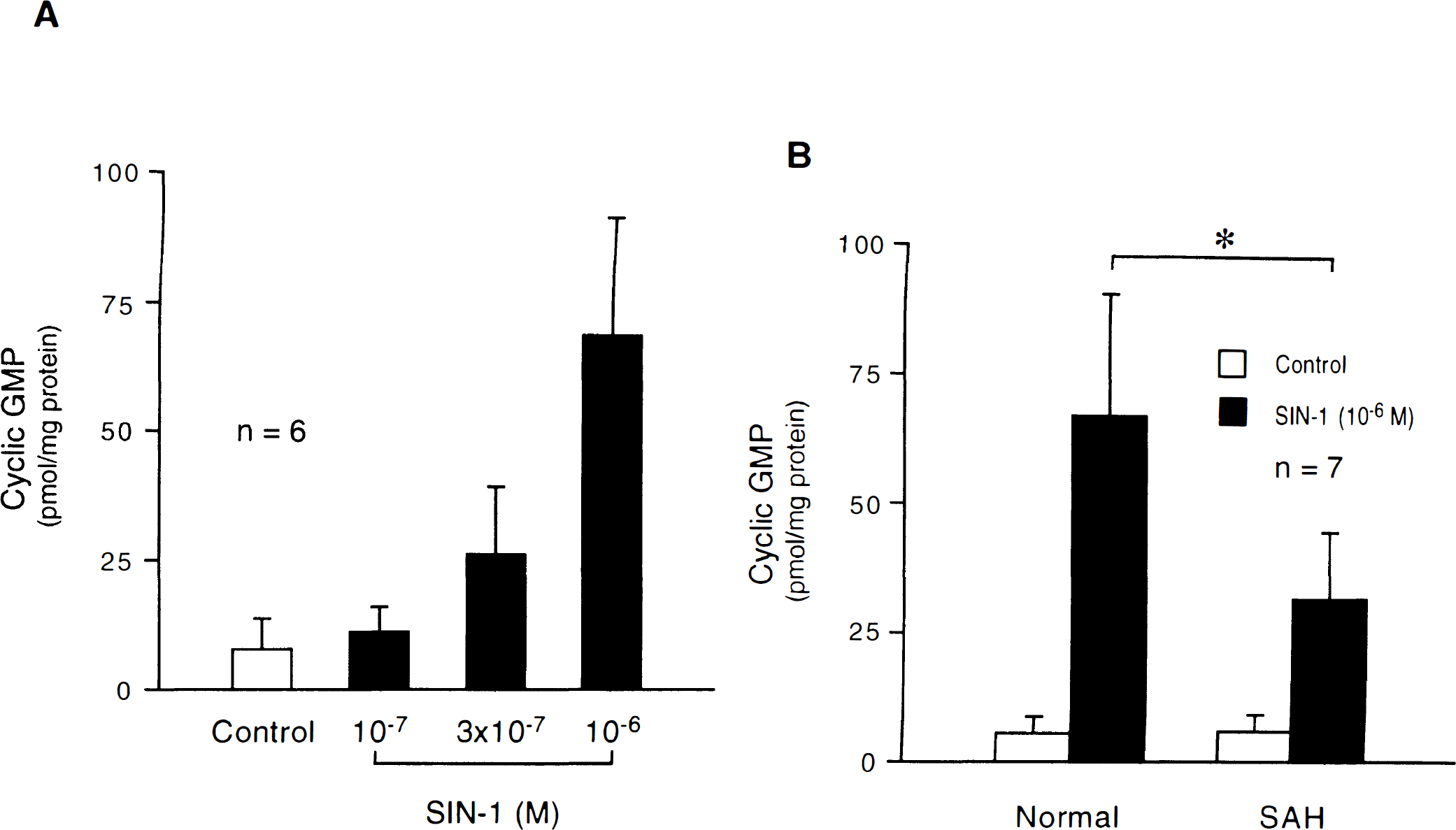
Effect of 3-morpholinosydnonimine (SIN-1) on guanosine 3′,5′-cyclic monophosphate (cyclic GMP) production in canine basilar arteries without endothelium obtained from untreated (normal) dogs (A), and effect of subarachnoid hemorrhage (SAH) on cyclic GMP production (B). Values are expressed as means ± SD. Significantly different (*); P < .05.
DISCUSSION
This is the first study to examine the effect of SAH on K+ channel functions involved in cerebral arterial relaxations to nitrovasodilators. The present results suggest that Ca2+-activated K+ channels may play a compensatory role in mediation of relaxations to nitric oxide in cerebral arteries exposed to SAH. This conclusion is based on the experiments showing that a selective large-conductance Ca2+-activated K+ channel inhibitor, CTX (Miller et al., 1985; Anderson et al., 1988; Gimenez-Gallego et al., 1988) caused a significantly greater reduction of relaxations to SIN-1 or SNP in arteries exposed to SAH as compared with normal arteries.
Recent evidence suggests that several types of K+ channels, including Ca2+-activated K+ channels, ATP-sensitive K+ channels, and voltage-dependent K+ channels are functional in cerebral blood vessels (Masuzawa et al., 1990; Bonnet et al., 1991; Asano et al., 1993; Kitazono et al., 1995). CTX reduced the relaxations to SIN-1 or SNP in normal canine basilar arteries, suggesting that Ca2+-activated K+ channels mediate relaxations to nitric oxide under physiologic conditions. However, our results show that the contribution of Ca2+-activated K+ channels to nitric oxide-induced relaxations is more important in spastic arteries. A similar enhanced role of Ca2+-activated K+ channels has been reported in mediation of endothelium-dependent relaxation in hypercholesterolemic rabbit carotid artery (Najibi et al., 1994), suggesting that Ca2+-activated K+ channels assume a more important role in regulation of vascular reactivity to nitric oxide in diseased blood vessels.
ATP-sensitive K+ channels are thought to play an important role in the control of cerebral arterial tone under pathologic conditions. Dilatation of rabbit cerebral arterioles (Taguchi et al., 1994) and rat middle cerebral arteries (Fredricks et al., 1994) caused by hypoxia is mediated by ATP-sensitive K+ channels, and dilatation of rat basilar artery in response to an ATP-sensitive K+ channel opener is enhanced after experimental SAH (Sobey et al., 1996). However, it is unlikely that ATP-sensitive K+ channels mediate the relaxations to nitric oxide in canine cerebral arteries even after SAH, because a selective ATP-sensitive K+ channel inhibitor, glyburide (Bean, 1992; Edwards and Weston, 1993), did not affect relaxations to SIN-1 or SNP in normal and spastic arteries.
It is well established that intracellular Ca2+ and membrane depolarization increase activity of Ca2+-activated K+ channels and voltage-dependent K+ channels, respectively (Nelson and Quayle, 1995; Kitazono et al., 1995). Previous studies have shown that SAH causes membrane depolarization of cerebral arterial smooth muscle cells (Waters and Harder, 1985; Harder et al., 1987). This, in turn, together with increased level of intracellular Ca2+, may activate Ca2+-activated K+ channels and voltage-dependent K+ channels, thereby increasing their contribution to maintenance of membrane potential and vascular tone of spastic arteries. In the present study, 4-aminopyridine, a voltage-dependent K+ channel inhibitor (Okabe et al., 1987; Robertson and Nelson, 1994; Nelson and Quayle, 1995), reduced the relaxations to nitrovasodilators only in cerebral arteries affected by SAH, suggesting that voltage-dependent K+ channels may also contribute to relaxations induced by nitric oxide under the pathologic condition. However, because 4-aminopyridine may have a nonselective effect, further studies are needed to determine whether voltage-dependent K+ channels participate in mediation of relaxations to nitric oxide after SAH.
Cyclic GMP production stimulated by a nitrovasodilator, SIN-1, was significantly reduced in arteries exposed to SAH. This finding is consistent with previously reported results obtained on arteries from animals with SAH (Kim et al., 1992; Pasqualin et al., 1992; Nakao et al., 1996). The increase in cyclic GMP levels induced by 10−6 mol/L SIN-1 in SAH arteries was significantly reduced and it was comparable to the increase induced by 3 × 10−7 mol/L of SIN-1 in arteries from untreated dogs. In contrast, the relaxations to nitrovasodilators, SIN-1 and SNP, were similar in normal and SAH arteries, suggesting that compensatory mechanisms might have improved relaxations mediated by nitric oxide-cyclic GMP pathway in spastic arteries. Our results suggest that K+ channels may contribute to the maintained normal relaxations to nitrovasodilators in SAH arteries despite decreased cyclic GMP production.
The relaxations to 8-bromo-cyclic GMP, an analogue of cyclic GMP, were not affected by SAH, suggesting that sensitivity in response to cyclic GMP is not altered in cerebral arteries after SAH. This finding is consistent with previous reports from other laboratories (Kim et al., 1992; Sobey et al., 1996). Furthermore, CTX or 4-aminopyridine did not affect the relaxations to exogenously applied 8-bromo-cyclic GMP in both normal and SAH arteries, demonstrating selectivity of these K+ channel inhibitors for the relaxations induced by nitrovasodilators. In contrast, our previous study showed that in normal canine cerebral arteries, the relaxations to zaprinast, which inhibits cyclic GMP phosphodiesterase and increases endogenous cyclic GMP, were sensitive to inhibition of K+ channels with CTX and barium chloride (Onoue and Katusic, 1997). This indicates that exogenous and endogenous cyclic GMP may activate different mechanisms of relaxation. We cannot exclude the possibility that exogenous cyclic GMP does not have access to the same molecular targets as authentic cyclic GMP generated after guanylate cyclase activation or phosphodiesterase inhibition. Therefore, it remains to be determined whether the enhanced K+ channel activation by nitrovasodilators after SAH was dependent (Williams et al., 1988; Robertson et al., 1993; Archer et al., 1994) or independent (Bolotina et al., 1994) on formation of cyclic GMP.
The results of the present study suggest that in canine cerebral arteries with developed vasospasm, Ca2+-activated K+ channels may play a compensatory role in mediation of relaxations to nitric oxide. This may help to explain the mechanisms of relaxations to nitrovasodilators in arteries with impaired production of cyclic GMP.