
Abstract
Ischemic preconditioning is a phenomenon that describes how a sublethal ischemic insult can induce tolerance to subsequent ischemia. This phenomenon has been observed after focal or global ischemia in different animal models. However, the hypothesis that bacterial infection might lead to neuronal tolerance to injury has not been investigated. To mimic cerebral bacterial infection, we injected bacterial lipopolysaccharide (LPS) in the right dorsal hippocampus, followed 24 hours later by an excitotoxic lesion using kainic acid in the mouse model. Quantification of lesion size after cresyl violet counterstaining revealed that LPS pretreatment afforded neuroprotection to CA3 neurons against KA challenge. To investigate the events underlying this protection, we studied the cytokine profile induced after LPS injection. Interleukin (IL)-1β and transforming growth factor beta 1 (TGF-β1) were the main cytokines expressed at 24 hours after LPS injection. Because IL-1β has been described as deleterious in acute injury, we decided to investigate the function of TGF-β1. An adenovirus expressing a constitutively active form of TGF-β1 was injected intracerebrally 1 week before the induction of excitotoxic lesion, and neuronal protection was observed. To confirm the neuroprotective role of TGF-β1, the TGF-β1 adenovirus was replaced by recombinant human TGF-β1 protein and total neuroprotection was observed. Furthermore, the antibody-mediated blocking of TGF-β1 action prevented the protective effect of pretreatment with LPS. We have demonstrated in vivo that the cerebral tolerance phenomenon induced by LPS pretreatment is mediated by TGF-β1 cytokine.
Ischemic preconditioning, a phenomenon describing how a brief episode of sublethal ischemia can induce tolerance to a subsequent ischemia, was first described in the heart (Murry et al., 1986). Using a canine model of myocardial infarction, the authors showed that the size of an infarct could be reduced to 25% when four episodes of 5 minutes of circumflex occlusions were followed by a sustained 40-minute occlusion. The tolerance or preconditioning phenomenon has also been observed in different organs including the brain. Cerebral ischemic preconditioning was first demonstrated in vivo by use of a global ischemia model in gerbils (Kitagawa et al., 1990). When two short sublethal ischemic episodes of 2 minutes were applied more than 24 hours before a longer ischemic insult of 5 minutes, protection of the CA1 hipoccampal neurons was observed, thus showing that a sublethal ischemia may confer ischemic tolerance in the brain. The acquisition of tolerance is a transient phenomenon, which requires at least 24 hours to be induced (Kitagawa et al., 1991). Cerebral ischemic tolerance was observed not only in the gerbil model but also in the C57BL/6J mouse (Wu et al., 2001) and in the rat (Liu et al., 1992; Nishi et al., 1993; Simon et al., 1993) after forebrain or focal cerebral ischemia.
The precise mechanism of cerebral tolerance is still not fully understood. However, many candidates have been suggested as possible inductors, transduction, and effectors of ischemic tolerance. The delay and the observed time window of brain preconditioning may reflect the underlying changes in cellular metabolism and gene expression. The suggested mechanisms of ischemic tolerance are as follows: (1) membrane stabilization and inhibition of excitability by an action on the ATP-sensitive K+ channel or the glucose receptor, therefore reducing the excitability in the hippocampus; (2) cell protection and inhibition of apoptosis by protein phosphorylation, nitric oxide production, cytokines, and glial cell proliferation; and (3) stress response (for review, see Kirino, 2002).
In an attempt to understand the molecular mechanisms of this neuroprotection, several groups have been able to mimic the preconditioning in vivo and in vitro with molecules such as ATP-sensitive K+ channels agonists or NMDA (Kasischke et al., 1996; Riepe et al., 1997), with agonists and antagonists of the adenosine A1 receptor (Heurteaux et al., 1995), electrically via the application of 2M KCl (Kawahara et al., 1995; Kobayashi et al., 1995) and by anoxia/hypoxia in various models (Pohle and Rauca, 1994; Simon et al., 1993), therefore inducing a cross-tolerance (for review, see Kirino, 2002). However, the hypothesis that a bacterial infection might also lead to cerebral tolerance has not been investigated extensively. When injected systematically to mimic peripheral infection, bacterial lipopolysaccharide (LPS) has been shown to induce tolerance to focal brain ischemia (Tasaki et al., 1997) through the production of the cytokines interleukin-1 (IL-1) and tumor necrosis factor-α (TNF-α). We have previously shown that the intracerebral (i.c.) injection of LPS leads to glial cell activation without inducing neuronal death (Andersson et al., 1992) and to local cytokine synthesis at 4 and 24 hours (Cunningham et al., 2002; Walsh et al., 2001). To test the hypothesis that LPS injection and glial activation could be protective against acute neuronal degeneration, we used a model of intracerebral endotoxin injection and investigated whether TGF-β1 might confer neuroprotection. Twenty-four hours after the i.c. injection of LPS, an excitotoxic lesion was induced by the injection of kainic acid (KA). The protective effect of LPS against the acute neurodegeneration was examined by measuring the lesion area. We have previously shown that transforming growth factor (TGF)-β1 is the principal cytokine expressed 24 hours after LPS injection in the brain (Cunningham et al., 2002). TGF-β1 is known to be multifunctional in many pathophysiologic processes, such as cell growth and differentiation (Massague et al., 1992), inflammation (Ruscetti and Palladino, 1991), and cell repair (Sporn et al., 1986). TGF-β1 is minimally expressed in the intact brain, but its expression increases after human stroke (Ata et al., 1997; Krupinski et al., 1996) and experimental ischemic lesions (Lindholm et al., 1992; Logan et al., 1992; Morgan et al., 1993; Wang et al., 1995; Wiessner et al., 1993). To evaluate the role of TGF-β1 in the neuroprotective process, we used an adenovirus expressing the active form of TGF-β1 gene (Sime et al., 1997).
METHODS
Animals
Male and female C57BL/6J mice aged 6 to 8 weeks were anesthetized with Avertin (2,2,2 tribromoethanol in tertiary amyl alcohol) at a dose of 0.1mL/5g body weight and injected stereotactically in the dorsal hippocampus via a pulled glass micropipette (coordinates: bregma, −1.9 mm; lateral, −1.5 mm; depth, −1.37 mm). The experiments were performed under Home Office Licence (UK).
Acute injury
The control groups were injected with a dose of 0.25 nM of KA acid (KA) (n = 6) or 1 μg/μL LPS (n = 3). A further group was injected with a dose of 1 μg/μL LPS, followed 24 hours later with a dose of 0.25 nM KA (n = 6). Another group was injected with a mixture of 1 μg/μL LPS and 0.25nM KA (n = 6). After surgery, the mice were allowed to recover under a warm lamp and then housed in IVC racks (Techniplast UK, Kettering). Five days after the KA injection or LPS alone, the mice were deeply anesthetized and transcardially perfused with fixative, and the brains were removed and embedded in OCT as previously described (Andersson et al., 1991a).
TGF-β1 cytokine
Protein
Recombinant TGF-β1 human (RhTGF-β1) protein (4 ng; R&D Systems Europe Ltd, Abingdon, UK) was injected 1 hour before the KA injection in one group of six mice.
Anti-human TGF-β1 RII antibody
Anti-human TGF-β1 type II receptor (αTGFβ1-RII) (500 ng; R&D Systems Europe Ltd, Abingdon, UK) was injected 24 hours after the injection of 1 μg/μL LPS in one group of eight mice. Another group of six mice were injected 24 hours after the LPS injection with 250 ng αTGFβ1-RII, followed immediately by the KA injection.
Quantification of the lesion
Sections measuring 10 μm were cut on a cryostat, and sections were collected every 50 μm throughout the dorsal hippocampus. The sections were stained with a Nissl stain to reveal the area of neuronal degeneration. The anterior limit of the lesion was defined as the first section presenting a minimum of 10 pyknotic neurons, probably undergoing apoptosis, in one microscopic field. Similarly, the caudal limit of the lesion was defined as the last section presenting a maximum of 10 pyknotic neurons within a field. The edges of the lesions were determined by the border between the healthy neurons and the pyknotic neurons (Fig. 2B). The extent of the neuronal loss was quantified by serial reconstruction of the dorsal hippocampus pyramidal fields with LEICA Qwin image analysis software (Leica Imaging Systems Ltd., Cambridge, UK). The lesion was always restricted to the CA3 region of the hippocampus. The area of neuronal loss between two sections was calculated according to the following mathematical formula: area = (a +b)×d ÷ 2, as illustrated in Fig. 1. The total area of neuronal loss of entire right dorsal hippocampus is the result of the sum of the lesion area calculated as described above.
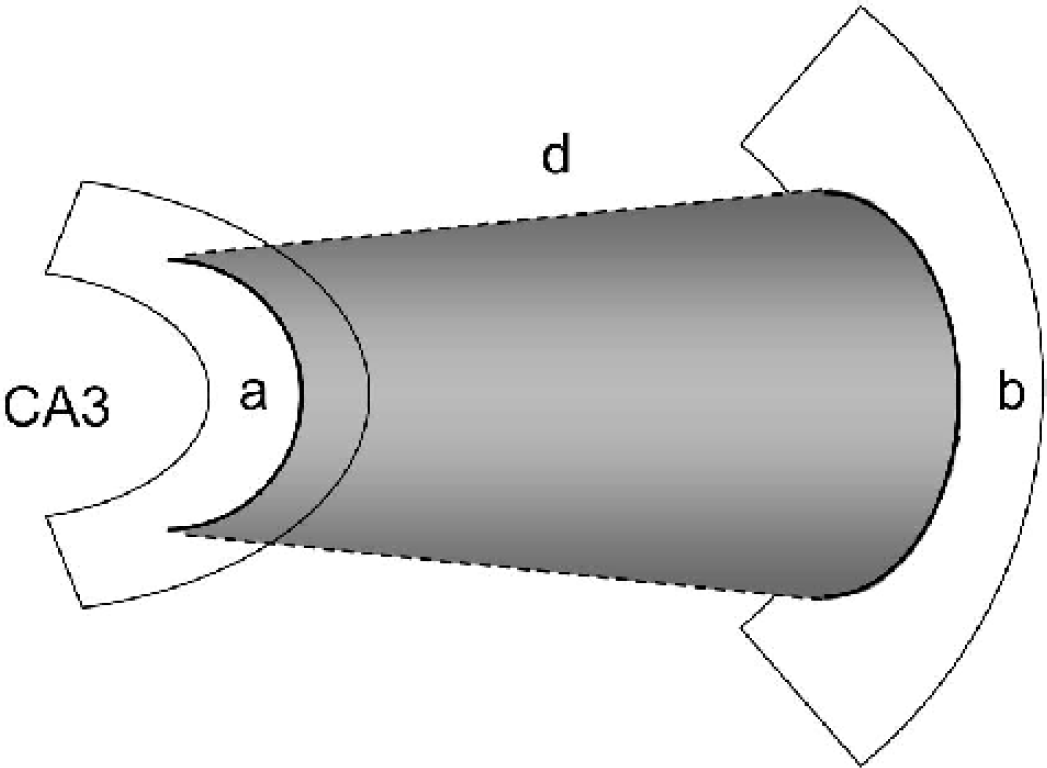
Representative diagram of the lesion measurement. The mathematical formula used to calculate the lesion between two sections is as follows: (a +b)×d ÷2, where d represents the distance between two sections (50 μm), and a and b represent the length of the lesion of two successive sections.
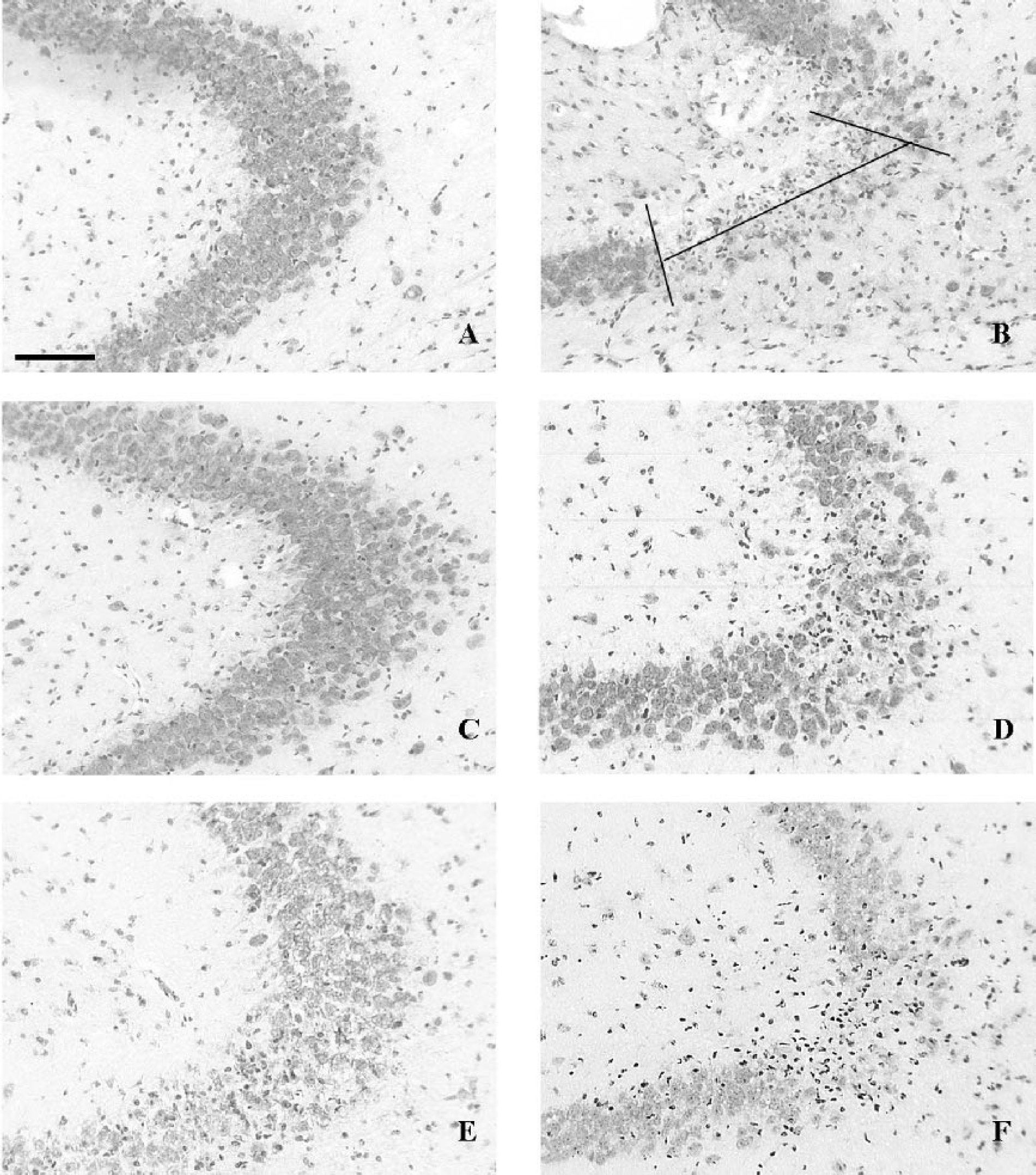
Detection of the neuronal injury by Cresyl Violet counterstaining of coronal sections in the region of the CA3 neurons. Animals were injected with
Immunohistochemistry
Frozen 10-μm coronal serial sections were cut on a cryostat and mounted on 3-aminopropyltriethoxysilane-coated slides. The sections were air-dried and fixed in ice-cold absolute ethanol for 10 minutes before staining. The primary antibodies, anti-CD68 mAb (FA-11) and anti-CD3 mAb (KT3) (Serotec, Oxford, UK), were incubated on the sections for 2 hours and detected using a biotinylated secondary antibody and the ABC Elite detection kit from Vector laboratories (Peterborough, UK) with 3,3 diaminobenzidine (DAB) as previously described (Walsh et al., 2001). Sections used for photographic purposes were counterstained with cresyl violet.
Determination of IL-1β, IL-10, TNF-α and TGF-β1 in brain homogenates by ELISA
The concentrations of IL-1β, IL-10, TNF-α and TGF-β1 in brain tissue were determined by ELISA. Animals were transcardially perfused with heparinized saline. Brains were removed and a thick (2mm) coronal section taken through the hippocampus. The hippocampus was then punched from this section, snap frozen in liquid nitrogen and homogenized using disposable pestle and mortars in phosphate buffered saline (PBS) containing the protease inhibitor cocktail Complete. Homogenates (0.1 g/mL) were centrifuged at 10,000g for 5 minutes at 4°C, and the supernatants were assayed according to the manufacturers instructions for IL-1β, IL-10, and TNF-α ELISA kits (Quantikine). All standards and samples were diluted in the extraction buffer and assayed in duplicate. For the determination of TGF-β1, homogenates were subjected to a pretreatment for the conversion of latent TGF-β1 to active TGF-β1. The conversion was effected by acidification with 2.5 M acetic acid and 10 M urea for 10 minutes, followed by neutralization with 2.7 M NaOH and 1 M HEPES and a final 1:25 dilution in wash buffer. After centrifugation, the samples were added to plates coated overnight with recombinant human TGF-β sRII/Fc chimera. After 3 hours incubation, the plates were washed, and biotinylated polyclonal anti-TGF-β1 antibody at 200 ng/mL was added. Bound TGF-β1 was detected as described previously (Cunningham et al., 2002).
To determine the in vivo time course of TGF-β1 expression by the adenovirus expressing the active form of TGF-β1 (AdTGFβ1223/225), six naive mice were injected in the right dorsal hippocampus at the concentration of 105 pfu/μL. Two animals were killed at each of 3 time points (3, 7 and 10 days), and the TGF-β1 was determined as described above. The limits of reliable quantitation of the assays for IL-10, TNF-α and TGF-β1 were less than 5 pg/mL, 4 pg/mL, and 15 pg/mL, respectively.
Statistics
The data are presented as the mean ± standard deviation of the mean. Where statistical analysis has been used, ANOVA test with Fischer's post hoc test was applied.
Reagents
Adenovirus
The construction of the adenoviral vector has been described previously in detail (Bett et al., 1994). We have used the AdTGFβ1223/225 adenovirus, expressing the active form of TGF-β1 and the AdDL70-3, the control virus with no exogenous cDNA insert. They were used at the concentration of 105 pfu/μL. The production of TGF-β1 by the AdTGFβ1223/225 construct in the brain was confirmed by ELISA.
LPS
LPS (Equine abortus) was from Sigma Chemicals (Poole, Dorset).
ELISA
IL-1β and TNF-α ELISA kits (Quantikine), recombinant human TGF-β sRII/Fc chimera, recombinant TGF-β1, and biotinylated polyclonal anti TGF-β1 antibodies used for the TGF-β1 ELISA were obtained from R&D Systems Europe Ltd. (Abingdon, UK).
RESULTS
LPS injection
An intracerebral injection of 1 μg/μL LPS was made into the right dorsal hippocampus of five C57BL/6J mice. The animals were killed 5 days later. To identify and quantify the lesion, the sections were counterstained with Cresyl Violet. Examination of the brain sections revealed no neuronal death (Fig. 2A).
Kainic acid injection
KA acid (0.25 nM) was injected into the right dorsal hippocampus of six mice. The animals were killed 5 days later. A clear lesion was evident with a specific neuronal loss in CA3 as previously reported (Andersson et al., 1991b). The lesion is illustrated in Fig. 2B. The extent of neuronal loss was quantified by serial reconstruction of the lesion in the dorsal hippocampus pyramidal fields (Fig. 3).
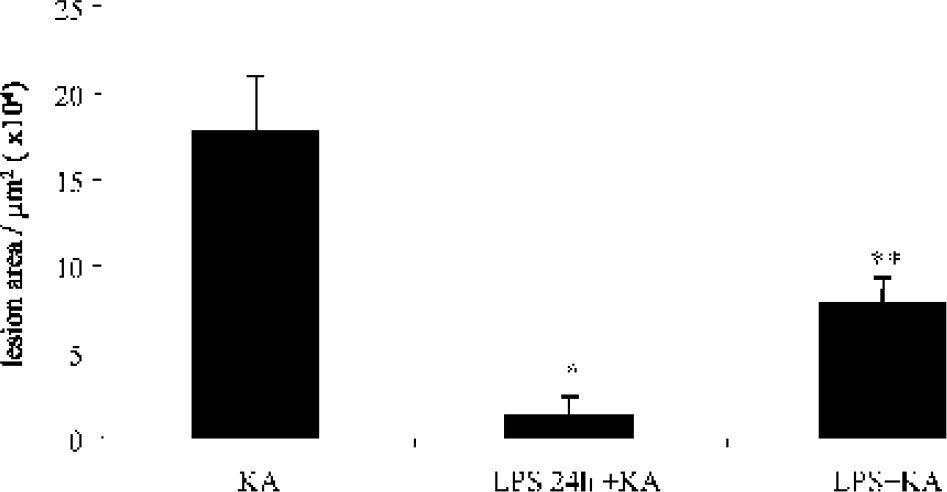
Quantification of the size of the lesion in animals injected with KA (n = 6), LPS followed 24 hours by KA (n = 6), and a mix of LPS and KA (n = 6) after Cresyl Violet counterstaining. ∗ P < 0.0001 for LPS injection followed 24 hours later by the KA versus unique KA injection by Fisher's test. ∗∗ P = 0.0017 for LPS and KA injected together versus unique KA injection by Fisher's test.
LPS/Kainic acid
An intracerebral injection of 1 μg/μL LPS in the right dorsal hippocampus was followed 24 hours later by KA acid injection. The KA injection was made in exactly in the same site as the LPS injection, using the same stereotaxic coordinates. Six mice were injected. The mice were killed 5 days later, and the lesions were quantified. A restricted area of neuronal death was present in the CA3 field of one animal, but in the other five animals, no lesion was visible (Fig. 1C). The difference between the two animal groups is significant (P < 0.0001) (Fig. 3).
Six mice were injected with a mixture containing 1 μg/μL LPS and 0.25 nM KA. The injection was made into the right dorsal hippocampus. The animals were killed 5 days after this injection. The combination of both LPS and KA acid induces a lesion that was smaller (7.73 × 104 ± 1.48 μm2) than that seen with KA acid injection alone (17.72 × 104 ± 3.18 μm2). The difference in lesion area between the injection of LPS and KA and the KA acid injection alone is significant (P = 0.0017). However, if we compare the lesion size following injection of the mixture of LPS and KA with the lesion following the injection of the LPS and KA with a delay of 24 hours, the lesion as a consequence of the single mixture injection of LPS and KA is seven fold bigger than the lesion resulting of the injection of LPS followed 24 hours later by KA. This difference between the two protocols is significant (P = 0.0299) (Fig. 3). There is clearly a difference in the neuroprotection in the group that received LPS 24 hours before the KA injection and the group that received the mixture of LPS and KA.
LPS-evoked cytokine profile
Because LPS appears to be protective, we were interested to know which cytokines were expressed in LPS-injected brains. The profile of cytokines in brains injected with LPS was determined by ELISA kit detection. The brains were injected in the right dorsal hippocampus with 2.5μg/μL LPS. The injected sites were dissected and prepared for cytokine detection by ELISA 24 hours after the LPS injection. Four cytokines were studied: two proinflammatory cytokines, IL-1β and TNF-α, and two antiinflammatory cytokines, TGF-β1 and IL-10. The results are illustrated in Fig. 4. All cytokines show significant differences between LPS and saline intra-cerebral injections (IL-1βP = 0.001; TNF-αP = 0.0001; TGF-β1, P = 0.0016; IL-10, P = 0.0010).
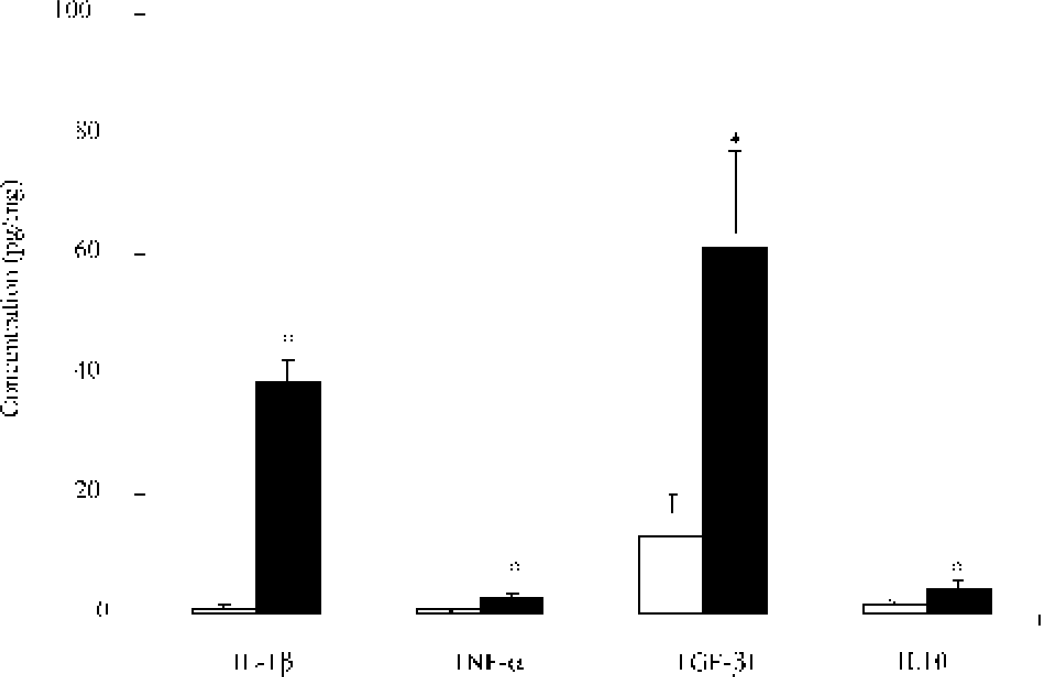
Quantification of IL-1β, TNF-α, IL-10, and TGF-β1 protein in animals injected with saline (n = 3) and LPS (n = 3) and killed 24 hours later by ELISA. ∗ P < 0.0001 for saline versus LPS injection by Fisher's test. Open box, saline-injected animals; Closed box, LPS-injected animals.
Adenovirus TGF-β1
The most likely cytokine to be protective was TGF-β1 (Henrich-Noack et al., 1996; Prehn et al., 1993a; Prehn et al., 1993b). We therefore examined whether the delivery of TGF-β1 was neuroprotective either when delivered as the cytokine alone or with an adenoviral vector system. The determination of the TGF-β1 expression by the AdTGF-β1223/225 adenovirus was performed after injection of the vector in the right dorsal hippocampus at the concentration of 105 pfu/μL. At 7 days postinoculation, a peak of TGF-β1 expression was observed by ELISA (564 pg/mg) (data not shown). At this time, the microglia stained by a CD68 marker shows a very weak activation of microglia without lymphocyte recruitment as observed by the CD3 staining (data not shown).
Seven days after the adenovirus injection, during the peak of cytokine expression, KA was injected in the same site in the dorsal hippocampus, and the animals were killed 5 days later. For the control animals, the AdTGFβ1223/225 was replaced by AdDL70-3. The brains from mice injected with AdDL70-3 followed 7 days later by the KA showed clear lesions in the CA3 area (9.37 × 104 ± 1.33 μm2). These mice showed a significant difference in lesion size compared with the animals injected with KA alone (17.72 × 104 ± 3.18 μm2) (Fig. 5) and with the animals injected with LPS followed 24 hours later by KA (7.73 × 104 ± 1.48 μm2) (P = 0.0088).
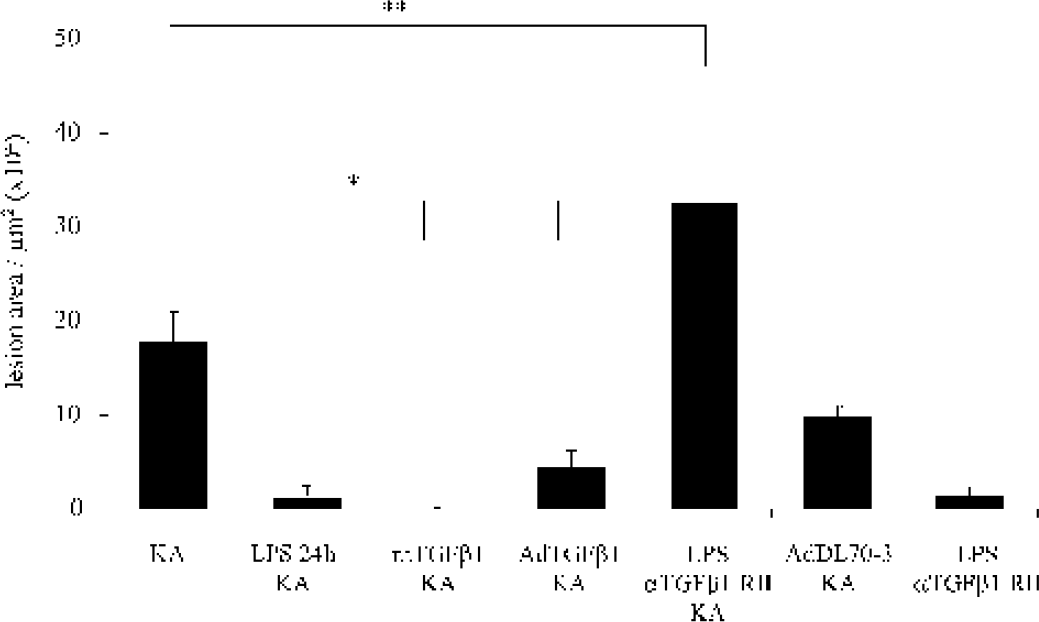
Quantification of the size of the lesion in animals injected with KA (n = 6), with LPS followed 24 hours by KA (n = 6), with a recombinant human TGFβ1 protein followed 1 hour later by KA (n = 5), with AdTGFβ1223/225 adenovirus followed 7 days later by KA (n = 7), with TGF-β1 RII antibody followed by KA and 24 hours after the LPS (n = 6), with Ad70-3 control adenovirus (n = 5) followed 7 days later by KA, and with TGF-β1 RII antibody 24 hours after the LPS (n = 8). ∗ P < 0.0001 for KA versus LPS 24 hours and KA, rhTGF-β1 and KA, and AdTGFβ1223/225 and KA. ∗∗ P = 0.048 for KA versus LPS and αTGFβ1-RII and KA.
The group with the AdTGFβ1223/225 shows a total neuroprotection for three mice and a clear reduction in lesion size for the other four animals (Fig. 2D). The mean size of the lesion in the seven mice is 4.35 × 104 ± 1.86 μm2. The difference in the size of the lesion between the animals with KA acid injection and the animals that have received AdTGFβ1223/225 adenovirus 7 days before the injury is significant (P < 0.0001), as shown in Fig. 5.
TGF-β1 cytokine
To further investigate the role of TGF-β1, two other experiments were conducted. Five animals were injected with 4 ng of human TGF-β1 recombinant protein in the right dorsal hippocampus 1 hour before KA acid injection. In four mice, there was no neuronal loss, and in one mouse there was a small lesion. However, the structure of the CA3 neurons in the animals with the absence of obvious lesion shows a disruption between the neuronal cells in the injected side by comparison with the noninjected side (Fig. 2E), but there were no apoptotic neurons. This group shows a significant difference of the size of the lesion in comparison to the five other groups (P < 0.0001) (Fig. 5). Additional experiments were performed to block the TGF-β1 neuroprotection by using a soluble TGF-β1 receptor II antibody (αTGFβ1-RII) as an antagonist. Six mice were injected 24 hours after the LPS injection with 250 ng of the αTGFβ1-RII, followed immediately by the 0.25 nM of KA. The animals were killed 5 days later. As a control, we injected eight mice with 500 ng of the αTGFβ1-RII 24 hours after the LPS injection. αTGFβ1-RII that was injected 24 hours after the LPS induced a small lesion (1.70 × 104 ± 0.9 μm2) (Fig. 5). The lesion induced by the injection of αTGFβ1-RII and KA 24 hours after the LPS pretreatment is greater than the lesion observed with KA alone (32.44 × 104 ± 13.19 μm2 versus 17.72 × 104 ± 3.18 μm2) (P = 0.047) (Fig. 5), and it is observed in the CA3 or CA1 area. This lesion is also significantly different to the lesion induced by KA after LPS activation (P < 0.0001).
DISCUSSION
The tolerance phenomenon and neuronal sparing induced by a sublethal ischemic lesion have been demonstrated in vivo by Kitagawa et al. (1990). However, the mechanisms leading to neuronal protection against an excitotoxic lesion have not been elucidated. The majority of animal models investigating tolerance concern sequential ischemic insults: (1) global-global ischemia, (2) focal-global ischemia, and (3) global-focal ischemia. No investigation on the effects of cerebral bacterial infection followed by a subsequent insult has been carried out. The mechanisms of ischemic tolerance or tolerance to excitotoxicity and the resulting preservation of neurons is likely to be important in a variety of neuropathologic diseases.
In this study, to mimic a bacterial infection, we injected the proinflammatory agent LPS into the dorsal hippocampus of mice. We observe that LPS affords protection of approximately 93% of CA3 neurons killed by this particular dose of KA. LPS gives the maximum neuroprotection when it is injected 24 hours before KA. The observed delay is necessary to obtain the protection; it has been described in the different models (Barone et al., 1998; Kitagawa et al., 1991; Perez-Pinzon et al., 1997a, 1997b) and is correlated with protein synthesis. To investigate the possible role of cytokines after the endotoxin injection, we have examined the cytokine profile in the hippocampus 24 hours after i.c. LPS injection. In this study, we have examined two proinflammatory cytokines, IL-1β and TNF-α, and two antiinflammatory cytokines, TGF-β1 and IL-10, because the balance between pro- and antiinflammatory cytokines in the brain may have a profound impact on the preservation of neuronal elements. We have shown that the levels of both IL-1β and TGF-β1 are significantly increased 24 hours after i.c. LPS injection, and this is associated with a protection of CA3 neurons.
To characterize the role of TGF-β1 in neuroprotection, we have used an adenovirus expressing the active form of TGF-β1 (Sime et al., 1997). The AdTGFβ1223/225 adenovirus does not induce an acute inflammation 7 days after the intracerebral injection; no leukocyte recruitment was detected, and only weak microglia activation was observed. The excitotoxic lesion produced by injection of KA 7 days after the AdTGF-β1223/225 adenovirus injection induces a neuronal death that is significantly smaller. To confirm that this neuroprotection can be attributed to TGF-β1, we have injected recombinant human TGF-β1 protein 1 hour before the KA injection. A total neuroprotection against excitotoxic lesion was observed. It is of interest that the control adenovirus AdDL70-3 also afforded small protection. An undetectable inflammation may occur after the i.c. AdDL70-3 injection and which may, in turn, induce protection. We have used a human TGF-β1 RII antibody as an antagonist of TGF-β1. The i.c. injection of TGF-β1 RII antibody 24 hours after the LPS injection and followed by the KA injection has produced a lesion that is twofold greater than that induced by KA alone. It is of interest to note that the observed lesion included both CA3 and CA1 subfields. It is possible that the blockage of endogenous TGF-β1 production can in some way sensitize CA1 neurons to the very low concentrations of KA used in this study. The blockage of TGF-β1 pathway inhibits the protection observed after the LPS injection. It is also of interest to note that the injection of the TGF-β1 RII antibody alone 24 hours after the LPS injection induces a small lesion. These two results confirm the neuroprotective role of TGF-β1 during an excitotoxic injury.
The expression of IL-1β, IL-10, and TNF-α is known to be rapidly inducible by experimental and clinical insults to the central nervous system (CNS), and it is clear that IL-1β and TNF-α exacerbate neurodegeneration or impaired neuronal function (Allan and Rothwell, 2001; Rothwell, 1999). In rodents, IL1-β mRNA cerebral expression is increased within 15 to 30 minutes, and protein is increased within 1 hour, of experimental cerebral ischemia (stroke), brain injury, or infusions of excitotoxins (Minami et al., 1992; Yabuuchi et al., 1994), all of which lead to neuronal death. Injection of recombinant IL-1β protein into the brain parenchyma of the rat exacerbates neuronal damage induced by cerebral ischemia, traumatic injuries, or excitotoxins (Lawrence et al., 1998; Loddick and Rothwell, 1996). TNF-α, like IL-1β, is also known to exacerbate neuronal death when delivered after cerebral ischemia (Barone et al., 1997) or traumatic brain injury (Arvin et al., 1996). In the current study, we also detected a small quantity of the antiinflammatory cytokine IL-10, which might participate in the neuroprotective effects. A drastic reduction of the infarct size 24 hours after middle cerebral artery occlusion has been reported after central administration of this cytokine (Spera et al., 1998), and we cannot exclude the participation of IL-10 in the neuroprotection observed after delivery of LPS. IL-10 might explain the neuroprotection observed by the injection of the mixture of LPS and KA before the TGF-β1 effect.
In contrast to the effect of IL-1β, TGF-β1 has been described as acting as a neuroprotectant in in vitro models of cerebral ischemia. Prehn et al. describe the neuroprotective effect of TGF-β1 on rat cortical neurons against neuronal degeneration caused by hypoxia (Prehn et al., 1993a). The same group observed neuroprotection mediated by an i.c. injection of TGF-β1 protein in vivo after transient forebrain ischemia (Henrich-Noack et al., 1996; Prehn et al., 1993b), although the observed neuroprotection was less significant than in the present study. TGF-β1 has also been shown to reduce infarct size in mice (Prehn et al., 1993b) and rabbits (Gross et al., 1993) subjected to focal cerebral ischemia. The use of the extracellular domain of the TGF-β1 type II receptor as a soluble TGF-β1 antagonist in a model of excitotoxic or ischemic reverses the neuroprotective effect of TGF-β1 (Ruocco et al., 1999).
TGF-β1 is likely to antagonize the effects of IL-1β through interactions with its transduction pathway. TGF-β1 exerts its biologic effects through a heteromeric complex of two receptor subunits, TGFβRI and TGFβRII. This complex in turn transduces the signal through activation of the SMAD family (Massague, 1998). However, TGF-β1 also uses other signal transduction pathways including MAPK (Allan and Rothwell, 2001), a pathway shared with IL-1β signaling. In vitro, IL-1β has been shown to be a potent inducer of TGF-β1 synthesis and release in vascular smooth muscle cells (da Cunha and Vitkovic, 1992; Yue et al., 1994). Furthermore, TGF-β1 has been shown to directly downregulate proinflammatory cytokine production such as IL-1β and TNF-α (Bogdan et al., 1992; Fadok et al., 1998); thus its presence before the excitotoxic insult may ensure a less aggressive acute inflammatory response.
TGF-β1 is also known to play a role in the regulation of the plasmin cascade through its upregulation of the serine protease inhibitor PAI-I (plasminogen activator inhibitor). PAI-1 has been shown in vitro in astrocyte culture to be overexpressed after TGF-β1 treatment (Buisson et al., 1998) and to mediate TGF-β1 associated neuroprotection. Indeed, the PAI-I inhibits tissue type plasminogen activator (t-PA), a serine protease that is involved in neuronal plasticity and cell death induced by excitotoxin. Mice deficient for t-PA are resistant to neuronal loss after the intrahippocampal injection of excitotoxins (Tsirka et al., 1997) or ischemic insult (Wang et al., 1998). TGF-β1 inhibits t-PA activity through expression of the inhibitor PAI-1 and thus prevents the degradation of the extracellular matrix (Docagne et al., 1999). In vitro, TGF-β1 can also acutely stabilize neuronal Ca2+ homeostasis under conditions of pathophysiologic Ca2+ overload, such as ischemia, and also produces a rapid neuronal induction of the survival gene Bcl2 (Prehn et al., 1994) that might suppress the detrimental effects of activated microglia on neighboring cells.
In conclusion, we have demonstrated that bacterial infection, mimicked by i.c. LPS injection, protects hippocampal neurons against an excitotoxic insult. This neuroprotection is strongly associated with the elevation of the anti-inflammatory TGF-β1. The in vivo injection of TGFβ1 protein or of an adenovirus expressing the active form of TGF-β1 afforded complete neuroprotection, whereas the blockage of TGF-β1 pathway by the injection of TGF-β1 RII antibody results in a dramatic enhancement of the area of the lesion. TGF-β1 is demonstrated in vivo as being an important neuronal survival factor. This protection may be through its downregulation of proinflammatory genes such as IL-1β and TNF-α or by its activation of PAI-1 and consequent inhibition of the plasmin cascade. TGF-β1 has been detected in necrotic human brain lesions (Ata et al., 1997) and also after ischemic stroke in human (Krupinski et al., 1996). These data suggest that TGF-β1 may play a role in antiinflammatory processes and in tissue remodeling after ischemic brain lesion and may be used for therapeutic purposes.