
Abstract
Apolipoprotein E (apoE, protein; APOE, gene) is the major lipid-transport protein in the brain and plays an important role in modulating the outcome and regenerative processes after acute brain injury. The aim of the present study was to determine if gene transfer of the ε3 form of APOE improves outcome in a murine model of transient focal cerebral ischaemia. Mice received an intrastriatal injection of vehicle, a second-generation adenoviral vector containing the green fluorescent protein gene (Ad-GFP) or a vector containing the APOE ε3 gene (Ad-APOE) 3 days before 60 mins focal ischaemia. Green fluorescent protein expression was observed in cells throughout the striatum and subcortical white matter indicating successful gene transfer and expression. ApoE levels in the brain were significantly increased after Ad-APOE compared with Ad-GFP or vehicle treatment. Ad-APOE treatment reduced the volume of ischaemic damage by 50% compared with Ad-GFP or vehicle treatment (13±3 versus 29±4 versus 27±5 mm3). The extent of postischaemic apoE immunoreactivity was enhanced in Ad-APOE compared with Ad-GFP or vehicle treated mice. These results show the ability of APOE gene transfer to markedly improve outcome after cerebral ischaemia and suggest that modulating apoE levels may be a potential strategy in human stroke therapy.
Introduction
Apolipoprotein E (apoE denotes protein; APOE denotes gene) is a 34 kDa glycoprotein that plays a critical role in the response to acute brain injury and in neurodegeneration. In humans, three isoforms of apoE are expressed: apoE2, E3, and E4, encoded by the corresponding alleles APOE ε2, ε3, and ε4. The APOE ε4 allele is the major genetic risk factor for Alzheimer's disease (Corder et al, 1993) and is associated with poorer outcome after traumatic brain injury (Teasdale et al, 1997) and intracranial haemorrhage (Alberts et al, 1995).
ApoE is markedly upregulated after experimental brain injury, and after stroke and global ischaemia (because of cardio-respiratory arrest) in humans (Horsburgh and Nicoll, 1996; Horsburgh et al, 1999, 2000a; Aoki et al, 2003). In addition, poorer pathological and neurological outcomes were evident in APOE deficient mice compared with wild-type littermates after focal and global ischaemia (Laskowitz et al, 1997; Sheng et al, 1999). These findings suggest that the availability of apoE is a key factor in protecting the brain after ischaemic brain injury. This is supported by reports showing neuroprotection after restoration of apoE levels in APOE deficient mice by administration of exogenous apoE (Masliah et al, 1997; Horsburgh et al, 2000b) or by intrathecal delivery of an apoE-derived peptide in rat pups after perinatal hypoxic—ischaemic injury (McAdoo et al, 2005). Collectively, these data indicate that augmenting endogenous apoE levels in the brain could improve outcome after cerebral ischaemia.
Aside from direct administration of apoE, another approach is to indirectly modulate apoE levels by viral vector-mediated gene transfer of APOE. Advantages of a gene delivery strategy include the potential for prolonged protein production and the ability to induce local tissue alterations in protein levels. Viral vector-mediated transfer of genes encoding various types of protein (e.g., neurotrophic factors, anti-apoptotic proteins, heat shock proteins, anti-inflammatory mediators) has been shown previously to have neuroprotective effects in models of cerebral ischaemia (Ooboshi et al, 2003). The efficacy of APOE gene transfer has not been investigated to date.
Adenoviral vectors are particularly suitable for gene transfer to the brain on account of their ability to transduce an extensive range of cell types (dividing and nondividing) (Glover et al, 2002). Adenoviral vector-mediated gene transfer of glial cell line-derived neurotrophic factor or interleukin-1 receptor antagonist reduces the extent of ischaemic damage after focal cerebral ischaemia (Betz et al, 1995; Kitagawa et al, 1999). These studies utilised conventional first-generation adenoviral vectors (E1 gene alone or E1 and E3 viral genes deleted), which are prone to ‘leaky’ expression of remaining viral genes and can trigger inflammatory and immune responses that can substantially diminish transgene expression (Wood et al, 1996). Second generation adenoviral vectors have been constructed, which have additional viral genes deleted (Amalfitano et al, 1998). These vectors are less immunogenic and promote more stable and prolonged transgene expression in comparison to conventional vectors (Thomas et al, 2000).
In the present study, we used a second generation adenoviral vector (viral E1, E3, and polymerase (E2b) genes deleted) containing the human APOE ε3 gene or green fluorescent protein gene (GFP) (Harris et al, 2002) to determine the effects of human APOE gene transfer to the brain on the outcome to transient focal ischaemia in mice. An APOE ε3-containing vector was used since studies in animal models of acute brain injury have shown a superior outcome in transgenic mice expressing human APOE ε3 compared with APOE ε4 (Horsburgh et al, 2000c). Mice express only one form of apoE that is structurally similar to human apoE4 but functionally related to human apoE3 (Weisgraber 1994; Strittmatter and Bova 2002). Therefore, vector-derived alterations in apoE levels will be of the apoE3 isoform. This Ad-APOE vector was previously administered by intramuscular or intravenous injection (to target the liver) where it was shown to promote regression of atherosclerotic pathology in susceptible APOE-deficient mice (Harris et al, 2002). In the present study, we show that adenoviral-mediated gene transfer of APOE ε3 to the brain (intrastriatal administration) markedly improves outcome after transient focal cerebral ischaemia.
Materials and methods
All experiments were performed in adult male C57Bl/6J mice (Charles River, UK) under an appropriate Home Office Licence and adhered to regulations as specified in the Animals (Scientific Procedures) Act (1986). Procedures involving adenoviral vectors were approved by the Health and Safety Executive and had local approval from the Advisory Committee on genetically modified work. All experiments were performed masked to treatment.
Adenoviral Vector
Construction of a second generation adenoviral vector containing the APOE ε3 gene (Ad-APOE) or the GFP gene (Ad-GFP) was described previously (Harris et al, 2002). Briefly, shuttle plasmid vectors containing the CMV promoter driving expression of either the apoE3 or eGFP cDNAs were linearised with PmeI and co-transformed with pAdEasy-Δpol (Amalfitano et al, 1998) into the BJ5183 strain Escherichia coli. Positive recombinants were linearised with PacI and transfected into C7 cells (Amalfitano and Chamberlain, 1997). Recombinant Ad-CMV/apoE and Ad-CMV/GFP vectors were isolated from individual plaques of transfected C7 cells, plaque purified once and grown to high titre stock (∼2 × 1012 vp/ml), purified by two rounds of CsCl banding, desalted using an Econo-Pac 10DG column (BioRad). The purified viruses were stored in 10 mmol/L Tris-HCL (pH 8), 2 mmol/L MgCl2 and 10% glycerol, and stored at −80°C.
Intrastriatal Gene Transfer
Mice were placed in a Perspex chamber and anaesthesia was induced with 3% halothane in a mixture of 30% oxygen and 70% nitrous oxide. Mice were then positioned on a stereotaxic frame and mechanically ventilated. Anaesthesia was maintained with approximately 2% halothane in the same mixture of oxygen and nitrous oxide for the duration of surgery. A skin incision was made above the parietal skull and a burr hole was drilled through the bone overlying the parietal cortex. Stereotaxic infusions (0.5 μL over 5 mins) of vehicle (10% glycerol in phosphate-buffered saline (PBS) (n=5), Ad-GFP (n=6) or Ad-APOE (n=6) were made in the dorsolateral caudate nucleus (2.5 mm lateral to midline at bregma level and 3 mm from the cortical surface) using a 2 μl Hamilton syringe. Adenoviral vectors were administered at a concentration of 3.4 × 1011 vp/mL in PBS containing 10% glycerol based on pilot experiments in which the most extensive distribution of transgene expression with minimal toxicity was observed at this dose. After infusion, the needle was left in place for 10 mins to prevent diffusion along the needle tract. The needle was withdrawn and the wound sutured before anaesthesia was discontinued and mice were returned to their cages.
Transient Focal Cerebral Ischaemia
Focal ischaemia was performed 3 days after adenoviral injection. Pilot experiments were undertaken to determine the optimal dose and expression of transgene and maximal transgene expression was found at 3 days postinjection. Transient focal ischaemia (60 mins) was induced by intraluminal filament occlusion of the right middle cerebral artery. Animals were anaesthetized and maintained with isofluorane (2%) in a mixture of 30% O2 and 70% N2O by facemask. Focal cerebral ischaemia was induced by occlusion of the left middle cerebral artery with an 8-0 nylon monofilament (Ethicon) coated with a mixture of silicone resin (Xantoprene, Bayer Dental, Osaka, Japan) and hardener (Elastomer Activator, Bayer Dental). Briefly, the left common carotid, external carotid and internal carotid arteries and their branches were exposed through a midline cervical incision. A 6-0 silk suture was tied around the common carotid proximal to the bifurcation of the external carotid and internal carotid and then a second suture was tied around the external carotid distal to the superior thyroid artery. The superior thyroid artery and occipital artery were closed by electrocoagulation. The silicone-coated monofilament (diameter 220 μm) was introduced into the common carotid via a small incision and advanced 10 mm distal to the carotid bifurcation so as to occlude the middle cerebral artery. Mice were then recovered from anaesthesia and placed in an incubator (30°C) for the duration of occlusion. The monofilament was completely withdrawn after 1 h to allow reperfusion. Wounds were sutured closed and anaesthesia discontinued. Mice were briefly re-anaesthetised and the filament withdrawn to induce reperfusion 60 mins after occlusion onset. Anaesthesia was discontinued and mice were returned to their cages.
Assessment of Neurological Deficit
After 24 h reperfusion, neurological status was assessed using a modification of a system described previously (Bederson et al, 1986), according to a neurological grading score of increasing severity of deficit: 0=no observable deficit; 1=failure to extend left forelimb; 2=spontaneous circling to left; 3=leaning/falling to left; 4=no spontaneous movement.
Measurement of Ischaemic Damage
Ischaemic damage was measured as described previously (McColl et al, 2004). Briefly, after neurological evaluation, mice were deeply anaesthetised and perfused transcardially with 4% paraformaldehyde. Brains were removed, postfixed and processed through paraffin. Sections (6 μm) were cut from paraffin-embedded brain and areas of ischaemic damage were identified on haematoxylin and eosin-stained sections at eight pre-determined coronal levels. Areas of ischaemic damage were transcribed on to scale diagrams and then measured using an MCID image-analysis system. Approximation of the total volume (mm3) of ischaemic damage was achieved by integration of areas (mm2) with the distance between each coronal level (mm) using GraphPad Prism software. The end points for integration were 2.9 mm (rostral limit) and 4.9 mm (caudal limit) with respect to bregma.
Measurement of apoE Levels by Enzyme-linked Immunosorbent Assay
ApoE levels were measured in mice receiving injections of vehicle, Ad-GFP or Ad-APOE (n=4 per treatment group) without subsequent focal ischaemia. Mice were deeply anaesthetised and perfused transcardially with approximately 20 ml chilled (4°C) 0.9% saline. The brain was removed and a 3 mm coronal slice of tissue containing the bilateral injection tracts was cut. Tissue around each injection tract (including striatum and cerebral cortex) was dissected out on ice and each tissue sample frozen in liquid nitrogen and stored at −80°C. Samples were homogenised in 5 × volume ice-cold homogenisation buffer (25 mmol/L Tris buffer pH 7.4 containing 3 mmol/L MgCl2, 100 mmol/L NaCl, 20% CHAPS with 1 μg/mL each of leupeptin, pepstatin A and aprotinin, and 100 mmol/L PMSF. Homogenates were stored on ice for 1 h and then centrifuged at 13,000 rpm for 5 mins at 4°C. Supernatant was collected and apoE concentration was determined by enzyme-linked immunosorbent assay. A 96-well plate was coated (100 μL/well) with the capture antibody (rabbit anti-human apoE (1:5000), Dako), diluted in 16 mmol/L carbonate/34 mmol/L bicarbonate buffer (pH 9.6) and incubated overnight at 4°C. After washes (3 ×) with PBS, blocking solution (2% bovine serum albumin in PBS) was added (150 μL/well) and the plate incubated for 1 h at 37°C. After washes (3 ×) in 1% PBS-Tween 20 (PBST), apoE standard (recombinant human apoE3, Chemicon) (0.25 to 40 ng/mL) and samples were diluted in sample diluent (1% bovine serum albumin in PBS) and added to wells (100 μL/well). The plate was then incubated for 2 h at 37°C. After further washes in PBST (3 ×), the detection antibody (goat polyclonal anti-apoE, Chemicon AB947, 1:2000 in sample diluent) was added (100 μL/well) and the plate incubated for 1 h at 37°C. Washes in PBST (3 ×) were followed by addition (100 μL/well) of secondary antibody (rabbit anti-goat horse radish peroxidase-conjugated IgG, Dako, 1:5000 in sample diluent) and the plate incubated for 1 h at 37°C. After washes in PBST (4 ×) and PBS (2 ×), colour was developed with phenylenediaminedihydrochloride (OPD) (100 μL/well) at room temperature. The colour reaction was terminated after 10 mins by addition of 1 mmol/L sulphuric acid (100 μL/well) and absorbance was read at a wavelength of 492 nm. ApoE concentration of unknown samples was derived from the standard curve. Determination of total protein concentration of samples (by BCA protein assay) enabled apoE concentration to be expressed as ng apoE/mg total protein.
Immunohistochemistry
Paraffin-embedded sections adjacent to those stained with haematoxylin and eosin for measurement of ischaemic damage were used for immunostaining. Sections were dewaxed in an oven at 60°C to 70°C for 20 mins and in xylene for 10 mins and then dehydrated in 100% alcohol (2 × 5 mins). Endogenous peroxidase was eliminated by incubating sections in 0.5% H2O2 in methanol for 30 mins. Sections were washed in running water for 15 mins and then PBS (2 × 5 mins). Nonspecific binding sites were blocked with 10% normal serum and 0.5% bovine serum albumin in PBS for 1 h. Sections were then incubated with primary antibody in blocking solution overnight at 4°C. ApoE was detected using polyclonal anti-apoE (1:5000, Chemicon AB947). This antibody detects endogenous mouse apoE and all human apoE isoforms. Astrocytes were detected with anti-GFAP (1:200, Chemicon MAB360) and microglia with anti-F4/80 (1:50, Caltag RM2900). After washes in PBS (2 × 5 mins), sections were incubated with biotinylated anti-goat diluted 1:100 in PBS for 1 h followed by further washes in PBS (2 × 5 mins). Sections were then incubated with an avidin-biotinylated peroxidase complex (Vectastain ABC kit) for 1 h. After further washes in PBS (2 × 5 mins), colour was developed by incubating sections in a 3′3′-diaminobenzidine tetrahydrochloride solution for 3 mins. The colour reaction was terminated after 3 mins by washing sections in running water. Sections were then dehydrated in a graded series of alcohols, cleared in xylene and cover-slipped.
Statistical Analysis
For all variables assessed, two comparisons were made: (1) vehicle versus Ad-GFP, (2) Ad-GFP versus Ad-APOE. Statistical significance was assessed using one-way ANOVA followed by unpaired Student's t-test with Bonferroni correction.
Results
Adenoviral Vector Transduction and Transgene Expression
Preliminary experiments were performed to confirm that the adenoviral vector could transduce cells in the brain and to assess the extent of transgene expression. These preliminary studies indicated that transgene expression was maximal at 3 days postinjection. In mice treated with Ad-GFP, GFP-positive cells were observed in the striatum adjacent and distant to the injection tract and also in the subcortical white matter 3 days after vector administration (Figure 1A). The majority of GFP-positive cells in the striatum displayed large, round cell bodies with processes of varying length (Figure 1B). GFP-positive cells in subcortical white matter displayed small, elongated cell bodies and were orientated along the plane of the fibres suggesting that oligodendrocytes were the predominant cell type expressing GFP in white matter (Figure 1C). It should be noted that although GFP expression provides an indication of adenoviral transduction, it is likely to be an underestimation since there may be additional cells transduced that are expressing GFP at undetectable levels or not at all.
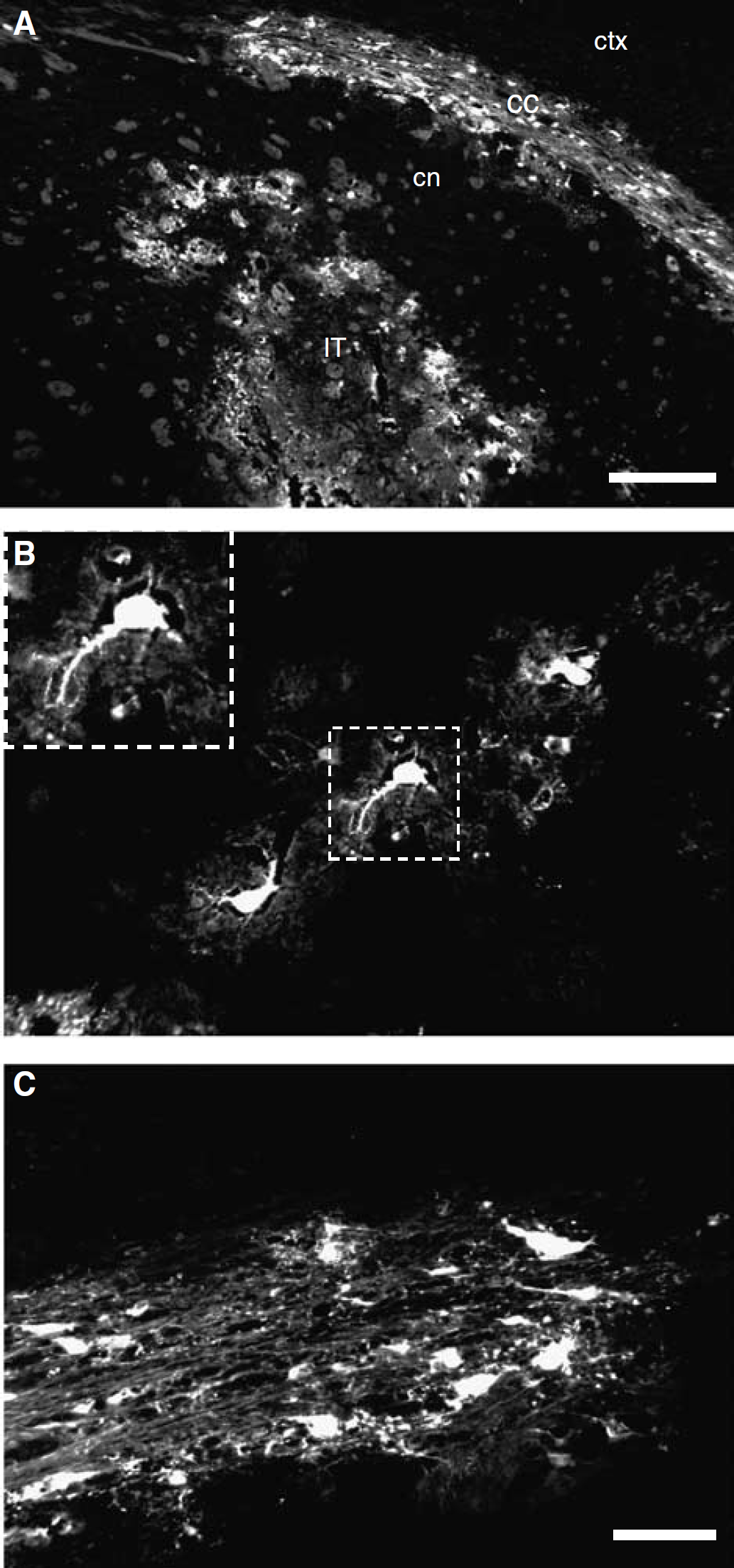
Distribution of adenoviral vector transduction. (
Sections stained with haematoxylin and eosin showed that, in areas of tissue containing GFP-positive cells, there was no histological evidence of cellular injury indicating that there was negligible direct cytotoxicity because of adenoviral vector transduction (Figure 2). In addition, there was no evidence of immunogenicity. The number of GFAP immunoreactive astrocytes and F4/80 immunoreactive microglia were similar after vehicle as compared to adenoviral vector injection (0.48+0.08 vs 0.46+0.11 astrocytes/mm2; 1.03±0.2 vs 0.75+0.14 microglia/mm2) (data not shown).
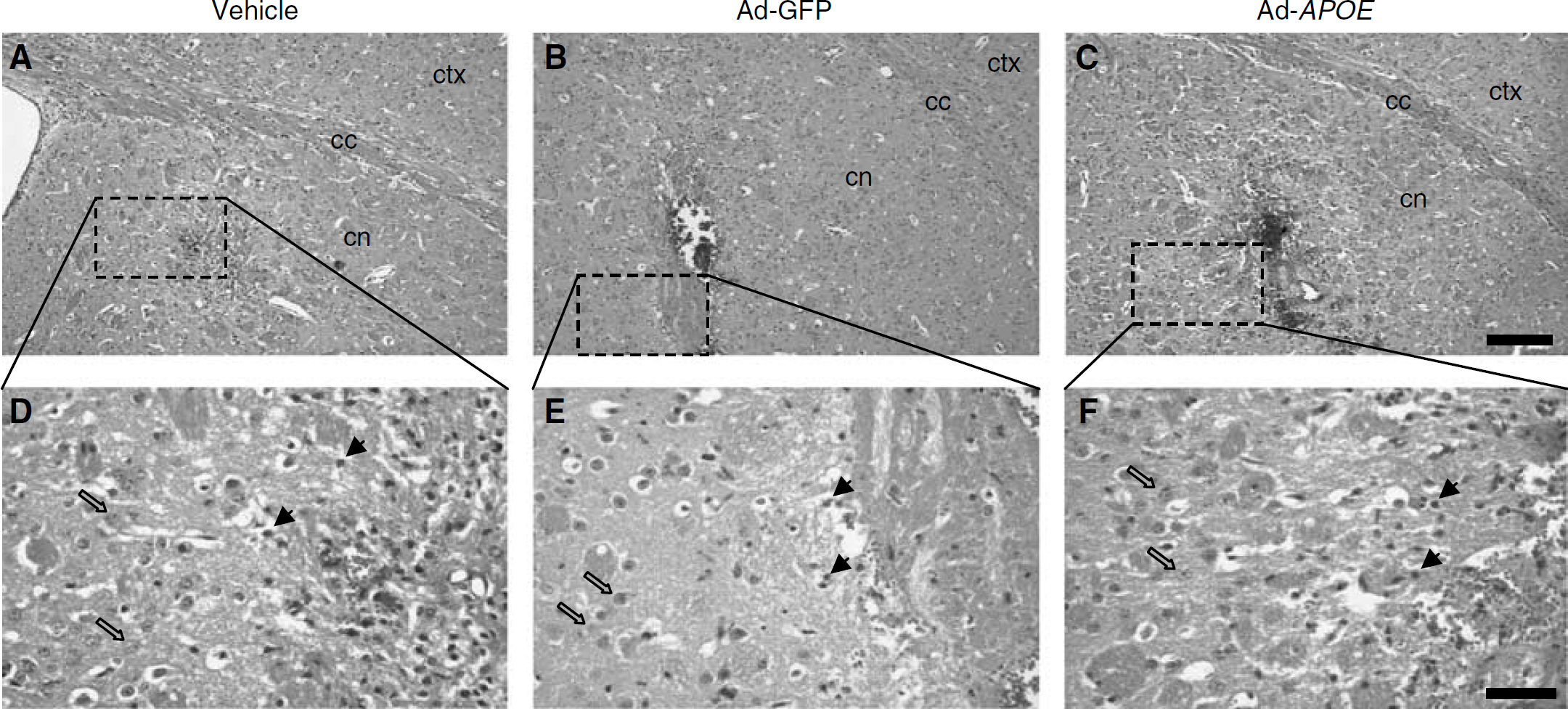
Minimal cytotoxicity of adenoviral vector transduction. (
Effect of Ad-APOE on apoE Expression
The distribution and level of apoE expression were investigated 3 days after injection in mice that did not undergo focal ischaemia. Ad-APOE administration resulted in markedly greater intensity and distribution of apoE immunoreactivity compared with vehicle or Ad-GFP treatment (Figure 3). ApoE immunoreactivity of the neuropil was extensive; there was a marked increase in apoE immunoreactivity encompassing most of the caudate nucleus, but notably this extended to the external capsule and deep layers of the cerebral cortex after Ad-APOE treatment (Figure 3C). In addition, intense cellular immunoreactivity was evident (Figures 3F–3H). ApoE immunoreactive cells were primarily glial in morphology (Figures 3G and 3H) although a few immunoreactive cells with neuronal morphology were observed (Figure 3F). Cellular apoE immunoreactivity adjacent to the injection tract is likely to be a response to mechanical damage caused by the needle since this was observed in all treatment groups.
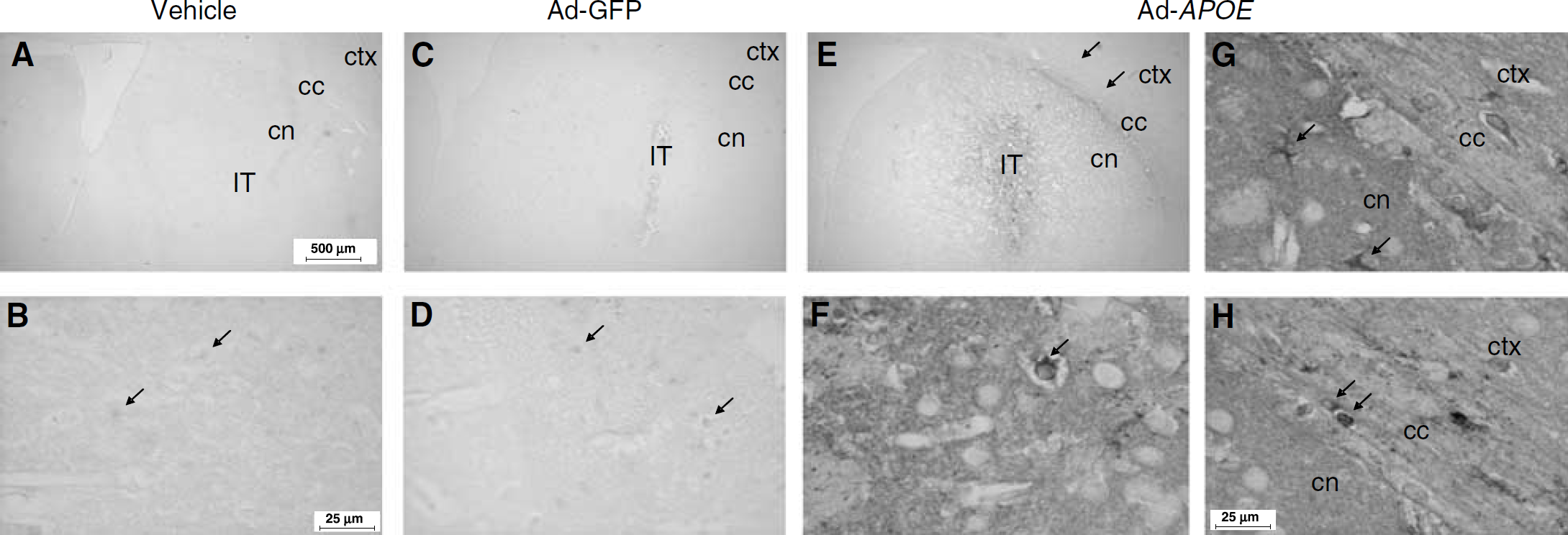
Effect of Ad-APOE on apoE immunoreactivity. (
ApoE levels were significantly increased by over 40-fold after Ad-APOE administration (Figure 4). ApoE levels were significantly greater in Ad-APOE compared with Ad-GFP treated mice (51±3 ng apoE/mg total protein versus 1.2±0.2 ng apoE/mg total protein, P<0.001). There was no significant difference in the level of apoE after administration of vehicle or Ad-GFP (1±0.4 ng apoE/mg total protein versus 1.2±0.2 ng apoE/mg total protein, P>0.05).
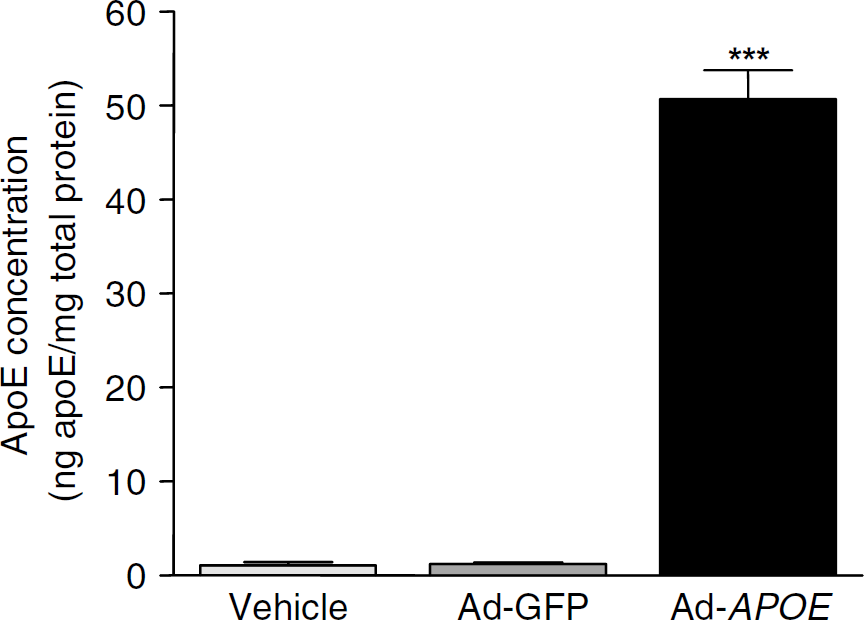
Effect of Ad-APOE on apoE levels. ApoE concentration was significantly elevated after Ad-APOE compared with Ad-GFP injection. The level of apoE was similar after vehicle or Ad-GFP injection. ∗∗∗P<0.001 for the comparison between Ad-GFP and Ad-APOE, one-way ANOVA followed by Student's unpaired t-test with Bonferroni correction.
Effects of Ad-APOE on Brain Damage And Neurological Deficit After Focal Ischaemia
Administration of Ad-APOE significantly reduced the total volume of ischaemic damage (by approximately 50%) in comparison to Ad-GFP treatment (13±3 versus 29±4 mm3, P=0.013) (Figure 5A) 24 h after occlusion. Administration of Ad-APOE also significantly reduced the volume of ischaemic damage in the cortex (P=0.012) and the striatum (P=0.010). Areas of ischaemic damage at coronal levels were also reduced by Ad-APOE administration and this was most marked at the level at which Ad-APOE was injected (Figure 5B). The total volume of ischaemic damage was similar in vehicle and Ad-GFP treated mice (27±5 versus 29±4 mm3, P>0.05). There were no significant differences in the volume of ischaemic damage in the cortex and striatum of vehicle and Ad-GFP treated mice (P>0.05). There was a trend toward a lower neurological deficit score in Ad-APOE treated mice, although this did not reach statistical significance (P>0.05) (Figure 5C).
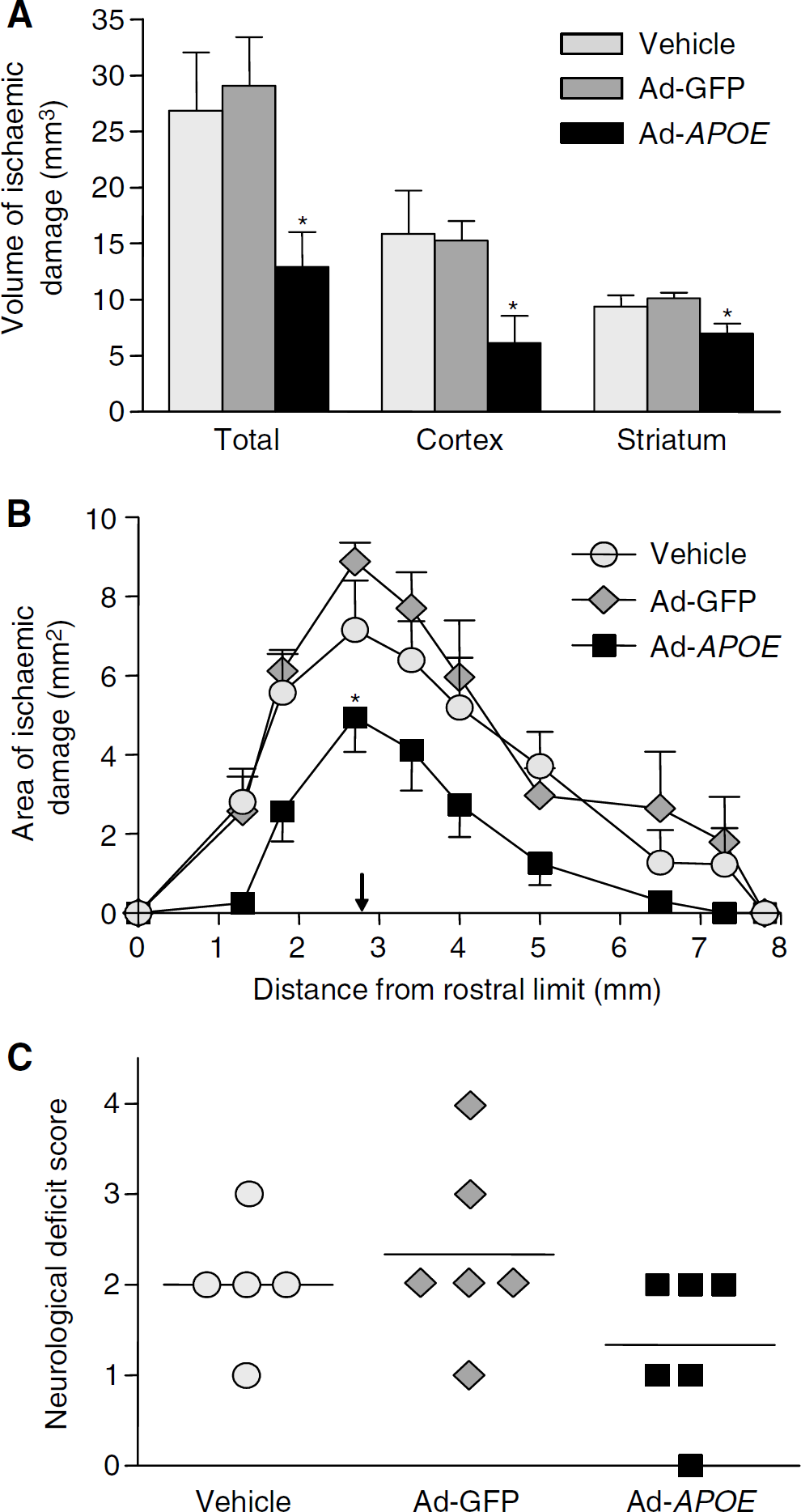
Effect of Ad-APOE on outcome after transient focal ischaemia. (
Effect of Ad-APOE on apoE Expression after Focal Ischaemia
In the hemisphere contralateral to injection and occlusion, apoE immunoreactivity was observed in astrocytes regardless of vehicle, Ad-GFP or Ad-APOE treatment (Figures 6A–6C). In the ipsilateral hemisphere, reactive glial cells localised to the injection tract were immunoreactive for apoE, likely a response to mechanical damage from needle penetration since no differences were observed among treatment groups (Figures 6D–6F). In all treatment groups, areas of ischaemic damage were associated with increased cellular apoE immunoreactivity (compared with nonischaemic hemisphere), notably in neurons with the morphological features of ischaemic cell change and in astrocytes. However, there were also marked differences in the distribution (Figures 6G–6I) and intensity (Figures 6J–6O) of apoE immunoreactivity in Ad-APOE treated mice in comparison to those receiving vehicle or Ad-GFP. First, in Ad-APOE treated mice, intense apoE immunoreactivity of the neuropil was evident at the injection level and at coronal levels rostral and caudal to this. In four of six mice, such intense immunoreactivity was distributed throughout the entire striatum and extended into the mid to deep layers of the fronto-parietal cerebral cortex at these levels (Figure 6I). Notably, in three of six mice, intense neuropil apoE immunoreactivity was not only localised to areas of ischaemic damage but also extended to undamaged tissue (no morphological evidence of ischaemic neurons) in the striatum and cerebral cortex. In contrast, intense neuropil apoE immunoreactivity was localised to the injection tract in vehicle and Ad-GFP treated mice (Figures 6G and 6H). Second, the intensity and distribution of cellular apoE immunoreactivity in areas of ischaemic damage, most notably in ischaemic neurons, was markedly greater in Ad-APOE treated mice (Figures 6J–6L). Intensely apoE-immunoreactive ischaemic neurons were distributed throughout the striatum of Ad-APOE treated mice whereas apoE-immunoreactive ischaemic neurons were most apparent at the ischaemic border of vehicle and Ad-GFP treated mice. Furthermore, intensely apoE-immunoreactive neurons (ischaemic and nonischaemic) were also evident in the cerebral cortex of Ad-APOE treated mice but not in those receiving vehicle or Ad-GFP (Figures 6M–6O).
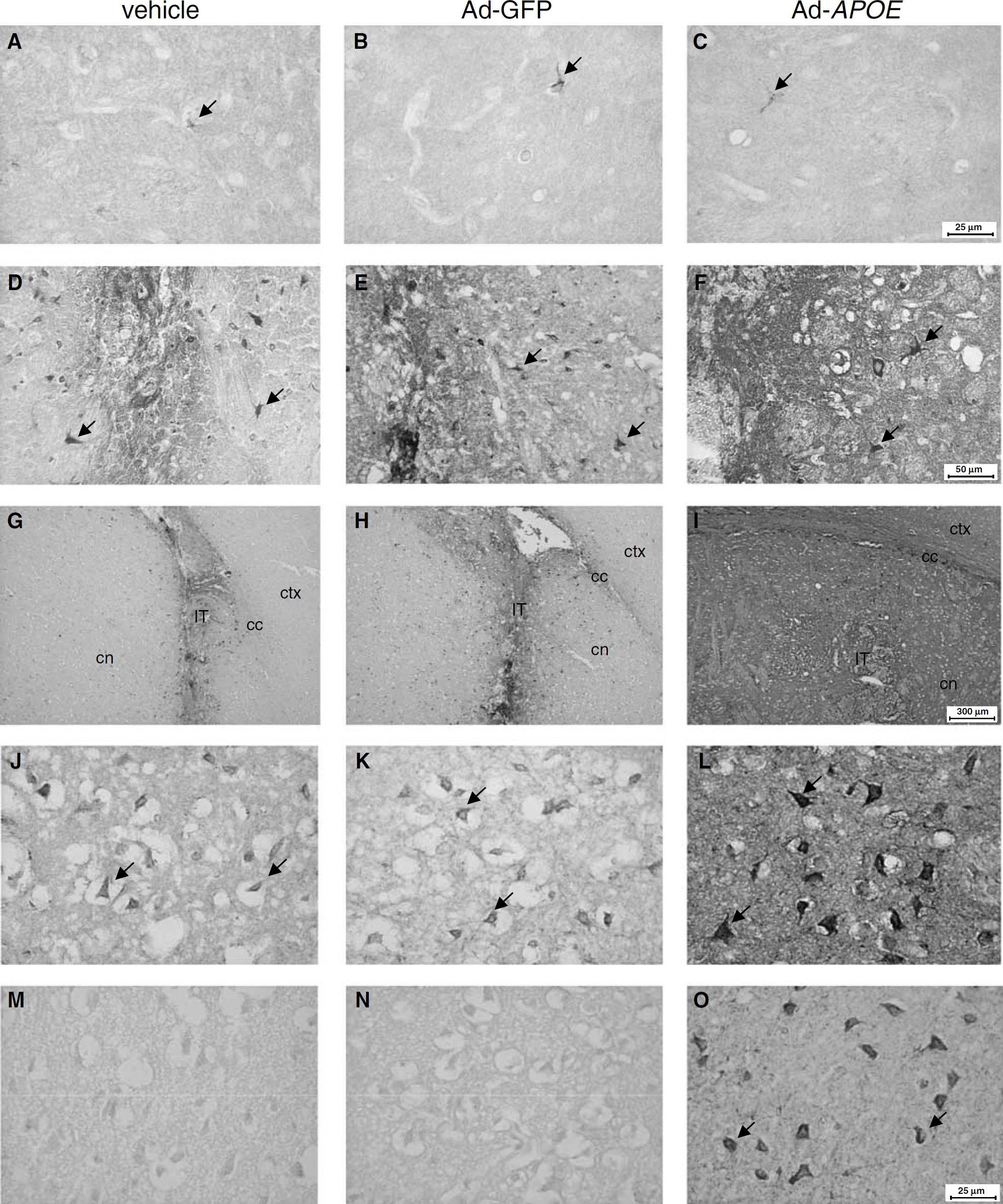
Effect of Ad-APOE on postischaemic apoE expression. (
Discussion
In the present study, we show that adenoviral-mediated gene transfer of human APOE ε3 significantly reduces the volume of ischaemic brain damage by approximately 50% after transient focal ischaemia in C57Bl/6J mice. To our knowledge, this is the first study to show neuroprotection after APOE gene transfer.
The present data support a protective role for apoE (specifically the apoE3 isoform) after ischaemic brain injury and are consistent with previous studies in which exogenous apoE delivery to the brain has been shown to promote neuroprotection. Intraventricular infusion of a physiological concentration of human plasma-derived apoE reduced the extent of selective neuronal damage after transient global ischaemia in APOE-deficient mice (Horsburgh et al, 2000b). Neurodegeneration and cognitive impairment were also attenuated by intraventricular infusion of recombinant apoE in APOE-deficient mice (Masliah et al, 1997). More recently, McAdoo et al (2005) have shown the protective effects of apoE-peptide delivery in perinatal hypoxic—ischaemic injury. Collectively, these results suggest that restoring and/or augmenting endogenous apoE levels may have beneficial effects on the outcome to ischaemic brain injury.
To modulate apoE levels, we used a second-generation adenoviral vector to deliver the APOE gene directly to the brain. The ability of the vector to transduce cells in the brain and mediate transgene expression was confirmed in mice treated with an adenoviral vector containing the GFP reporter gene. GFP-expressing cells were distributed throughout a large part of the striatum and in the overlying subcortical white matter. The extent of GFP expression was similar to that observed previously after intrastriatal injection of similar doses of first generation adenoviral vectors (Hermann et al, 2001). Most GFP-expressing cells were glial cells, which is in accordance with previous in vivo and in vitro studies in which the CMV promoter has been used to drive adenoviral transgene expression. Although adenoviral vectors transduce both neurons and non-neuronal cells, glial transduction is more efficient (Hermann et al, 2001; Kugler et al, 2001). In addition, CMV promoter activity and transgene expression in glial cells may suppress promoter activity in neurons (Kugler et al, 2001). Efficent transgene expression in Ad-APOE treated mice was confirmed by enzyme-linked immunosorbent assay, which indicated a robust increase in apoE levels. It is unlikely this was a response to adenoviral transduction since apoE levels were minimal after Ad-GFP treatment and similar to those detected after vehicle treatment. Interestingly, apoE immunoreactivity in Ad-APOE treated mice was observed in a few glial cells, but predominantly in the neuropil, suggesting that the majority of apoE is secreted after adenoviral transduction and transgene expression. Thus, apoE may diffuse to areas distant to adenoviral transduction sites, which would explain why apoE immunoreactivity extends beyond areas that contain GFP-expressing cells.
It was also noted that pretreatment of mice with Ad-APOE increased expression of apoE in ischaemic brain. The marked enhancement of postischaemic apoE immunoreactivity by Ad-APOE may represent induction of vector-derived apoE expression after ischaemia or may reflect increased levels of apoE already present in the brain before ischaemia. Although it is not possible to completely dissociate these two possibilities, the extent of apoE immunoreactivity appeared even greater in ischaemic compared with nonischaemic mice, suggesting postischaemic induction of apoE. Such a scenario would require activation of the CMV promoter driving apoE expression from the vector. There is considerable evidence that the CMV promoter is ubiquitously active and further activated under conditions of stress to host cells, as would occur during cerebral ischaemia (Speir et al, 1996; Loser et al, 1998; Wheeler et al, 2000). The CMV promoter contains three consensus binding sites for the transcription factor NFκB and CMV-linked transgene expression has previously been shown to be activated by NFκB (Wheeler et al, 2000). NFκB is activated after ischaemia, particularly by cytokines, and therefore it seems likely that CMV driven transgene expression could be induced after ischaemia.
Despite injection into the striatum, we observed markedly greater protection of cortical compared with striatal tissue after Ad-APOE. In common with most neuroprotective strategies, vector-derived apoE is likely to protect tissue at risk (penumbra) of which this area is larger in cortex than striatum in this murine MCAo model (McColl et al, 2004). Several mechanisms may contribute to this effect. The secretion and dispersal of apoE will enable it to act distant to the striatal injection and transduction areas, and therefore cortical neuroprotection may result from the diffusion of apoE. In support of this, apoE immunoreactivity was distributed throughout the striatum and extended into the cerebral cortex after focal ischaemia in Ad-APOE treated mice. Intense cortical apoE immunoreactivity was observed in undamaged tissue suggesting that augmentation of apoE levels in these areas may prevent ischaemic cell death. Adenoviral transfer of genes encoding other diffusible proteins, such as GDNF and CNTF, also reduced infarct volume after focal ischaemia (Kitagawa et al, 1999; Hermann et al, 2001). In contrast, viral vector-mediated overexpression of intraneuronal proteins that are not secreted (e.g., Bcl-2, calbindin D28K) have generally failed to reduce infarct volume, although have protected against local neuronal loss in the striatum and infarct margin (Yenari et al, 2001; Zhao et al, 2003). Cortical neuroprotection may also be secondary to actions of apoE in the striatum. ApoE has antiexcitotoxic and antioxidant properties (see below also), thus striatal-initiated spreading depression and oxidative stress, both of which are mechanisms that can have distant effects on the conversion of penumbra to infarct, may be attenuated by Ad-APOE.
The present data further show the acute neuroprotective effects of apoE. A poorer outcome in APOE deficient mice has been shown in various models of acute ischaemic or traumatic brain injury (Horsburgh et al, 2000a). It has been proposed that apoE scavenges and redistributes cholesterol and lipids to damaged neurons where they are internalised and used to promote neuronal survival and repair (Poirier 1994; Vance et al, 2000). Neuronal accumulation of apoE after ischaemia, as observed in this and previous studies, may reflect this process. In addition, apoE may exert neuroprotective effects independent of its lipid-transport properties. ApoE and apoE-derived peptides can attenuate NMDA-mediated excitotoxic damage in cultured neurons (Aono et al, 2002). Putative interactions between the major neuronal apoE receptor, low-density lipoprotein receptor-related protein, and the NMDA receptor via adaptor proteins such as PSD-95 have been described and may provide a pathway by which apoE could alter glutamate receptor function and excitotoxicity (Bacskai et al, 2000). Antioxidant actions of apoE have also been shown after global ischaemia (Horsburgh et al, 2000b). Significantly, isoform-specific antioxidant effects of apoE have been shown, with the apoE3 isoform more effective (Buttini et al, 1999). ApoE3 also has a greater ability to attenuate the acute inflammatory response (Lynch et al, 2003) and apoptotic cell death (DeKroon et al, 2003). Furthermore, apoE3 binds with greater affinity to various cytoskeletal components than apoE4, which may promote greater cytoskeletal stability and neuronal survival after injury (Fleming et al, 1996). In the present study, we administered an APOE ε3-containing vector and so the various ‘beneficial’ effects of this isoform may contribute to the protection we observed. Alternatively, the concentration of apoE available (regardless of isoform) may be the key factor underlying neuroprotection. In support of this, intraventricular administration of human plasma-derived apoE of mixed isoform reduced neuronal damage after global ischaemia (Horsburgh et al, 2000b). Intraventricular administration of recombinant apoE3 or apoE4 both reduced cognitive impairment and neuronal degeneration similarly in aged APOE deficient mice (Masliah et al, 1997). In humans, APOE ε4 carriers have lower plasma levels of apoE than ε2 and ε3 carriers (Schiele et al, 2000) and some studies have shown lower plasma and brain levels of apoE in APOE ε4 patients with Alzheimer's disease (Bertrand et al, 1995; Panza et al, 2003). Lower apoE levels in APOE ε4 carriers has been suggested to underlie their increased susceptibility to Alzheimer's disease and poorer recovery from acute brain injury (Bertrand et al, 1995). Regardless of the mechanism of neuroprotection, however, the present data indicate the clinical potential of modulating apoE3 levels in cerebral ischaemia.
A possible limitation of an adenoviral-mediated gene transfer approach in human stroke is the potential triggering of an inflammatory/immune response to adenoviral transduction, which not only has safety implications but also can substantially suppress transgene expression (Wood et al, 1996). However, second-generation adenoviral vectors, as used in the present study, are less immunogenic and promote more stable and long-term transgene expression than conventional first-generation vectors (Thomas et al, 2000; Zou et al, 2000). In the present study, there was negligible cellular damage (other than mechanical tissue disruption) and minimal astrocytic and microglial reactivity after adenoviral administration suggesting that the vector had minimal, cytotoxicity and immunogenicity. This is in accordance with a previous report in which a gutless, second-generation vector was substantially less toxic than a first-generation vector (Zou et al, 2000).
In the present study, we observed abundant apoE immunoreactivity in Ad-APOE treated mice after focal ischaemia, suggesting there was preservation of transgene expression and protein synthesis. Further studies are warranted to determine if the neuroprotective effects of APOE gene transfer are also evident when vectors are administered in the postischaemic phase. In particular, it will be important to determine if the residual protein synthesis capacity after ischaemia can support sufficient levels of transgene expression. In this regard, viral transduction of glial cells, as observed with our vector, may have important benefits since protein synthesis inhibition in glial cells is less severely affected than in neurons (Thilmann et al, 1986). Studies have shown that expression of the β-galactosidase reporter gene is possible when vectors are administered after ischaemic brain injury (Abe et al, 1997). Furthermore, adenoviral-mediated GDNF gene transfer immediately after focal ischaemia in rats was shown to significantly reduce infarct volume (Zhang et al, 2002).
In summary, our data indicate that neuroprotection after focal ischaemia can be achieved by modulating levels of the ‘beneficial’ apoE3 isoform in the brain. This may represent a potential therapeutic strategy to pursue in human stroke, particularly in view of the multiple mechanisms by which apoE3 can exert its protective effects, an inadequacy of many compounds that have failed to reproduce preclinical efficacy in human stroke trials.