
Abstract
Monocyte chemoattractant protein-1 (MCP-1) is expressed in the ischemic cortex after focal brain ischemia and appears to exacerbate ischemic damage. The authors examined the effect of gene transfer of dominant negative MCP-1, called 7ND, 90 minutes after induction of focal brain ischemia in hypertensive rats. Adenoviral vectors encoding mutant MCP-1 (Ad7ND; n = 11), or Escherichia coli β-galactosidase (AdlacZ; n = 17) as control were injected into the lateral ventricle of male spontaneously hypertensive rats. Both AdlacZ (n = 12) and Ad7ND (n = 6) administration provided transgene expression as early as 6 hours after injection and the expression further increased on day 1, followed by a sustained detection on day 5. Five days after ischemia, infarct volume (75 ± 13 mm3, n = 5, mean ± SD) significantly reduced to 72% of control (104 ± 22 mm3, n = 5, P < 0.05) by 7ND gene transfer. Numbers of leukocytes in the vessels (48.3 ± 32.9/cm2) and macrophage/monocyte infiltration (475.2 ± 125.5 /mm2) of the infarct area in the Ad7ND group were significantly less than those measured in the AdlacZ group (143.8 ± 72.1/cm2 and 671.8 ± 125.5/mm2, P < 0.05, respectively). In summary, the postischemic gene transfer of dominant negative MCP-1 attenuated the infarct volume and infiltration of inflammatory cells, suggesting potential usefulness of the anti–MCP-1 gene therapy.
Gene transfer is an attractive intervention for studies of basic mechanisms of neurobiology, and potentially for therapy of cerebrovascular disease (Heistad and Faraci, 1996; Verma and Somia, 1997). Although several studies have shown usefulness of gene transfer to the brain to protect against ischemic damage, vectors were introduced in the brain before induction of brain ischemia (Betz et al., 1995; Lawrence et al., 1996; Linnik et al., 1995; Yenari et al., 1998). To rationalize gene therapy for brain ischemia, it is important to show efficacy of gene transfer even when vectors are administered after induction of brain ischemia. We have recently shown that gene transfer can be accomplished after induction of brain ischemia either to brain parenchyma or ventricular walls (Ooboshi et al., 2001; Takada et al., 2002; Kumai et al., 2003).
Monocyte chemoattractant protein-1 (MCP-1) is a member of the C-C chemokine subfamily of chemokines (Rollins, 1996), and has chemoattractant properties for monocytes, memory T-cells, and natural killer cells (Allavena et al., 1994; Carr et al., 1994; Matsushima et al., 1989; Valente et al., 1996; Yoshimura et al., 1989). In experimental brain ischemia model, MCP-1 messenger RNA and MCP-1 protein in the ischemic cortex are expressed after focal brain ischemia (Che et al., 2001; Kim et al., 1995; Wang et al., 1995). Recent studies using genetic models have reported that MCP-1 deficiency attenuates infarct volume in the murine stroke model (Hughes et al., 2001), and overexpression of MCP-1 causes larger infarct volume and more chemoattraction of monocytes and macrophages into ischemic region than wild-type control (Chen et al., 2003), suggesting that elevated MCP-1 levels might lead to an increased influx of monocytes and evolution of size in brain infarction.
We have recently reported that an N-terminal deletion mutant of human MCP-1 gene (7ND), which lacks the N-terminal amino acids 2 to 8, acts as a dominant negative inhibitor for MCP-1 and blocks the MCP-1/CCR2 signal pathway in vivo (Egashira et al., 2000; Egashira, 2003). This mutant MCP-1 and normal MCP-1 form a heterodimer, which binds to the MCP-1 receptor (CCR-2) and completely inhibits MCP-1–mediated monocyte chemotaxis in vitro (Rollins, 1996). In this study, we delivered the adenoviral vector encoding 7ND into the cerebral ventricle 90 minutes after focal brain ischemia, and examined whether anti–MCP-1 gene therapy would protect against focal brain ischemia.
MATERIALS AND METHODS
Adenoviral vectors
We used replication-deficient recombinant adenoviral vectors expressing Escherichia coli β-galactosidase (AdlacZ) or mutant MCP-1 (Ad7ND). An N-terminal deletion mutant of human MCP-1, called 7ND, lacks the amino-terminal amino acids 2 to 8. The 7ND was constructed by recombinant polymerase chain reaction using a wild-type human MCP-1 cDNA as the template and cloned into BamHI (5′) and NotI (3′) sites of the pcDNA3 expression vector (Egashira et al., 2000, Ni et al., 2001). The DNA constructs of vectors composed of a full-length copy of the adenovirus genome of approximately 36 kb, from which the early region 1 gene (E1) was replaced by the CAG (cytomegalovirus enhancer, chicken β-actin enhancer-promoter and rabbit β-globin poly-A signal) promoter and a cDNA for lacZ or 7ND. Recombinant viruses were grown in human embryonic kidney (HEK) 293 cells that complemented the E1 early viral promoters, and were triple plaque purified to assure that viral suspensions were free of wild-type viruses. Viral titer was determined by plaque assay on HEK 293 cells. After purification, the virus was suspended in phosphate-buffered saline (PBS) with 3% sucrose, and was kept at −80°C until further use.
Animals
All animal procedures were approved by the Animal Care and Use Review Committee at the Kyushu University (12-053-0). Twenty-eight male spontaneously hypertensive rats (SHR), aged 5 to 10 months and weighing 320 to 400 g, were used. Eighteen rats were semiquantitatively or quantitatively analyzed for transgene expression of β-galactosidase or 7ND, and 10 rats were used for the brain ischemia study.
Histochemical analysis of gene expression
Twelve male SHR were quantitatively analyzed for transgene expression of β-galactosidase. Briefly, rats were anesthetized with pentobarbital (65 mg/kg, intraperitoneal injection) and mounted on a stereotaxic head holder in the prone position. A 2-cm incision was made vertically midway. Rectal and head temperature was maintained at 37°C and 36°C, respectively, by means of a warming lamp and a heating pad. For the injection of adenoviral vectors into the left ventricle, a small burr hole was made in the parietal region (1.5 mm posterior and 1.0 mm lateral to the bregma) with a dental drill. A 27-G needle on a Hamilton syringe was stereotaxically inserted into the left lateral ventricle (4.5 mm deep), and 30 μL of AdlacZ (1.3 × 109 plaque forming units per milliliter, n = 12) was injected over 10 minutes. Efficacy of transgene expression to the brain was assessed 6 hours (n = 1), 12 hours (n = 2), 1 day (n = 3), 3 days (n = 2), 5 days (n = 3), and 7 days (n = 1) after injection of AdlacZ. After the designated survival periods, the rats were killed with an intraperitoneal injection of pentobarbital and perfused transcardially with 2% paraformaldehyde and 0.2% glutaraldehyde in PBS. The brain was removed and washed thoroughly with PBS. The brain was cut into coronal sections at intervals of 2 mm and incubated in 5-bromo-4-chloro-3-indolyl-β-
Measurement of 7ND
Six male SHR, weighing 370 to 400 g, were quantitatively analyzed for transgene expression of 7ND. In this experiment, procedures for operation were similar to those described for the previous experiment, except for the type of adenoviral vector. Thirty microliters of AdlacZ (1.3 × 109 plaque forming units per milliliter, n = 3) or Ad7ND (1.3 × 109 plaque forming units per milliliter, n = 3) was injected over 10 minutes. Six hours, 1 day, and 5 days after injection of vector, rats were anesthetized with pentobarbital (65 mg/kg, intraperitoneal injection) and the CSF was withdrawn (Ooboshi et al., 1995, 1997). The CSF concentration of 7ND released by transfected ependyma was measured by using sandwich enzyme-linked immunosorbent assay (ELISA) of human MCP-1. The ELISA kit (R&D Systems, Minneapolis, MN, U.S.A.) with the monoclonal antibody was used as reported previously according to the manufacturer's instructions (Kohara et al., 2002). The antibody does not cross-react with rat MCP-1.
Brain ischemia
Ten male SHR were used for the brain ischemia study. Briefly, rats were anesthetized with halothane (3% for induction; 1.5% during the surgical preparation, with a facemask; 0.75% after intubation; and 0.5% for maintenance) in a mixture of 70% nitrous oxide and 30% oxygen. The right femoral artery and vein were cannulated using PE-50 tubing. The rats were endotracheally intubated with PE-240 tubing. Pancuronium bromide (an initial dose of 0.3 mg followed by 0.1 mg every 30 minutes) was intravenously injected, and the rats were mechanically ventilated. Mean arterial pressure was continuously monitored. Physiologic variables were measured before and 1 hour after the distal middle cerebral artery (MCA) occlusion. Rectal and head temperature was maintained at 37°C and 36°C, respectively, by means of a warming lamp and a heat pad.
The rat was mounted on a stereotaxic headholder in the prone position, and a 2-cm incision was made vertically midway between the right orbit and the right external auditory canal. The temporal muscle was separated and, under an operating microscope, a burr hole 3 mm in diameter was made 1 mm posterior to the anterior junction of the zygoma and squamosal bone, revealing the distal segment of MCA above the rhinal fissure. The dura was left intact. Cerebral blood flow (CBF) before and during ischemia at the parietal cortex was measured by laser Doppler flowmetry. A burr hole, 2 mm in diameter, was made in the parietal cortex at 4 mm lateral and 1.5 mm posterior to the bregma in the ipsilateral to ischemic side. The resting CBF value was regarded as baseline and changes after induction of brain ischemia were expressed as percentages of the resting value.
Brain ischemia was produced by photochemical occlusion of the distal MCA of SHR as described previously (Yao et al., 1996). A krypton laser operating at 568 nm (Innova 301, Coherent Inc., Santa Clara, CA, U.S.A.) was used to irradiate the distal MCA at a power of 20 mW. The laser beam was focused with a 30-cm focal length cylindrical lens (CKX 300; Newport Corporation, Irvine, CA, U.S.A.) and positioned with a mirror onto the distal MCA. The photosensitizing dye, rose bengal (15 mg/mL in 0.9% saline; Wako Pure Chemical), was administered intravenously to a body dose of 20 mg/kg over 90 seconds simultaneously with 4 minutes of laser irradiation.
For the injection of adenoviral vectors into the lateral ventricle contralateral to the ischemic side, a small burr hole was made in the parietal region as the above experiments. Ninety minutes after induction of ischemia, 30 μL of viral suspension of AdlacZ (1.3 × 109 plaque forming units per milliliter, n = 5) or Ad7ND (1.3 × 109 plaque forming units per milliliter, n = 5) was injected into the lateral ventricle over 10 minutes. Two hours after the distal MCA occlusion, the head wound was closed and the catheters were removed. The rats were carefully weaned from the respirator and returned to the home cage after regaining the ability to breathe independently. After the injection of vectors, the rats were housed for 5 days.
Quantification of brain infarction and infiltration leukocyte
Five days after brain ischemia, rats were anesthetized with pentobarbital. The brain was removed, washed thoroughly with PBS, and cut into coronal sections at intervals of 2 mm followed by postfixation with 4% formaldehyde. The fixed tissue was then processed for paraffin embedding, and sections (5 μm thick) were cut from the block with microtomes for hematoxylin-eosin staining. Morphometric determination of infarct volume by the direct method has been described previously (Liu et al., 1989). The cross-sectional area of infarct was measured with NIH Image software (version 1.63) and infarct volume of each rat was calculated.
The number of polymorphonuclear and mononuclear leukocytes in blood vessels at the infarct area was determined in the coronal slice at the level of caudate-putamen by hematoxylin-eosin staining. The number of leukocytes was divided by infarct area, and was expressed as number per square centimeter.
Immunohistochemistry of macrophage
For immunohistochemistry, paraffin slices 5 μm thick at the caudate-putamen level were preincubated with 3% skim milk to decrease nonspecific binding. Sections were incubated overnight at 4°C with the mouse anti–rat macrophage/monocyte antibody (ED1, Serotec, Oxford, U.K.) diluted 1:1000, or nonimmune mouse IgG (Santa Cruz Biotechnology Inc., Santa Cruz, CA, U.S.A.) diluted 1:1000 as negative control. The slides were washed and incubated with biotinylated, affinity-purified rabbit anti-mouse IgG (Nichirei Corporation, Tokyo, Japan) as the secondary antibody. After avidin-biotin amplification, the slides were incubated with 3′,3′-diaminobenzidine. For quantification, the number of ED1-positive cells at the ischemic area was analyzed with NIH Image software (version 1.63), and was expressed as number per square millimeter. The slides were counterstained with hematoxylin for nuclear staining.
Statistical analysis
Data are presented as means and standard deviations. Differences in physiologic variables, infarct volume, and number of leukocytes and ED1-positive cells between groups were analyzed by unpaired t-test. Differences in amount of 7ND were analyzed with repeated measure one-way analysis of variance followed by Bonferroni post hoc t-test. P < 0.05 was regarded as statistically significant.
RESULTS
Transgene expression of β-galactosidase
Expression of the reporter gene was consistently detected at the periventricular areas since 6 hours to 7 days after gene transfer (Fig. 1A). X-Gal staining was not observed in the cortex, as reported previously (Kumai et al., 2003). The time course of semiquantitative analysis for transgene expression at the periventricular area is shown in Fig. 1B. Transgene expression was observed at the ependyma as early as 6 hours after gene transfer, and peaked at day 1 followed by gradual decreases.
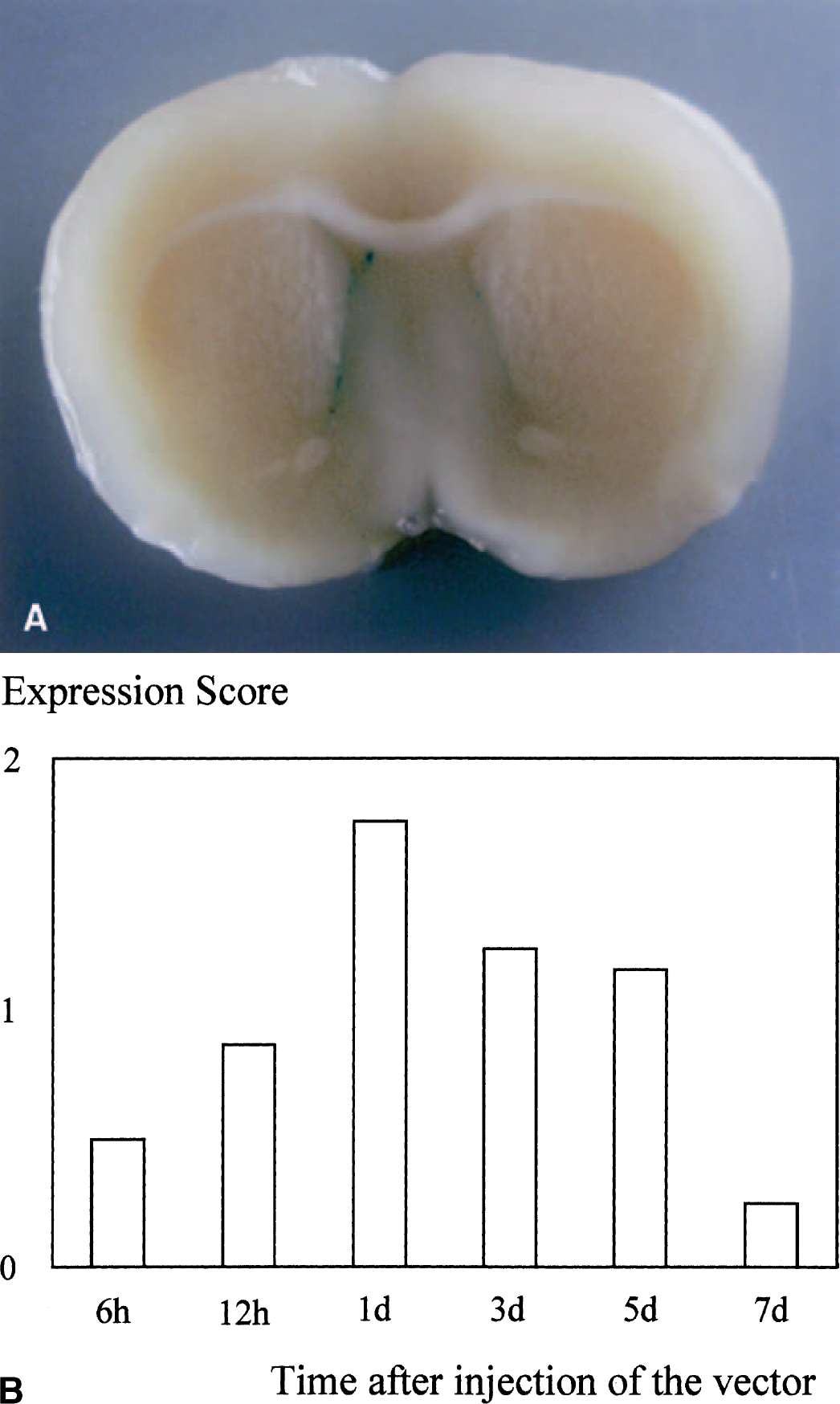
Transgene expression in rat brains after gene transfer. (
Measurement of 7ND
Values of ELISA for 7ND in the CSF from the Ad7ND group are shown in Fig. 2. A marked amount of 7ND was detected 6 hours after gene transfer (8,010 ± 1,965 pg/mL). Amounts of dominant negative MCP-1 were significantly increased at day 1 and day 5 (43,600 ± 866 and 19,467 ± 5,105 pg/mL, respectively) as compared with those seen at 6 hours (P < 0.01). In the AdlacZ group and in normal rats, 7ND was undetectable in the CSF.
Physiologic variables
Physiologic variables before and after ischemia in AdlacZ and Ad7ND groups are shown in Table 1. There were no significant differences in physiologic variables before and after ischemia between the two groups. Blood flow to the cortex on the occlusion side began to decrease within 10 minutes after focal ischemia and lasted for more than 60 minutes. CBF reductions in AdlacZ and Ad7ND group at 60 minutes were −62% ± 17% and −69% ± 10%, respectively. Changes in CBF were not significantly different between the two groups (Fig. 3).
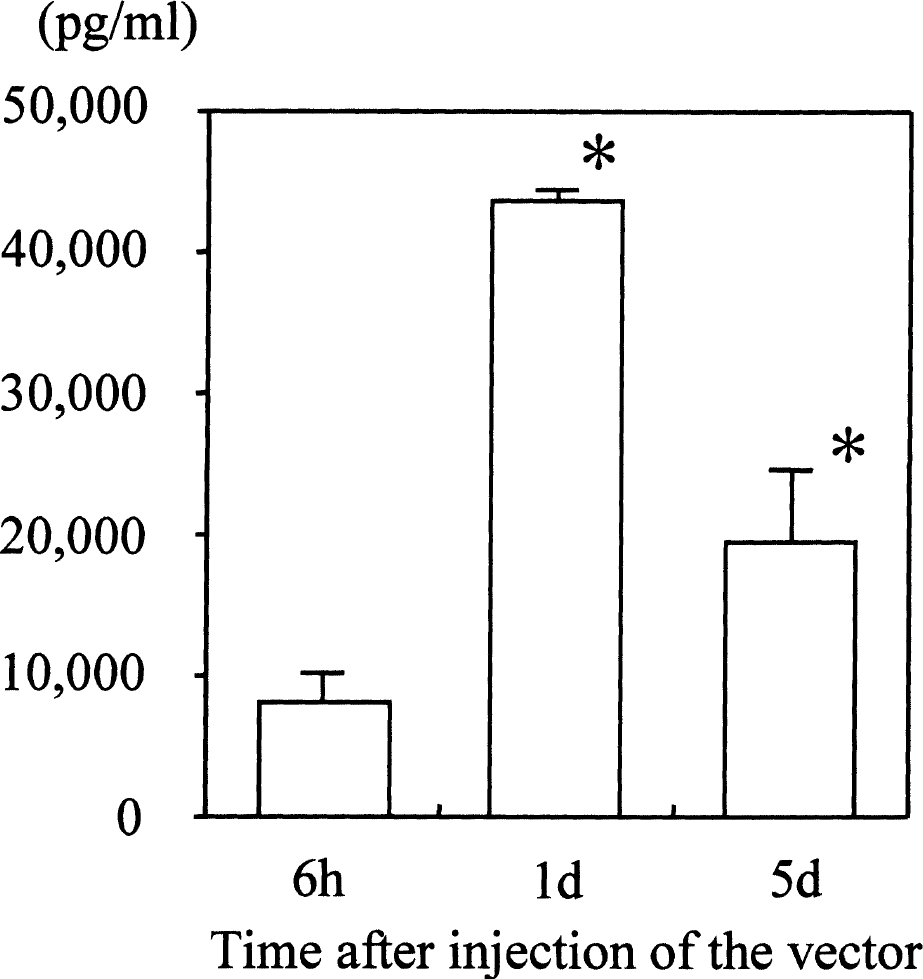
Amount of dominant negative MCP-1 (7ND) in the CSF after gene transfer. A marked amount of 7ND was detected 6 hours after gene transfer. The level of 7ND at days 1 and 5 was significantly increased as compared with that at 6 hours (P < 0.01). Values are mean ± SD. *P < 0.01 versus 6 hours.
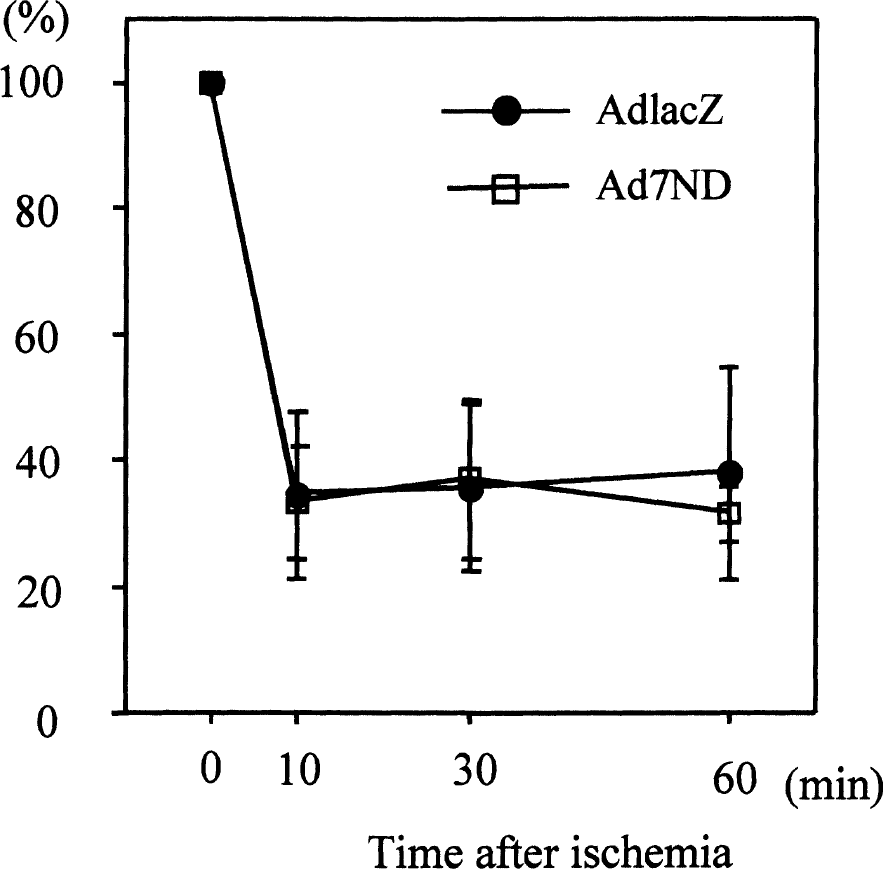
Changes in cerebral blood flow at parietal cortices during distal middle cerebral artery occlusion. Blood flow to the cortex on the occlusion side began to decrease within 10 minutes after focal ischemia and lasted for more than 60 minutes. Changes in CBF were not significantly different between the two groups. Values are mean ± SD.
Infarct volume and leukocyte infiltration
Infarct volume in the AdlacZ and Ad7ND groups is shown in Fig. 4. Infarct volume in the Ad7ND group (75 ± 13 mm3) was significantly smaller (by 28%) than that observed in the AdlacZ group (104 ± 22 mm3, P < 0.05). Numbers of total and mononuclear leukocytes in the vessels at the infarct area in the Ad7ND group were 48.3 ± 32.9/cm2 and 35.3 ± 27.7/cm2, and were significantly smaller than those measured in the AdlacZ group (143.8 ± 72.1/cm2 and 89.6 ± 40.9/cm2, respectively; P < 0.05) (Fig. 5). Numbers of polynuclear leukocytes in the vessels at the infarct area tended to be lower in the Ad7ND group (13.0 ± 10.1/cm2) than in the AdlacZ group (54.2 ± 13 /cm2, P = 0.07).
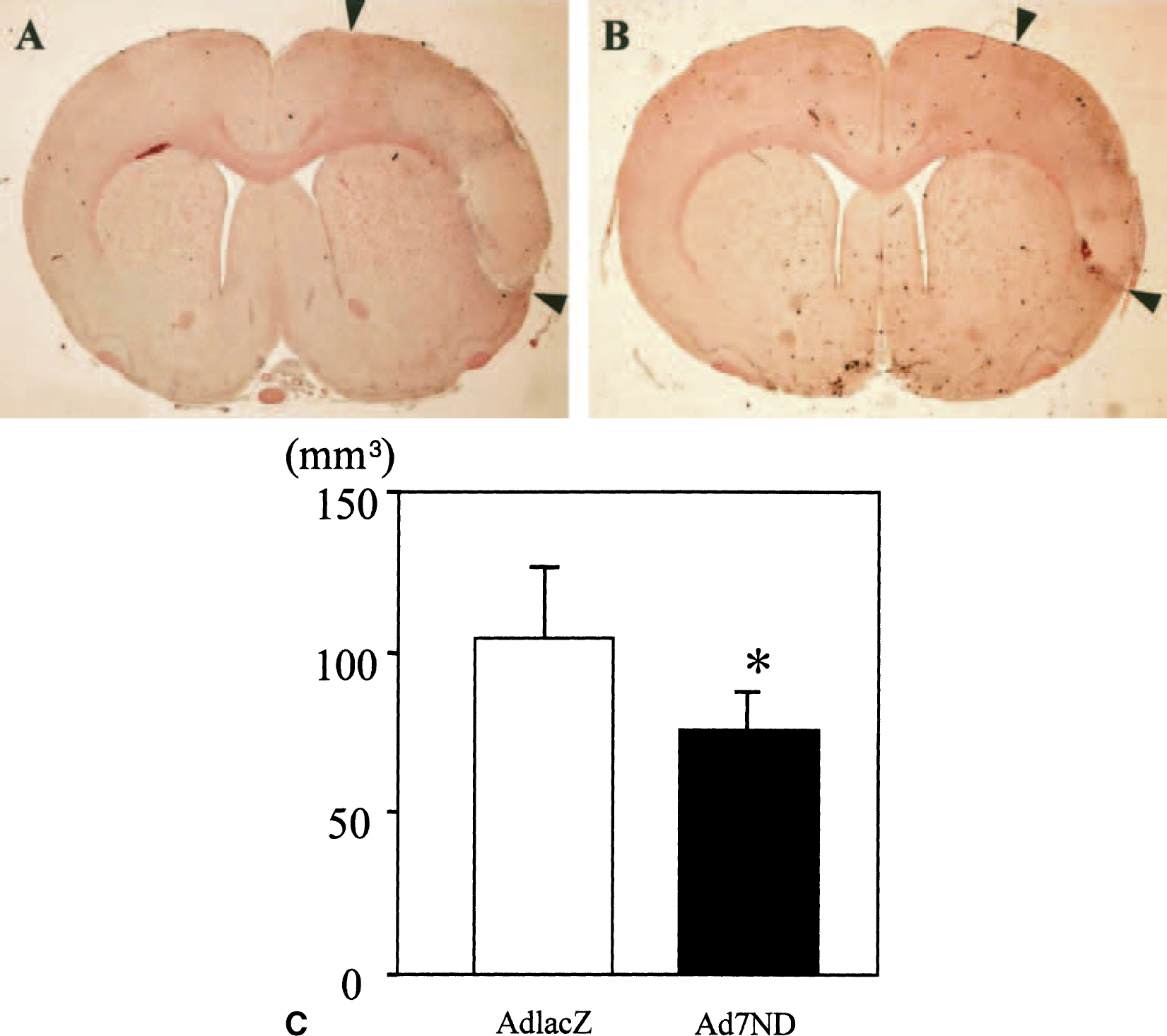
Brain infarction after gene transfer. Hematoxylin-eosin staining of rat brains 5 days after brain ischemia and gene transfer (
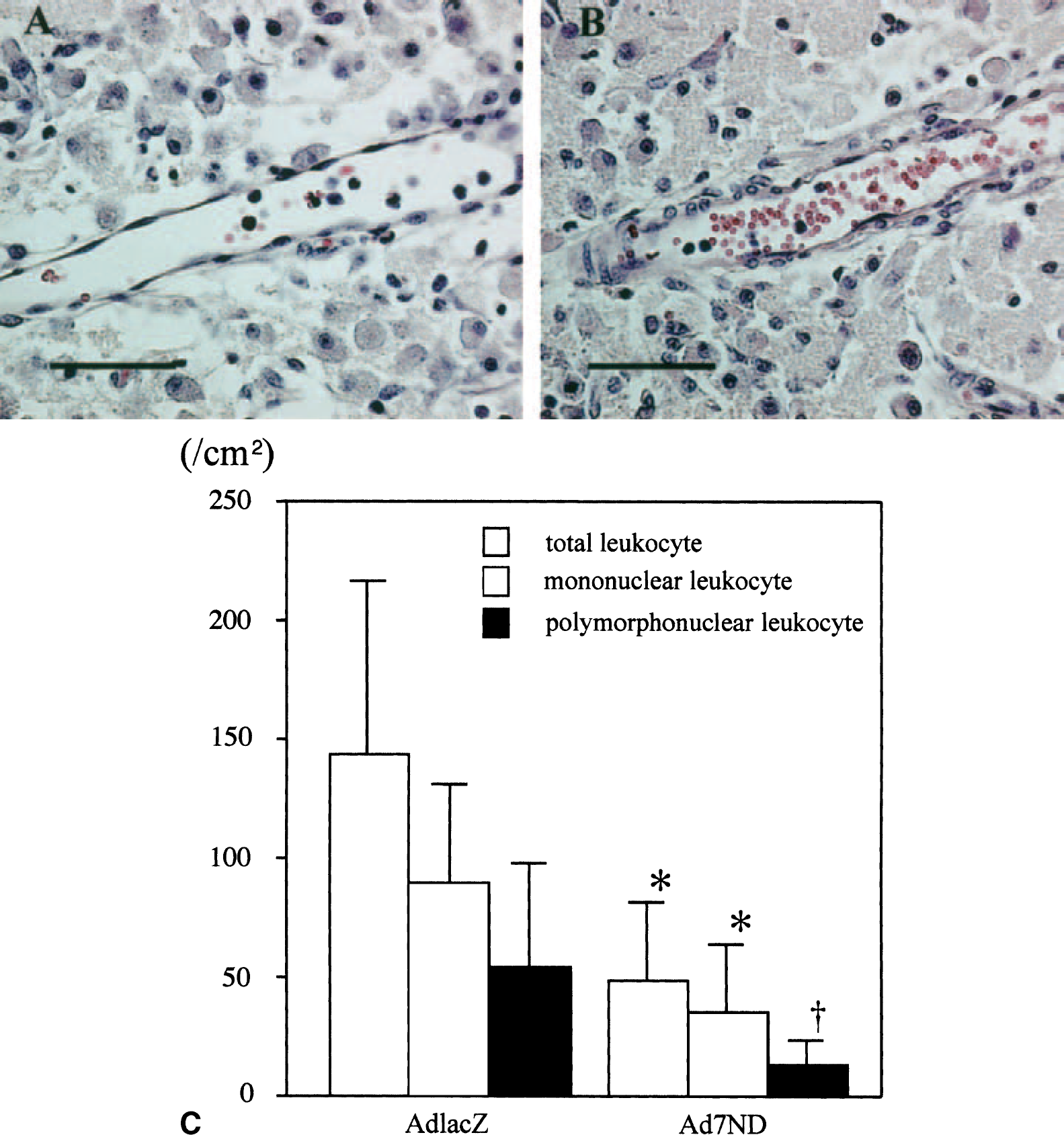
Leukocyte infiltration at the ischemic area after gene transfer. Hematoxylin-eosin staining of rat brains 5 days after brain ischemia and gene transfer (
Physiologic variables
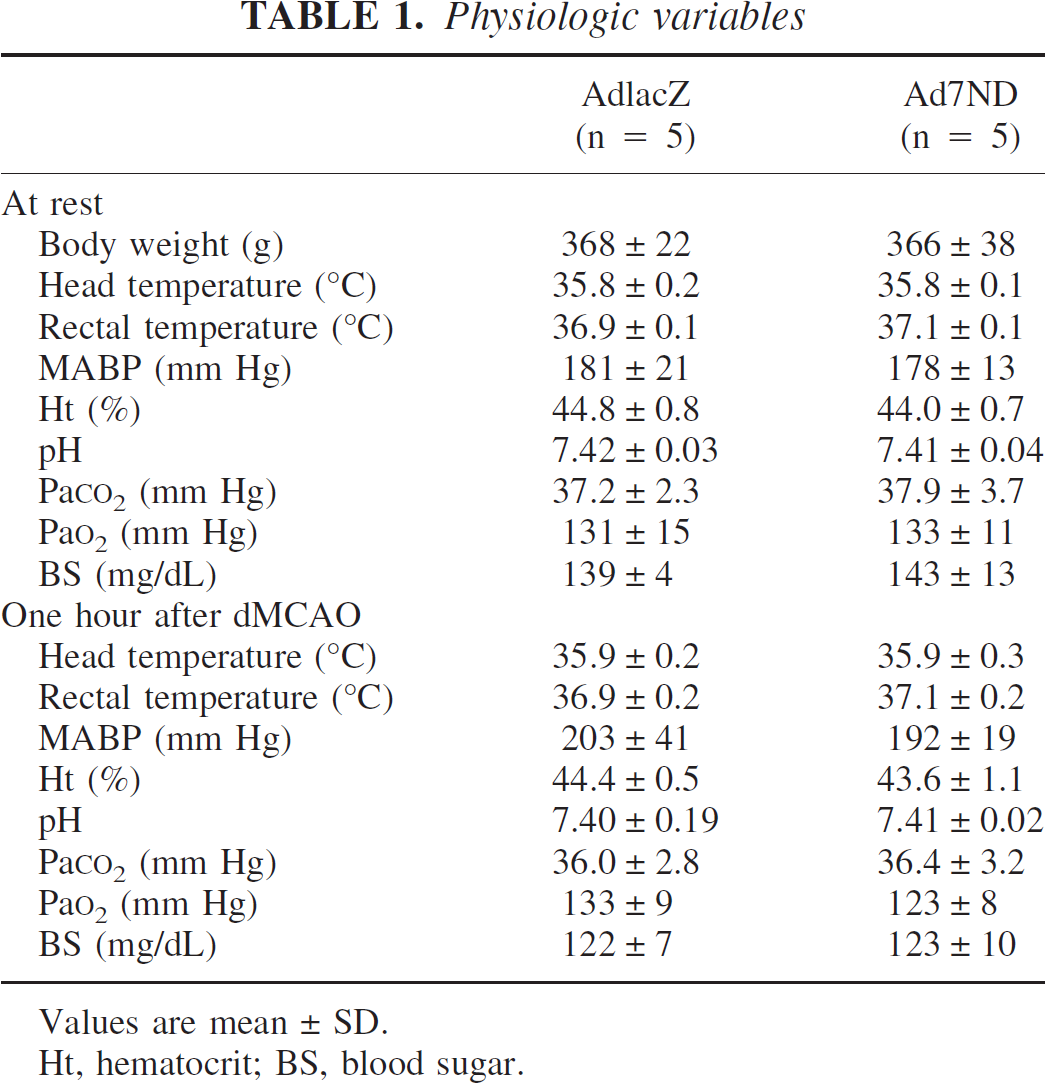
Values are mean ± SD.
Ht, hematocrit; BS, blood sugar.
Immunohistochemistry of macrophage
ED1-positive cells were predominantly located in the border of the ischemic area (Figs. 6A and 6B, brown). There were fewer ED1-positive cells in the infarct area of the Ad7ND group (475.2 ± 125.5/mm2) than in the AdlacZ group (671.8 ± 125.5 /mm2, P < 0.05) (Fig. 6E).
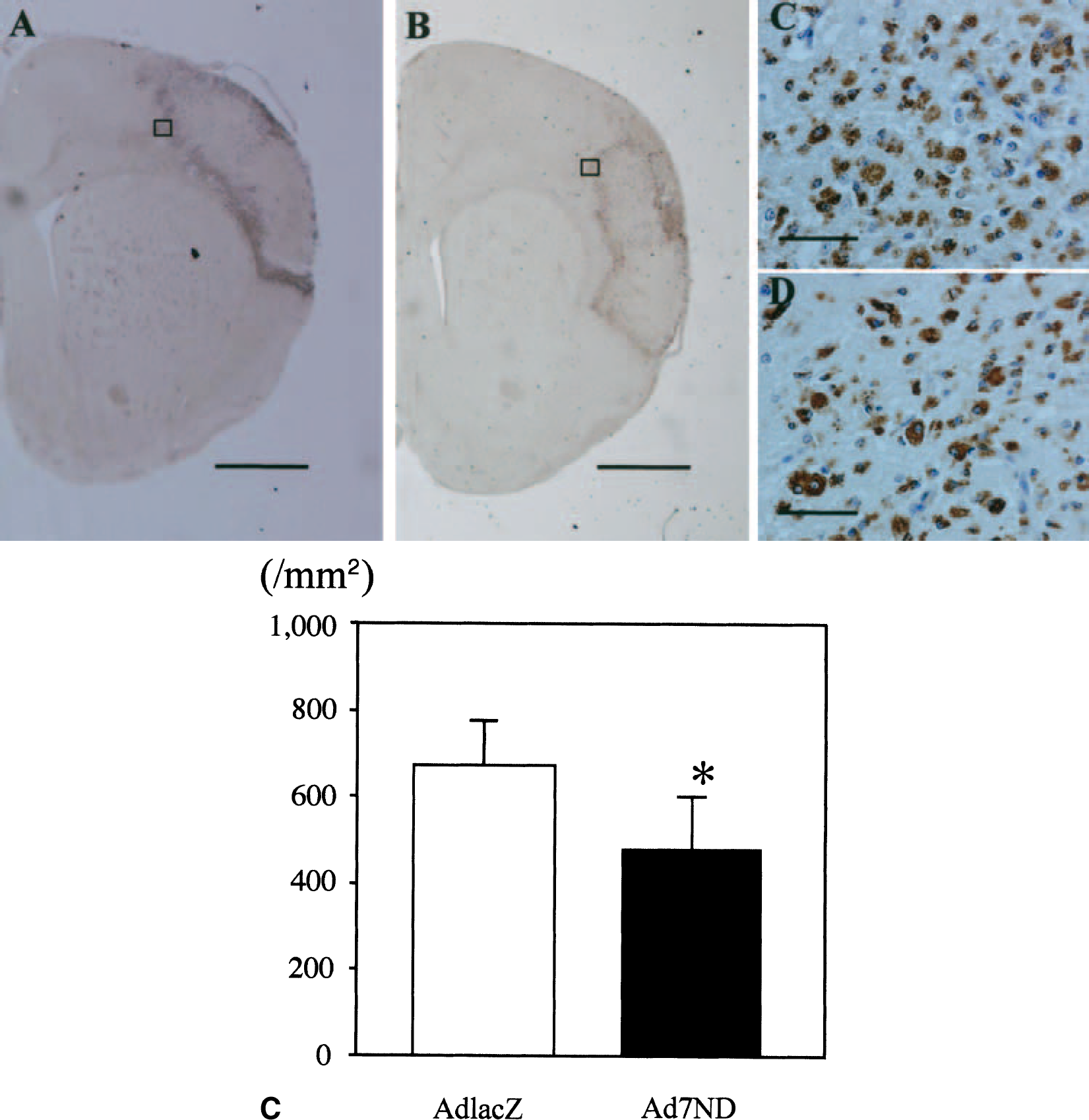
Macrophage infiltration 5 days after brain ischemia and gene transfer. Sections were immunochemically stained with antimacrophage Ab, ED1 (
DISCUSSION
In this study, gene transfer with adenoviral vectors into the lateral ventricle provided marked expression and release of transgene products in the CSF as early as 6 hours after gene transfer. Gene transfer of dominant negative MCP-1 reduced infarct volumes even when vectors were delivered after induction of focal brain ischemia, and the reduction of infarct size was associated with attenuations of both macrophage/monocyte and leukocyte infiltrations. Therefore, we clearly demonstrated the protective effect of anti–MCP-1 gene therapy against focal brain ischemia.
MCP-1 is involved in several inflammatory diseases, including rheumatoid arthritis (Koch et al., 1992), nephritis (Panzer and Stahl, 1999), infections (Dawson et al., 2000; Sato et al., 1999), and atherosclerosis (Egashira, 2003; Yla-Herttuala et al., 1991). These data indicate that MCP-1 is one of the major proinflammatory cytokines. In the central nervous system, MCP-1 was detected in the serum and CSF of patients with multiple sclerosis and the ischemic stroke (Franciotta et al., 2001; Losy and Zaremba, 2001). Previous studies have reported that MCP-1 deficiency in genetically altered mice and the nonpeptide C-C chemokine receptor antagonist TAK-779 reduced infarct volume and macrophage accumulations in the stroke model (Hughes et al., 2001; Takami et al., 2002), and that anti–MCP-1-neutralizing antibody attenuated N-methyl-
Recently, anti–MCP-1 gene transfer with dominant negative gene has shown protective effects in several experimental models, including rat vascular injury induced by chronic blockade of nitric oxide synthases (Egashira et al., 2000), atherosclerosis in apolipoprotein E–knockout mice (Ni et al., 2001), restenosis after coronary intervention in rats and monkeys (Usui et al., 2002), pulmonary hypertension in rats (Ikeda et al., 2002), renal ischemia reperfusion injury in mice (Furuichi et al., 2003), and myocardial infarction in mice (Hayashidani et al., 2003). Our study clearly shows a protective effect of dominant negative MCP-1 gene transfer in the focal ischemia model. Thus, anti–MCP-1 gene transfer may be useful in the treatment of acute brain ischemia.
One of the major mechanisms of neuroprotection by anti–MCP-1 gene therapy is the inhibition of monocyte/macrophage infiltrations. MCP-1 is involved in monocytic recruitment in several inflammatory setting in vivo, including the brain (Bell et al., 1996; Lu et al., 1998). Forty-eight hours after focal brain ischemia, transgenic mice overexpressing MCP-1 had more monocyte/macrophage infiltrations than control mice (Chen et al., 2003). We showed that 7ND gene transfer significantly attenuated monocyte/macrophage activity 5 days after stroke, which was associated with smaller infarcts as compared with the control group. Therefore, the beneficial effect appeared to be attributable to inhibition of monocyte/macrophage recruitment and activation.
We showed that anti–MCP-1 gene transfer significantly attenuated total and mononuclear leukocyte counts and tended to attenuate polynuclear leukocyte counts in the vessels at the infarct area. In a previous study, MCP-1–transgenic mice were reported to have a trend toward an increase in neutrophils in a distinct area surrounding intraparenchymal blood cells in the ischemic brain (Chen et al., 2003). Furthermore, anti–MCP-1-neutralizing antibody prevented neutrophil influx in newborn hyperoxia-exposed rats, and had a trend toward a reduction of cytokine-induced neutrophil chemoattractant (Vozzelli et al., 2003). Thus, the decrease in leukocyte infiltrations may also have contributed to neuroprotection in our study.
Proinflammatory cytokines, such as interleukin 1β (IL-1β) and tumor necrosis factor-α (TNF-α), are induced in response to brain ischemia (Arvin et al., 1996, Rothwell and Luheshi, 2000). Several studies have reported that injection of IL-1β in brain ischemia exacerbates brain damage (Loddick and Rothwell, 1996; Yamasaki et al., 1995). In contrast, IL-1β blockade and IL-1 deficiency cause a significant reduction of ischemic damage (Boutin et al., 2001; Yamasaki et al., 1995). TNF-α is another important mediator after brain ischemia, and may act as a pleiotropic peptide to elicit the production of IL-1β and adhesion molecules in brain ischemia (Ellison et al., 1999). Adhesion molecules are induced by IL-1β and TNF-α (Hess et al., 1994) and play a pivotal role in leukocyte infiltrations (Danton and Dietrich, 2003). IL-1β was induced by MCP-1 in human monocytes (Jiang et al., 1992), and the induction after focal brain ischemia was significantly reduced in MCP-1–knockout mice (Hughes et al., 2001). 7ND gene transfer significantly reduced several cytokines, including IL-1β, TNF-α, interleukin 6, transforming growth factor β, and MCP-1 in aorta (Inoue et al., 2002, Ni et al., 2004) and TNF-α, transforming growth factor β, and MCP-1 in ischemic heart from 1 to 7 days after myocardial infarction (Hayashidani et al., 2003). Therefore, the anti-MCP-1 approach appears to suppress the induction of proinflammatory cytokines and thereby inhibit the upregulation of adhesion molecules and inflammatory cell infiltrations, which would result in attenuation of tissue injury by brain ischemia.
In this study, 7ND gene transfer into cerebral ventricle reduced the infarct volume and provided marked expression of transgene in the CSF as early as 6 hours after gene transfer. Administration of the adenovirus into the lateral ventricle has produced extensive expression in the ependymal cells (Bajocchi et al., 1993; Ooboshi et al., 1995), and preinjection of adenovirus carrying IL-1 receptor antagonist into the cerebral ventricle reduced infarct volume (Betz et al., 1995; Yang et al., 1997). Therefore, the ependyma may be a good target for gene therapy of stroke. In other studies using 7ND gene transfer, intramuscular transfection of the 7ND resulted in marked secretions of 7ND protein into the circulating blood, which bound to the MCP-1 receptor on monocytes or target cells and, thus, achieved an effective blockade of MCP-1 activity in remote organs (Egashira, 2003). Although 7ND gene transfer to skeletal muscle has obstacles of the blood–brain barrier, the delivery method may be applicable to ischemic stroke where the barrier is disturbed.
In our study, gene transfer initiated 90 minutes after stroke reduced the volume of brain infarction. Although several studies reported protective effects of postischemic gene therapy (Hayashi et al., 2001; Hoehn et al., 2001; Lawrence et al., 1997; Shimazaki et al., 2000; Zhang et al., 2002), those studies were examined with the transient brain ischemia model, and the report that achieved reductions of brain infarct volume in the permanent ischemia model was limited. The therapeutic time window of postischemic gene transfer lasted up to 90 minutes in our study, but it did not by 150 minutes after ischemia in another study (Zhang et al., 2002). Therefore, it is necessary to examine a longer therapeutic time window in our model. Combination of gene therapy and protein may increase the therapeutic time window, because neurotrophic peptide may have a neuroprotective effect up to 150 minutes after ischemia (Zhang et al., 2001).
The focal brain ischemia produced by photochemical occlusion of the distal MCA of SHR provides small variations in infarct volume without extensive surgery (Yao et al., 2003). However, one of drawbacks in our model is difficulty in the assessment of neurologic function, because this model shows small neurologic deficit due to the relatively confined infarction to the cortex. Although the pathologic features of our model are similar to those of other models (Yao et al., 1996) and suitable for examination of cytoprotective drugs (Cai et al., 1998), further examination, including the use of primate models, is inevitable before clinical trials can be started.
In conclusion, postischemic gene transfer of dominant negative MCP-1 protects against focal brain ischemia, which is related to the reduction of both macrophage/monocyte and leukocyte infiltrations. Anti–MCP-1 gene therapy may be a promising approach for the treatment of brain ischemia.