
Abstract
Glucose had long been thought to fuel oxidative metabolism in active neurons until the recently proposed astrocyte-neuron lactate shuttle hypothesis (ANLSH) challenged this view. According to the ANLSH, activity-induced uptake of glucose takes place predominantly in astrocytes, which metabolize glucose anaerobically. Lactate produced from anaerobic glycolysis in astrocytes is then released from astrocytes and provides the primary metabolic fuel for neurons. The conventional hypothesis asserts that glucose is the primary substrate for both neurons and astrocytes during neural activity and that lactate produced during activity is removed mainly after neural activity. The conventional hypothesis does not assign any particular fraction of glucose metabolism to the aerobic or anaerobic pathways. In this review, the authors discuss the theoretical background and critically review the experimental evidence regarding these two hypotheses. The authors conclude that the experimental evidence for the ANLSH is weak, and that existing evidence and theoretical considerations support the conventional hypothesis.
Until recently, glucose had stood unchallenged as the principal metabolic substrate of the mature brain (for example, see McIlwain and Bachelard, 1985; Sokoloff, 1989). The primacy of glucose, though, has been questioned recently by some investigators who view lactate as a substrate that active neurons prefer over glucose (Magistretti, 1999; Magistretti et al., 1999). The brain can consume lactate as a substrate, as has been demonstrated by studies showing that the brain uses lactate during hypoglycemia or during periods of elevated blood lactate (Ide et al., 2000; Nemoto et al., 1974). Also, some in vitro studies have shown that lactate supports neural activity in brain tissues in the absence of glucose (e.g., Izumi et al., 1997; Schurr et al., 1988). However, because lactate does not pass through the blood-brain barrier nearly as well as glucose (e.g., McIlwain and Bachelard, 1985), lactate cannot serve the brain as a blood-borne substrate the way glucose does. In the mid 1990s, an astrocyte-neuron lactate shuttle hypothesis (ANLSH) was proposed that assigned a major metabolic role to brain-derived lactate (Pellerin and Magistretti, 1994; Tsacopoulos and Magistretti, 1996). According to this hypothesis, lactate is produced in an activity-dependent and glutamate-mediated manner by astrocytes and is then transferred to and used by active neurons (Pellerin et al., 1998a). In greater detail, the ANLSH postulates that neuronal activation increases the extracellular concentration of brain glutamate, which astrocytes take up via high affinity, Na+-dependent transporters. Increases in glutamate and Na+ in astrocytes activate glutamine synthetase and Na+-K+ ATPase, respectively. Activation of Na+-K+ ATPase spurs astrocytic ATP consumption. This leads to activation of anaerobic glycolysis 1 in astrocytes, and the resulting lactate is transported out of astrocytes and into neurons, where it fuels the activity-related energy needs of neurons (for reviews, see Bouzier-Sore et al., 2002; Magistretti, 1999, 2000; Magistretti and Pellerin, 1999; Magistretti et al., 1999). As currently expressed, the ANLSH applies only to glutamatergic neurons and does not address how activity-related energy demands are met in nonglutametergic neurons.
Early support for the ANLSH came from a study by Pellerin and Magistretti (1994) that showed that glutamate increased 2-deoxy-D-[1,2-3H]glucose uptake and lactate release in cultured astrocytes. Later, Magistretti and colleagues (Bittar et al., 1996) proposed that the distribution of lactate dehydrogenase (LDH) isoforms in brain cells might favor lactate oxidation in neurons. Also, results from several other studies were used as evidence that lactate may be preferred over glucose as an energy substrate for the brain (e.g., Hu and Wilson, 1997; Larrabee, 1995; Poitry-Yamate et al., 1995; Schurr et al., 1999). In 1998, Shulman and coworkers (Sibson et al., 1998) found a 1:1 stoichiometry between oxidative glucose metabolism and glutamate cycling. Magistretti and coworkers (Magistretti, 1999; Magistretti and Pellerin, 1999; Magistretti et al., 1999) used this finding to suggest that glucose is used predominantly, if not exclusively, by astrocytes and that neurons use lactate released from astrocytes as their primary substrate during activity. In 2002, Magistretti and colleagues (Bouzier-Sore et al., 2002) reviewed the evidence supporting the ANLSH and proposed again that astrocytes provide lactate as an energy substrate for neurons, especially during periods of enhanced synaptic activity.
The conventional view of glucose metabolism (Fig. 1) and the ANLSH (Fig. 2) differ most noticeably in (1) the sites and modes of glucose use, (2) the sites of lactate production and use, and (3) the timing of lactate use. The conventional hypothesis contends that activity-induced energy demand is met predominantly by metabolizing glucose oxidatively (e.g., Chih et al., 2001b; Dienel and Hertz, 2001; McIlwain and Bachelard, 1985; Sokoloff, 1989). In the conventional view, increased Na+-K+ ATPase activity in brain cells resulting from neural activation causes increased ATP use, which activates the glycolytic pathway and increases oxidative metabolism in both neurons and astrocytes. Production of lactate in the brain during neural activity is considered to be a result of glycolytic activity transiently exceeding the rate of oxidative metabolism (e.g., Prichard et al., 1991). In this view, a buildup of lactate is viewed as potentially detrimental (e.g., Prichard et al., 1991; Siesjö, 1982); therefore, lactate must be either removed via the circulation or consumed by inactive brain cells after neural activity (e.g., Chih et al., 2001a, 2001b). The conventional hypothesis also maintains that both neurons and astrocytes can produce and use lactate (Dringen et al., 1993a; Chih et al., 2001b; Dienel and Hertz, 2001). However, lactate is used only when the prevailing glycolytic rate is low.
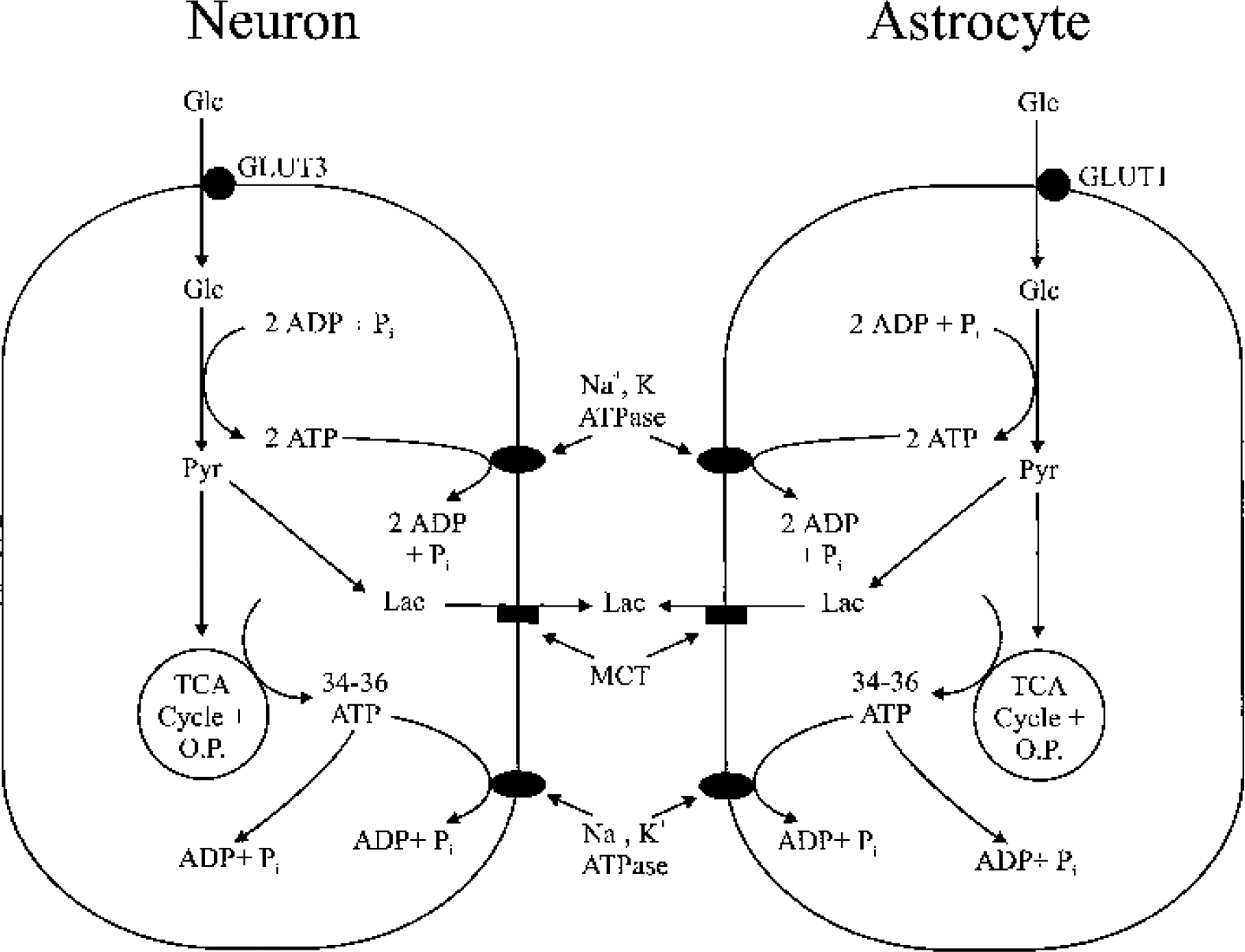
Schematic illustration of glucose metabolism during neural activity in neurons and astrocytes according to the conventional hypothesis. According to this hypothesis, glucose use increases in both neurons and astrocytes during activity. Heightened Na+-K+ ATPase activity in both cell types during neural activity increases ATP consumption and stimulates glucose use by activating glycolytic enzymes. Glucose is transported into neurons and astrocytes via the glucose transporters GLUT3 and GLUT1, respectively. Glucose is metabolized to pyruvate (hereafter designated as glycolysis) to produce 2 ATPs in either cell type. Pyruvate is either converted to lactate (anaerobic glycolysis) by LDH or taken up by mitochondria, where it enters the TCA cycle and is metabolized via oxidative metabolism. Lactate produced by both neurons and astrocytes is transported to the extracellular space via MCT. Increased energy demands resulting from neural activity are met by ATP generated from both glycolysis and oxidative metabolism. The conventional hypothesis does not ascribe any particular fraction of glucose metabolism to either the aerobic or anaerobic pathways. O.P., oxidative phosphorylation; Glc, glucose; Pyr, pyruvate; LDH, lactate dehydrogenase; Lac, lactate; MCT, monocarboxylate transporters.
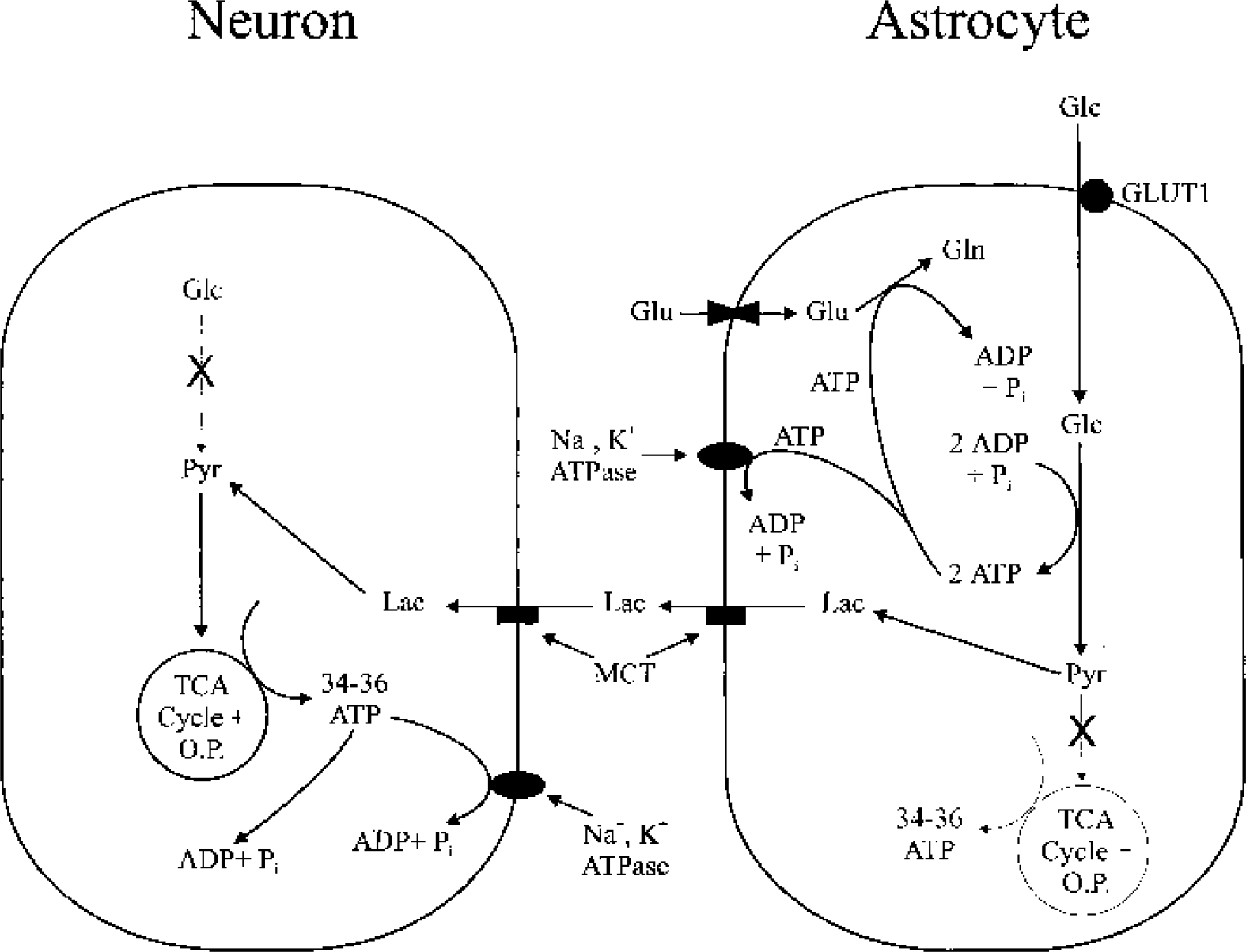
Schematic illustration of glucose metabolism during neural activity in neurons and astrocytes, according to the ANLSH. Activation of neurons causes release of glutamate, which is then taken up by astrocytes. Cotransport of Na+ with glutamate into astrocytes (not shown) activates astrocytic Na+-K+ ATPase, which in turn stimulates ATP consumption and activates glycolytic enzymes in astrocytes. According to the ANLSH, activity-induced glucose uptake occurs predominantly, if not exclusively, in astrocytes, where glucose is metabolized anaerobically to lactate. The two ATPs produced by anaerobic glycolysis are used for glutamine synthesis and Na+-K+ ATPase activity. Lactate produced by astrocytes is released to the extracellular space and taken up by neurons to fuel neuronal oxidative metabolism. One stipulation of the ANLSH is that glutamate cycling, which includes uptake of glutamate and conversion of glutamate to glutamine, is fueled entirely by astrocytic anaerobic glycolysis, and is the driving force for glucose use during neural activity. The ANLSH also contends that active neurons prefer to use lactate released from astrocytes rather than ambient glucose as an energy substrate. X's in metabolic pathways emphasize that, for the ANLSH, increased neural activity does not activate neuronal glycolysis, and that activity-induced increases in astrocytic energy metabolism are nonoxidative. ANLSH, astrocyte-neuron lactate shuttle hypothesis; Glu, glutamate; Gln, glutamine; O.P., oxidative phosphorylation; Glc, glucose; Pyr, pyruvate; LDH, lactate dehydrogenase; Lac, lactate; MCT, monocarboxylate transporters.
In contrast, the ANLSH proposes that activity-induced increases in glucose uptake occur mainly in astrocytes and that astrocytes consume glucose anaerobically to produce lactate (Magistretti, 1999; Magistretti et al., 1999). The ANLSH postulates that neural activity increases anaerobic glycolysis in astrocytes without enhancing the neuronal glycolytic pathway (Magistretti et al., 1999) and that, during activity, astrocytes release lactate destined to fuel oxidative metabolism in active neurons. Thus, with regard to lactate use, the two hypotheses not only differ in the timing of lactate use and in the cell types using lactate, but also in the rationale for why lactate is produced and used in the brain.
The features of both hypotheses deserve a deeper examination, given the fundamental importance of glucose use to our understanding of brain energy metabolism and the widespread acceptance of the ANLSH by neuroscientists. In this review, we discuss the theoretical background and critically review the experimental evidence regarding the conventional hypothesis and the ANLSH. We find little convincing experimental support for the ANLSH and that existing evidence and theoretical considerations support the conventional hypothesis that brain cells, including neurons, consume glucose directly to meet their activity-related energy needs. In Part I we explain, from the perspectives of enzyme kinetics, thermodynamics, and substrate availability, that glucose is probably the primary substrate used by neurons during activity. In Part II we critically examine current experimental evidence to evaluate the validity of the two hypotheses. In Part III we summarize the role of lactate as an energy substrate for the brain.
PART I: COMPARISON OF GLUCOSE AND LACTATE AS ENERGY SUBSTRATES FOR NEURONS DURING ACTIVITY
Availability of glucose and lactate to neurons during activity
The contention of the ANLSH that neural activity does not stimulate the neuronal glycolytic pathway is valid if glucose is not available to neurons during activity or if neurons have little capacity to use glucose. However, although neuronal processes are not directly coupled to the bloodstream as astrocyte processes are, much indirect evidence suggests that glucose is readily available to and easily used by neurons. First, synaptic membranes have glucose transporters in abundance (Leino et al., 1997; McCall et al., 1994). Second, neurons have GLUT3 as their dominant glucose transporter. GLUT3 transports glucose seven times faster than GLUT1, which is the dominant glucose transporter in astrocytes (Vannucci et al., 1997). Third, glucose, which is evenly distributed between the intra- and extracellular compartments of the brain (Pfeuffer et al., 2000), is present at extracellular concentrations that are well above the Km for hexokinase (0.04 mM) (Lowry and Passonneau, 1964). This is true whether the brain is at rest or stimulated in vivo (see data of Fray et al., 1996; Hu and Wilson, 1997). For example, resting extracellular glucose concentrations measured directly with glucose-sensitive microelectrodes (Hu and Wilson, 1997; Silver and Erecinska, 1994) are 2.4 to 2.6 mM. Fourth, neurons have high activities of glycolytic enzymes (Cimino et al., 1998; Lai et al., 1999; Kao-Jen and Wilson, 1980; Wilson, 1972). Finally, a recent in vivo study (Wittendorp-Rechenmann et al., 2001) showed that 14C-labeled 2-deoxyglucose (2-DG) was taken up approximately equally by both astrocytes (42% to 58%) and neurons (54% to 62%). Many in vitro studies also have shown that both neurons and astrocytes can use glucose as a substrate (Itoh et al., 2003; Peng et al., 1994; Vicario et al., 1993; Walz and Mukerji, 1988) and that neurons may increase drastically their glucose use during periods of heightened energy demand (Peng et al., 1994; Walz and Mukerji, 1988). Thus the existing evidence suggests that neurons use glucose directly during neural activity in situ.
Neurons and astrocytes also possess the cellular machinery necessary for using lactate. For example, both cell types have LDH (Bittar et al., 1996). In addition, both neurons and astrocytes have monocarboxylate transporters (MCTs) (Broer et al., 1997; Pierre et al., 2002). However, as discussed below, the lack of a mechanism to activate LDH and slow increases in lactate levels during activity hinder the use of lactate during activity.
Responses of glycolytic enzymes and LDH to increases in energy demand during neural activity
Lactate must compete with glucose for use if glucose is readily available to neurons. Because energy demands fluctuate rapidly between active and inactive states in neurons, the key question regarding substrate use is whether glucose or lactate metabolism offers a quicker response to sudden increases in energy demand. Glucose metabolism via the glycolytic pathway appears well adapted to meet increased cellular energy demands because it has the important characteristic of being tightly coupled to energy demand (Clarke et al., 1989). For example, during periods of low energy demand, the glycolytic rate is low because key glycolytic enzymes, including hexokinase and phosphofructokinase, are inhibited by high ATP levels (Clarke et al., 1989). For brain hexokinase, this inhibition is 97% for resting conditions (Clarke et al., 1989). This basal inhibition gives neurons the potential to respond quickly to increased energy demands. When neurons are active, increased intracellular Na+ and extracellular K+ stimulate neuronal ATP use via Na+-K+ ATPase. The resulting increases in adenine nucleotide and phosphate levels activate the glycolytic pathway and oxidative metabolism at several key control points. The arrangement of these control points allows increased rates of glucose use without increases in glucose concentrations (Peng et al., 1994; Walz and Mukerji, 1988).
In contrast to the responsiveness of glycolytic enzymes to energy status, increased energy demand does not activate LDH. Thus, increased rates of lactate dehydrogenation require either increases in lactate levels or decreases in pyruvate levels. Given that pyruvate levels increase during elevated neural activity (Ferrendelli and McDougal, 1971; Goldberg et al., 1966), extracellular lactate levels must first go up for neuronal lactate use to increase. This requirement has been demonstrated in an in vitro study of cultured cerebellar neurons (Peng et al., 1994). In this study, substrate concentrations were held constant, and energy demand was increased by raising the extracellular K+ concentration. The rate of lactate oxidation in the cultured neurons did not increase significantly with increased K+. In contrast, both glucose and pyruvate oxidation increased significantly with elevated K+, despite unchanged ambient glucose and pyruvate levels. Thus, rapid increases in extracellular lactate must occur if lactate is to fuel the increased neuronal oxidative metabolism seen with elevated neural activity.
However, increases in extracellular lactate in situ lag behind neural activity (Fellows et al., 1993; Fray et al., 1996; Hu and Wilson, 1997) and are sometimes not seen until after activity has ended (Hu and Wilson, 1997). For example, extracellular lactate does not increase in the rat brain for the first 2.5 minutes of a 5 minute tail pinch stimulation (Fellows et al., 1993). By the end of the 5 minute stimulation, lactate levels had increased 40% to 50% and did not peak (55% to 80%) until approximately 5 minutes later. When extracellular lactate changes were examined in the hippocampus with lactate-sensitive electrodes (Hu and Wilson, 1997), lactate levels began increasing only 10 to 12 seconds after 5 seconds of electrical stimulation had ended. Lactate peaked (40% to 100% above control) after another 50 to 60 seconds.
The increases in extracellular lactate noted in the preceding studies are far too slow to meet cellular metabolic needs resulting from neural activity. For example, the half-life for the clearance of extracellular K+ in hippocampal slices after they have been intensely activated electrically is only approximately 5 seconds (Roberts, 1993; Roberts and Feng, 1996). Also, as noted from hemoglobin desaturation, oxygen use during neural activity in situ increases rapidly within the first 1 to 3 seconds of activity (Malonek and Grinvald, 1996; Malonek et al., 1997). Because it rises slowly, extracellular lactate probably cannot fuel these rapid responses. Thus, the available physiological evidence tends not to support lactate's use as an energy substrate early during neural activity.
Unlike lactate, glucose use in neurons can rise rapidly because key glycolytic enzymes are highly sensitive to declines in cellular energy status, and rate changes only require the diffusion of small metabolites within the cell. Many studies have shown that neurons can increase their glycolytic rates in response to increased energy demands (e.g., Peng et al., 1994; Sokoloff, 1999; Walz and Mukerji, 1988). Measurements of oxygen-glucose metabolism in the human visual cortex also suggest that, during visual stimulation, oxidative metabolism and glycolysis in neurons increase concurrently (Gjedde and Marrett, 2001; Gjedde et al., 2002). Therefore, the available evidence suggests that neural activity should activate the glycolytic pathway in both neurons and astrocytes and not in astrocytes alone, as the ANLSH proposes (Magistretti, 1999; Magistretti et al., 1999).
Suppression of lactate oxidation by increased glycolysis
The above analysis suggests that the faster activation of the glycolytic pathway makes glucose more likely than lactate to be the substrate used during the initial stages of neural activity. An additional question is whether lactate can become an important energy source for neurons during prolonged activation when lactate levels increase. Successful use of lactate during prolonged activation would come at the expense of glucose because both lactate dehydrogenation and glycolysis need NAD+ (Fig. 3). Also, the cotransport of lactate and H+ (Schneider et al., 1993) into neurons would cause acidification, which may inhibit glycolysis.
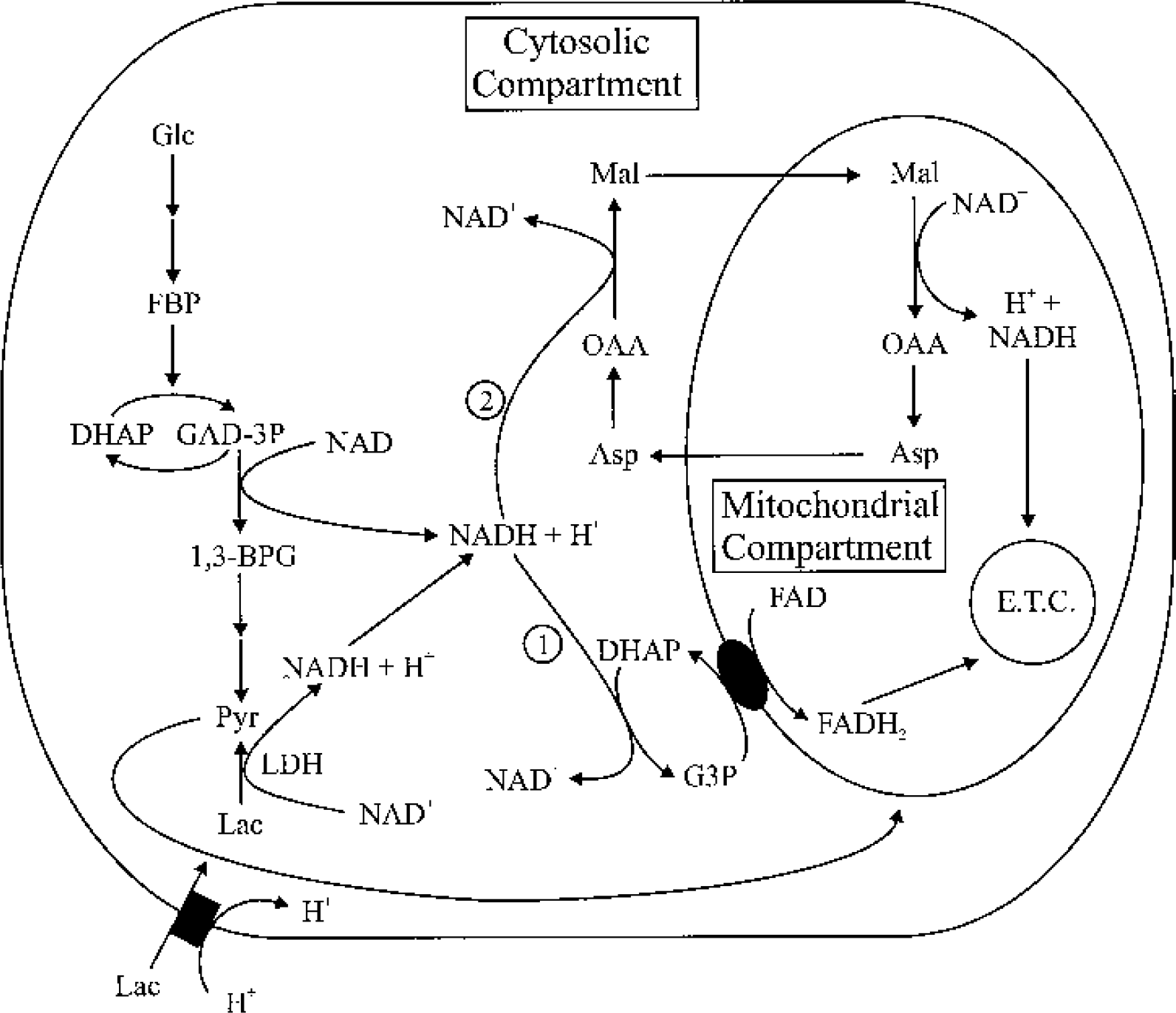
Schematic illustration of mechanisms for maintaining the redox balance when lactate is the energy substrate. NADH generated from lactate dehydrogenation can only be reoxidized to NAD+ via (1) the G3P shuttle and (2) the malate shuttle. In areas where mitochondria are rare, such as in dendritic spines (see text), NADH generated by LDH must diffuse to areas where mitochondria are present. When energy demand increases, no mechanism is available to increase the activity of the G3P shuttle if lactate is the energy substrate. Also, NADH generated by LDH must compete with NADH generated via the action of glyceraldehyde-3-phosphate dehydrogenase for access to the two NADH shuttles. G3P, glycerol-3-phosphate; LDH, lactate dehydrogenase; Asp, aspartate; E.T.C., electron transport chain; FBP, fructose-1,6-bisphosphate; Mal, malate; OAA, oxaloacetate; GAD-3P, glyceraldehyde-3-phosphate; 1,3-BPG, 1,3-biphosphoglycerate; DHAP, dihydroxyacetone phosphate.
The key issue for brain cells using lactate is the thermodynamic feasibility for conversion of lactate to pyruvate. In the initial stages of neural activation, as glycolytic rates increase, pyruvate (Ferrendelli and McDougal, 1971; Goldberg et al., 1966) and H+ (Chesler and Kaila, 1992; Hochachka and Mommsen, 1983) levels go up, and the cytosolic NAD+/NADH ratio decreases (Clarke et al., 1989). These changes drive the LDH-catalyzed reaction toward lactate production rather than lactate use. Also, changes in extracellular lactate during activity may fall within a range of only 0% to 135% (see Fellows et al., 1993; Fray et al., 1996; Hu and Wilson, 1997) above its resting levels (0.6 to 1.37 mM) (Harada et al., 1992; Prichard et al., 1991) during activity. Thus, even if lactate increases during neural activity, whether it rises to levels that can create conditions thermodynamically favorable for driving the LDH-catalyzed reaction in neurons toward lactate oxidation remains unclear. Even if conditions were favorable for lactate oxidation in neurons during activity, LDH must still compete with glyceraldehyde-3-phosphate (P) dehydrogenase (GAPDH) for NAD+ (Fig. 3). GAPDH, like LDH, is present at high concentrations in brain cells (McIlwain and Bachelard, 1985). The increased glyceraldehyde-3-P levels that occur with elevated glycolytic activity increase the activity of GAPDH (Williamson, 1965). Because the conversion of glucose to pyruvate, which produces two net ATPs, is more favorable energetically than the conversion of lactate to pyruvate, it is doubtful that lactate use will occur at the expense of glucose use during periods of increased energy demand.
As discussed in Part II, little unequivocal experimental evidence exists for active brain areas using lactate during neural activity. Also, if lactate levels are high enough to inhibit glycolysis, then glycolysis in both neurons and astrocytes would be inhibited, leaving open the possibility that both cell types would use lactate. Indeed, both neurons and astrocytes can use lactate as an energy substrate (Peng et al., 1994; Vicario et al., 1993). In fact, the high sensitivity of LDH-1, the dominant LDH isoform in neurons (Bittar et al., 1996), to pyruvate product inhibition (Stambaugh and Post, 1966; also see Part I, The LDH isoform distribution) may help suppress lactate use in neurons when glycolytic rates increase and pyruvate levels go up. Thus, if lactate is being used during activity, it may be more likely that astrocytes are using it because astrocytes have both LDH-1 and LDH-5 and because LDH-5 has a lower sensitivity to pyruvate product inhibition than LDH-1.
As indicated in Part II, little unambiguous experimental evidence exists for the shuttling of lactate from astrocytes to neurons during neural activity. The observation that lactate levels diminish only after neural activity in most in vivo studies supports the idea that lactate is used in the brain mainly after neural activity instead of during activity, as postulated by the ANLSH (Bouzier-Sore et al., 2002; Pellerin et al., 1998a).
Thermodynamics of glucose oxidation and lactate oxidation
ANLSH supporters also have proposed that lactate oxidation is favored over glucose oxidation in neurons because ATP consumption at the hexokinase (HK) and phosphofructokinase (PFK) steps of glycolysis imposes an energetic disadvantage (Magistretti, 1999; Schurr and Rigor, 1998). However, the reactions catalyzed by HK and PFK are thermodynamically very favorable and are essentially irreversible under most physiological conditions (Lehninger et al., 1993). In contrast, as seen in the preceding section, the thermodynamics of conversion of lactate to pyruvate may not favor the occurrence of this reaction during neural activity. Also, declining ATP levels greatly activate both HK and PFK (Clarke et al., 1989) but not LDH. In addition, conversion of glucose to pyruvate produces two net ATPs, whereas conversion of lactate to pyruvate produces no ATP. These factors favor glucose use during times of increased ATP consumption. Furthermore, because HK and PFK have Km levels for ATP (0.13 mM for HK and 0.025 mM for PFK) (Lowry and Passonneau, 1964) that are well below resting ATP levels (approximately 3 mM) (McIlwain and Bachelard, 1985), fluxes through these two reactions are affected only if ATP drops to extremely low levels, such as under pathological conditions. Thus, from a thermodynamic standpoint, neurons gain an energetic advantage by oxidizing glucose instead of lactate once increased energy demand has activated key glycolytic enzymes. In fact, many cell types usually respond to increased energy demands by activating glycolysis, including cultured neurons (Peng et al., 1994; Walz and Mukerji, 1988) and astrocytes (Pellerin and Magistretti, 1994).
Glycolysis as an essential energy source for localized areas of brain cells
Glycolysis may generate ATP locally in both neurons and astrocytes for ion pumping and other processes (Wu et al., 1997). Glycolytically derived ATP may be particularly important for synapses. This is especially true for dendritic spines, which rarely have mitochondria (Sorra and Harris, 2000; Wu et al., 1997). In the hippocampus, mitochondria are found only in the large, complex spines found in subfield CA3 (Chicurel and Harris, 1992). Sometimes, mitochondria are absent presynaptically. For example, in the CA3 to CA1 projection in the hippocampus, mitochondria occur with low frequency in axonal varicosities and are in fact absent in 50% of synaptic boutons (Shepherd and Harris, 1998). Glycolytic enzymes have been clearly identified in dendritic spines, where they are bound to postsynaptic densities (PSDs) (Wu et al., 1997). PSDs contain not only neurotransmitter receptors and ion channels but also protein kinases (Wu et al., 1997). These protein kinases consume ATP generated in PSDs via glycolysis (Wu et al., 1997). The presence of glycolytic enzymes, lactate dehydrogenase (Wu et al., 1997), and the monocarboxylate transporter (Bergersen et al., 2001) in PSDs, and the rarity of mitochondria in dendritic spines, suggests that at least some glucose may be used anaerobically and that the lactate produced via anaerobic glycolysis in PSDs may be released via the monocarboxylate transporter in active neurons. The rarity of mitochondria in dendritic spines and in synaptic boutons in some regions of the brain may help explain at least partially the increased lactate production during heightened neural activity in such brain areas, even when there is no shortage of oxygen (e.g., Fox et al., 1988). Similarly, the peripheral processes of astrocytes may require glycolysis for energy generation because they are essentially bereft of mitochondria, which may be too large to fit into these processes (Dienel and Cruz, 2003).
Moreover, energy generated from glycolysis has been linked to ion transport via Na+-K+ ATPase. Studies of erythrocytes (Proverbio and Hoffman, 1977), smooth muscle (Paul et al., 1979), and cardiac muscle (Weiss and Hiltbrand, 1985) have provided compelling evidence that glycolytically generated ATP is linked to Na+-K+ ATPase. Although no direct evidence for a similar link has been identified yet in the brain, both glycolytic enzymes and Na+-K+ ATPase are bound at high specific activity within synaptosomal membranes (Knull, 1978; Lim et al., 1983). Also, several studies have shown that ion transport is less robust when glucose is replaced either by lactate or by pyruvate (Lipton and Robacker, 1983; Roberts, 1993; Silver et al., 1997). The association of glycolytic enzymes with Na+-K+ ATPase in synaptic membranes (Knull, 1978; Lim et al., 1983) suggests that glucose, rather than lactate, is used as the substrate to provide energy for the Na+-K+ pump in active neurons.
Maintenance of the cytoplasmic oxidation/reduction balance
A factor that argues against the use of lactate as a substrate by neurons is the cytoplasmic oxidation/reduction (redox) balance. With glucose as the substrate, the redox balance in neurons can be maintained by NADH shuttles and by converting pyruvate to lactate, which regenerates NAD+ (Fig. 4). In contrast, when lactate is the energy substrate, the redox balance is maintained by NADH shuttles alone (Fig. 3). The fact that lactate is produced during neural activity suggests that activation of NADH shuttles in the brain may lag behind increases in energy demand. Also, as mentioned in Part I, Glycolysis as an essential energy source for localized areas of brain cells, mitochondria are extremely rare in dendritic spines (Sorra and Harris, 2000; Wu et al., 1997) and infrequent in the synaptic boutons of some brain areas, such as the hippocampal CA3 to CA1 projection (Shepherd and Harris, 1998). In those synaptic elements containing no mitochondria, reliance on NADH shuttles to maintain the redox balance may not be possible. This may constitute another disadvantage for metabolizing lactate to meet increased cellular energy demands during neural activation.
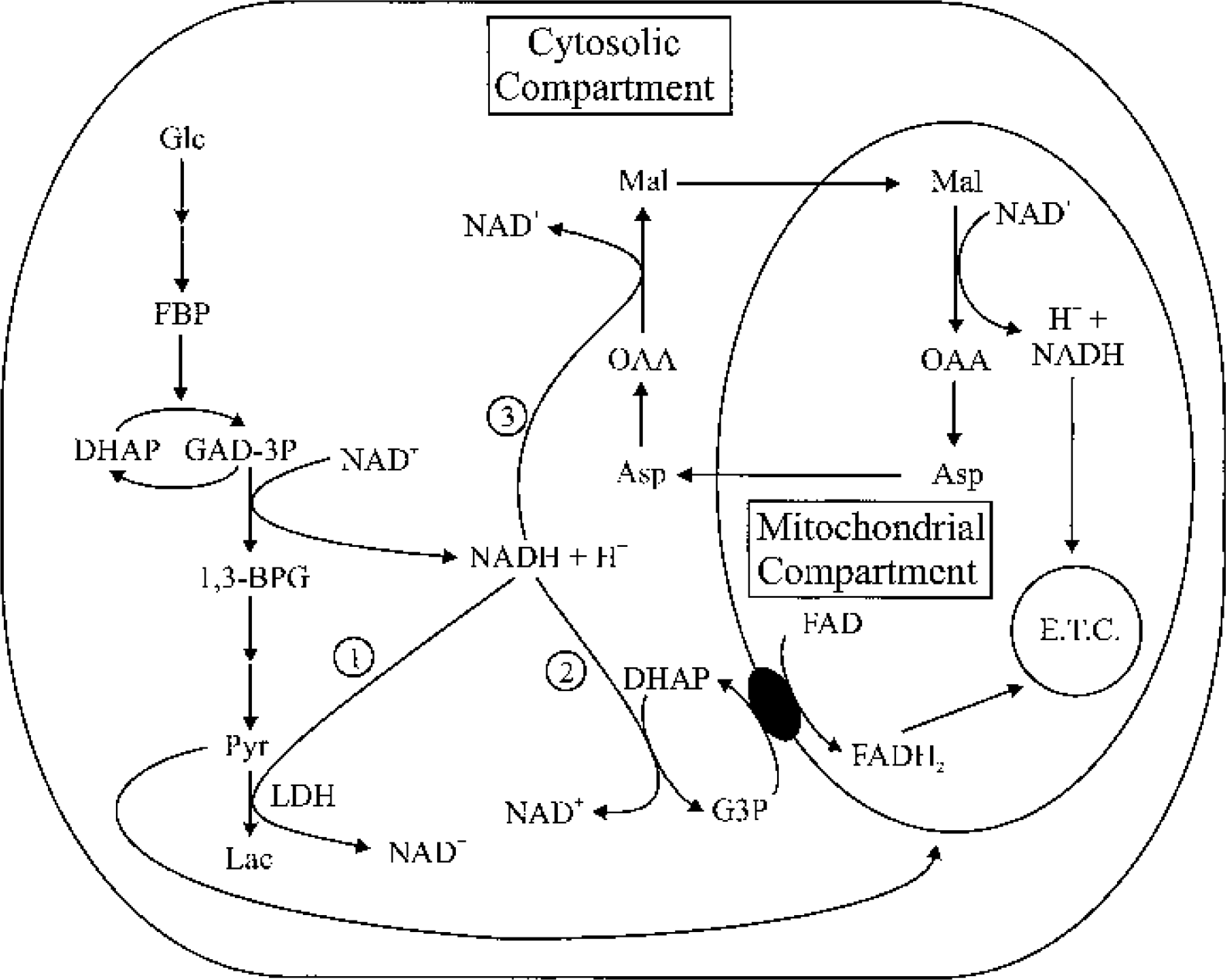
Schematic illustration of cellular mechanisms used to maintain the redox balance when glucose is the energy substrate. NADH generated by the conversion of GAD-3P to 1,3-BPG (catalyzed by glyceraldehyde-3-phosphate dehydrogenase) in the glycolytic pathway can be reoxidized to NAD+ via (1) LDH, which converts pyruvate to lactate, (2) the G3P shuttle, which transfers reducing equivalents from cytosolic NADH to FAD inside mitochondria, or (3) the malate shuttle, which transfers reducing equivalents from cytosolic NADH to NAD+ inside mitochondria. In areas where mitochondria are rare, such as dendritic spines (see text), lactate formation may be the dominant mechanism for maintaining the redox balance because diffusion of NADH to where mitochondria are may be slow. During increased glycolytic activity, the activity of the G3P shuttle may be accelerated by increased levels of DHAP, which is also a glycolytic intermediate. GAD-3P, glyceraldehyde-3-phosphate; 1,3-BPG, 1,3-bisphosphoglycerate; G3P, glycerol-3-phosphate; DHAP, dihydroxyacetone phosphate; Asp, aspartate; E.T.C., electron transport chain; FBP, fructose-1,6-bisphosphate; Mal, malate; OAA, oxaloacetate.
Another advantage of glucose oxidation is that accelerated activity in at least one of the NADH shuttles can occur via activation of the glycolytic pathway. NADH generated from glycolysis via GAPDH can be reoxidized by both glycerol-3-P dehydrogenase (glycerol-3-P shuttle) and malate dehydrogenase (malate-aspartate shuttle) (Cheeseman and Clark, 1988; McKenna et al., 1993) (Fig. 4). Each NADH shuttle system depends upon the presence of carrier metabolites (Berry et al., 1992). The glycolytic intermediate dihydroxyacetone-3-P is the carrier for the glycerol-3-P shuttle. Dihydroxyacetone-3-P levels rise as the glycolytic rate increases (Williamson, 1965), so the rate of NAD+ regeneration via the glycerol-3 phosphate shuttle can increase (Berry et al., 1992) to maintain a high glycolytic rate. In contrast, no mechanism is available to increase dihydroxyactone-3-P levels with lactate use; therefore, NAD+ regeneration may be slower when lactate, instead of glucose, is used as the substrate.
Also, LDH may not have as good an access to the glycerol-3-P shuttle as GAPDH does. For example, in hepatocytes, NADH produced by lactate dehydrogenation failed to reach the glycerol-3-P shuttle, possibly because the enzymes for glycolysis are segregated from the enzymes for gluconeogenesis (Berry et al., 1992). Thus, NADH shuttles may limit lactate oxidation, as suggested in an important study by Hertz and colleagues (Peng et al., 1994). In this study, cultured cerebellar granule cells exposed to high K+ concentrations doubled their rates of glucose and pyruvate oxidation but experienced no statistically significant changes in lactate oxidation (Peng et al., 1994). The finding that pyruvate oxidation, but not lactate oxidation, increased with high energy demand points out that lactate oxidation is limited at the step where lactate is converted to pyruvate, possibly because of a limited supply of NAD+. Thus, neurons face difficulties in increasing lactate oxidation during heightened energy demand even when glucose is absent. This may explain why in some studies lactate is less efficient than glucose in supporting ion pumping activity in brain tissue (Lipton and Robacker, 1983; Roberts, 1993; Silver et al., 1997). The findings of Hertz and colleagues serve as strong evidence against lactate competing with glucose as a substrate during elevations in energy demand.
The LDH isoform distribution
The distribution of LDH isoforms in neurons and astrocytes often has been used to support the ANLSH. LDH-1 is the main LDH isoform in neurons, whereas astrocytes have both LDH-1 and LDH-5 (Bittar et al., 1996; Tsacopoulos and Magistretti, 1996). ANLSH supporters suggest that the dominant presence of LDH-1 in neurons means that neurons are sites of lactate use, whereas astrocytes are sites of lactate production (Bittar et al., 1996; Pellerin et al., 1998a), because (1) the higher sensitivity of LDH-1 to pyruvate inhibition drives the reaction catalyzed by LDH-1 toward pyruvate production (Bittar et al., 1996), and (2) the lower Km of LDH-1 for lactate (Bittar et al., 1996) makes LDH-1 better suited than LDH-5 for lactate oxidation. However, the free energy of a biochemical reaction determines its direction, not the enzyme catalyzing it (Lehninger et al., 1993), so the dominant presence of LDH-1 in neurons does not influence the direction of the LDH reaction. Also, the lower Vmax of LDH-1 can offset the effects of its lower Km when the enzyme activity of LDH-1 is compared with that of LDH-5 (Nitisewojo and Hultin, 1976). Of greater importance is that LDH-1 is more sensitive to pyruvate product inhibition (Ki = 0.18 mM for LDH-1 and 0.28 mM for LDH-5) (Stambaugh and Post, 1966). Because pyruvate levels are between 0.1 to 0.2 mM in brain tissue (McIlwain and Bachelard, 1985), the higher sensitivity of LDH-1 to pyruvate product inhibition implies that LDH-1 has greater difficulty oxidizing lactate than LDH-5 in most physiological situations, particularly when pyruvate levels increase. Thus, LDH-1 may help to inhibit lactate oxidation in neurons during neural activity when the glycolytic pathway is activated and pyruvate levels increase.
The prevalence of LDH-1 in neurons also may help neurons oxidize glucose fully. This is because of the higher sensitivity of LDH-1 to pyruvate substrate inhibition and lactate product inhibition, which helps direct glycolytically derived pyruvate to the TCA cycle instead of toward lactate production (Cahn et al., 1962). The fact that less lactate is produced at rest in cultured neurons than in cultured astrocytes (Schousboe et al., 1997; Walz and Mukerji, 1988) supports this conclusion. LDH-1 is also the dominant isoform in other highly aerobic cells, such as cardiac muscle cells, where glucose is often fully oxidized, while LDH-5 is the dominant isoform in cells where anaerobic glycolysis (or glycogenolysis) is a major mode of energy production, such as in skeletal muscle cells. Therefore, the presence of both LDH-1 and LDH-5 in astrocytes means that astrocytes may metabolize glucose either aerobically or anaerobically.
The prevailing glycolytic rate probably influences lactate use more than the LDH isoform. For example, in skeletal muscle in vivo, where LDH-5 is the predominant isoform, lactate produced and released from active muscle, where the rate of glycogenolysis is high, is used by inactive muscle, where the rate of glycogenolysis is low (Bangsbo et al., 1995). Thus, the dominant presence of LDH-1 in neurons does not support the claim of the ANLSH that neurons are the primary users of lactate.
Distribution of monocarboxylate transporters in neurons and astrocytes
The distribution of monocarboxylate transporters in the brain is heterogeneous because neurons have mainly monocarboxylate transporter 2 (MCT2), whereas astrocytes have MCT1, MCT2, and MCT4 (Bergersen et al., 2001; Gerhart et al., 1998; Pellerin et al., 1998b). This heterogeneous distribution has been cited as additional evidence for the ANLSH (Bergersen et al., 2001; Broer et al., 1997; Pellerin et al., 1998b; Pierre et al., 2002). Of the different MCT isoforms, MCT2 has the highest affinity for lactate (Bergersen et al., 2001). Based on this, Bergersen et al. (2001) concluded that MCT2 allows movement of lactate from astrocytes into neurons. However, the gradient for lactate across brain cell membranes determines the direction of lactate transport, not the type of MCT present. For example, when glycolysis is activated, lactate efflux from cultured neurons can equal that of astrocytes (Walz and Mukerji, 1988). Also, the association of MCT, LDH, and glycolytic enzymes with PSDs in dendritic spines (Bergersen et al., 2001; Wu et al., 1997), where mitochondria are rare, suggests that lactate is being produced and transported out of dendritic spines during heightened synaptic activity (also see Part I, Glycolysis as an essential energy source for localized areas of brain cells). In addition, the activity of the MCT is determined not only by its Km, but also by its Vmax. Thus, a higher affinity of MCT2 for lactate does not necessarily imply a higher rate of lactate uptake. Indeed, the initial rates of lactate uptake are similar in cultured astrocytes and neurons for lactate concentrations of 1 to 5 mM (Dienel and Hertz, 2001; Dringen et al., 1993c; Tildon et al., 1993). Therefore, it seems unlikely that differences in MCT isoforms significantly influence lactate transport in either neurons or astrocytes.
SUMMARY
The characteristics of glycolytic enzymes and the kinetics of metabolite changes suggest that glucose is the primary energy substrate in both neurons and astrocytes during neural activation. Factors arguing against active neurons using lactate as an energy substrate include (1) the lack of a mechanism to activate LDH during increased energy demand, (2) the slow increase of brain lactate levels during neural activity, (3) the rapid activation of the glycolytic pathway by increased energy demand, and (4) the likelihood that activation of glycolysis may suppress lactate oxidation. The observed decline of lactate after physiological brain stimulation suggests that use or removal of lactate occurs mainly after neural activity.
PART II: EXPERIMENTAL EVIDENCE REGARDING GLUCOSE AND LACTATE USE
Sites of glucose uptake
1:1 coupling of oxidative glucose consumption to glutamate cycling
A study by Shulman and coworkers (Sibson et al., 1998) regarding the coupling between oxidative glucose use and glutamate cycling is thought to provide key support for the ANLSH (Magistretti and Pellerin, 1999; Magistretti et al., 1999). These investigators used 13C-NMR to follow the fate of 13C-glucose injected into the rat brain in vivo when different levels of neural activity were simulated with various levels of anesthesia. They concluded that a 1:1 stoichiometry existed between increases in oxidative glucose consumption and increases in glutamate cycling in astrocytes (measured as increases in glutamine synthesis) as neural activity increased. ANLSH supporters have suggested that this 1:1 ratio means that glutamate cycling in astrocytes is the principal driving force behind brain glucose metabolism.
The support this study gives the ANLSH is debatable for two major reasons. First, the calculated rate of glutamate cycling may not be applicable in situ. A critical assumption of the Sibson et al. (1998) study was that the rate of glutamine synthesis via the anaplerotic pathway is unchanged when the rate of total glutamine synthesis goes up with increased neural activity. This assumption has no obvious foundation and may seriously underestimate the stoichiometry between oxidative glucose consumption and glutamate cycling. For example, if the rate of glutamine synthesis via the anaplerotic pathway during normal activity had been 30% of total glutamine synthesis, as others have observed (Kunnecke et al., 1993; Lapidot and Haber, 2000), then the stoichiometry between oxidative glucose consumption and glutamate cycling would be roughly 1.5:1 rather than 1:1 (estimated from Sibson et al., 1998). Thus, the stoichiometry between oxidative glucose metabolism and glutamate cycling may vary greatly depending on the assumptions used. Additional assumptions made in the Sibson et al. study also may bring into question how applicable the results of this study are in situ (also see Gruetter, in press; Roberts and Chih, in press).
Second, even if the data did support a 1:1 stoichiometry between oxidative glucose metabolism and glutamate cycling, other models besides the ANLSH, including the conventional hypothesis, may explain the data. Because the sites of glucose uptake and modes of glucose consumption in astrocytes and neurons were not determined in the Sibson et al. study, the calculated amount of glucose consumed by neurons and astrocytes could vary greatly depending on the percentage of glucose metabolized aerobically in neurons and astrocytes. If activity-induced increases in astrocyte glucose metabolism were partially aerobic instead of entirely anaerobic as assumed by the ANLSH, glutamate cycling would require much less glucose consumption in astrocytes. This is due to the large difference in efficiency between aerobic and anaerobic glucose metabolism (34 to 36 ATP versus 2 ATP). For example, if as little as 6% of glucose metabolism in astrocytes had been aerobic, the total glucose needed for the measured glutamate cycling in astrocytes would be only 50% of measured glucose consumption. This means neurons could have consumed the other 50% of glucose aerobically. Likewise, if 20% of astrocytic glucose metabolism had been aerobic, then neurons would have accounted for 75% of total glucose consumption in the Sibson et al. study. Thus, for the results of Sibson et al. to suggest that glucose is predominantly consumed in astrocytes, the assumption of the ANLSH that glutamate cycling in astrocytes is supported entirely by anaerobic glucose metabolism needs verification.
However, this assumption is controversial based on existing evidence. Although uptake of exogenous glutamate is associated with lactate production in cultured astrocytes (Pellerin and Magistretti, 1994) and in the brain in vivo (Demestre et al., 1997), the percent of glutamate uptake in astrocytes fueled by anaerobic glycolysis is unknown. Many studies have shown that glutamate uptake in astrocytes can be sustained at least partially by oxidative metabolism (e.g., Hertz et al., 1998; Swanson, 1992). In fact, Hertz et al. (1998) concluded that energy for glutamate uptake in cultured astroglia was largely provided by oxidative metabolism of accumulated glutamate. Also, astrocyte cultures increase their oxygen consumption when exposed to 100 μM glutamate (Eriksson et al., 1995), which suggests that glutamate cycling in astrocytes is at least partially supported by oxidative metabolism. In addition, studies of mammalian astrocytes have shown no preferential use of energy produced by either glycolysis or oxidative phosphorylation for the support of Na+-K+ pump activity (Silver and Erecinska, 1997). Moreover, Rothman and coworkers have recently determined that the astroglial TCA cycle accounts for 14% of brain oxidative metabolism (Lebon et al., 2002). Other researchers have estimated a higher percentage of glucose metabolism via the glial TCA cycle based on the relative activity of pyruvate carboxylase and pyruvate dehydrogenase (Hassel et al., 1995; Qu et al., 2000). Thus, glutamate probably activates both anaerobic glycolysis and oxidative metabolism in astrocytes. Because aerobic metabolism is more efficient than anaerobic metabolism, the glucose consumed by astrocytes should be much less than the observed total glucose consumption in the Sibson et al. study. Therefore, this calculated 1:1 stoichiometry should not be used to draw conclusions regarding the relative contributions of neurons and astrocytes to glucose consumption in the brain during neural activity.
Uptake of 2-deoxyglucose
Activity-induced 2-DG uptake has been found to occur primarily in the neuropil, a region enriched in axon terminals, dendrites, and synapses (see Kadekaro et al., 1985). Because the membranes of synaptic terminals contain a high density of the glucose transporter GLUT3 (Leino et al., 1997; McCall et al., 1994), the pattern of 2-DG uptake suggests that active neurons do take up glucose. A recent in vivo study (Wittendorp-Rechenmann et al., 2001), in which a microautoradiographic imaging procedure was used also showed that 54% to 62% of 14C-2-DG was picked up by microtubule-associated protein 2 (MAP2)-positive cells (neurons), compared with 42% to 58% by glial fibrillary acidic protein (GFAP)-positive cells (astrocytes). Thus, these findings call into question the idea of the ANLSH that activity-induced glucose uptake occurs predominantly in astrocytes.
Consumption of glucose in cultured neurons and astrocytes
Many studies have shown that both neurons and astrocytes use glucose as a substrate (Peng et al., 1994; Walz and Mukerji, 1988). The fact that more glucose is consumed by cultured astrocytes than by cultured neurons (Lopes-Cardozo et al., 1986; Waagepetersen et al., 2000) has been cited as evidence that glucose is preferentially consumed in astrocytes (e.g., Magistretti, 1999; Magistretti et al., 1999). However, neurons tend to oxidize glucose fully instead of metabolizing glucose to lactate. This is seen in a higher rate of glucose oxidation (Itoh et al., 2003; Vicario et al., 1993) and in a smaller production of lactate by cultured neurons compared with cultured astrocytes (Waagepetersen et al., 2000; Walz and Mukerji, 1988). The dominant presence of LDH-1 in neurons may help direct pyruvate generated from the glycolytic pathway toward the TCA cycle instead of toward lactate production (also see Part I, The LDH isoform distribution). Thus, cultured neurons may need less glucose than cultured astrocytes because they may rely less than astrocytes do on anaerobic glycolysis for energy production. In vivo, the actual consumption of glucose by neurons and astrocytes would depend on the relative masses and energy requirements of these brain cells.
Sites of lactate release
Lactate release from cultured neurons and astrocytes
As noted in the previous section, under conditions considered normoxic for cell cultures, both cultured neurons and astrocytes generate lactate from glucose, but neurons typically produce less lactate then astrocytes (Itoh et al., 2003; Walz and Mukerji, 1988). Consistent with this finding is the observation that neurons typically have a higher rate of glucose oxidation than astrocytes (Itoh et al., 2003; Vicario et al., 1993). These observations suggest that glucose is more fully metabolized in neurons than in astrocytes (also see Part II, Sites of glucose uptake, Consumption of glucose in cultured neurons and astrocytes). However, observations of lower lactate production in cultured neurons at rest do not mean neurons have a smaller glycolytic capacity than astrocytes. This is because, when oxidative metabolism is blocked, neurons can increase lactate production as much as astrocytes (Walz and Mukerji, 1988).
Lactate release during neural activity in vivo
Two microdialysis studies (Demestre et al., 1997; Fray et al., 1996) have been cited by ANLSH proponents as providing evidence for lactate release specifically from astrocytes during stimulated neural activity (Bouzier-Sore et al., 2002). These studies showed that the competitive glutamate uptake blockers threo-hydroxyaspartate (THA) and L-trans-pyrrolidine-2,4-dicarboxylate (tPDC) blocked stimulation-induced increases in lactate (Demestre et al., 1997; Fray et al., 1996). From these results, it was concluded that stimulated release of lactate is dependent on the uptake of glutamate (Demestre et al., 1997). However, the uptake blockers themselves elevated lactate before sensory stimulation as much as sensory stimulation did in the absence of uptake inhibitors. Thus, the lack of an effect of sensory stimulation on lactate release may have been because the glycolytic rate was maximal or near maximal before stimulation. Also, the two glutamate uptake blockers used are not specific for glial glutamate transporters and would inhibit glutamate uptake into either glia or neurons (e.g., Danbolt, 2001), given that glutamate is taken up by pre- and postsynaptic neurons as well as by astrocytes (Conti and Weinberg, 1999; Danbolt, 2001). Thus, the results from the microdialysis studies do not establish that lactate was produced specifically by glia, as has been suggested by some investigators (e.g., Bouzier-Sore et al., 2002). As in cultured neurons, neurons in vivo may produce less lactate during neural activity because they may oxidize glucose more fully (see above).
Sites of lactate use
Comparison of lactate use in neuronal and astrocyte cultures
The ability of both cultured neurons and astrocytes to use lactate has been well established (Itoh et al., 2003; McKenna et al., 1993; Peng et al., 1994; Vicario et al., 1993). Some studies have shown that neurons and astrocytes have similar rates of lactate oxidation (Peng et al., 1994; Vicario et al., 1993), whereas others have reported that rates of lactate oxidation are higher in neurons or isolated synaptic terminals than in astrocytes (Itoh et al., 2003; McKenna et al., 1993). Neurons and astrocytes may use lactate via other metabolic pathways besides oxidation. Tabernero et al. (1993) reported that the rate of lipogenesis from lactate is approximately twofold greater in astrocytes than in neurons. Dringen et al. (1993b) showed that cultured astrocytes can incorporate lactate into glycogen. Thus, both astrocytes and neurons can use lactate.
The higher rates of lactate oxidation in neurons compared with astrocytes observed in some studies (Itoh et al., 2003; McKenna et al., 1993) are probably due to generally higher rates of oxidative metabolism in neurons because the rates of glucose oxidation observed in neurons are also higher than those in astrocytes (Itoh et al., 2003; Vicario et al., 1993). It is interesting to note that both cultured neurons and astrocytes oxidize lactate much faster than they do glucose. In astrocytes, rates of lactate oxidation are approximately 4 to11 times greater than rates of glucose oxidation, whereas in neurons, rates of lactate oxidation are approximately 3 to 10 times greater than rates of glucose oxidation (Itoh et al., 2003; McKenna et al., 1993; Vicario et al., 1993). This may be caused in part by the fact that some glucose is metabolized anaerobically to lactate to provide energy in cultured brain cells. Also, as mentioned previously (see Part I, Responses of glycolytic enzymes and LDH to increases in energy demand during neural activity), the glycolytic pathway is controlled mainly by energy demand (Sokoloff, 1989), not by glucose concentration, and so is greatly suppressed at rest. In contrast, LDH catalyzes a reaction near equilibrium at rest and can proceed rapidly when the ratio of its substrates and products levels are favorable. Other substrates, such as glutamine and 3-hydroxybutyrate, were also oxidized at a much higher rate than glucose in isolated synaptic terminals and astrocytes (McKenna et al., 1993).
Sites of lactate use in vivo
Two 13C-NMR spectroscopy studies of metabolic substrate turnover in the brain in vivo (Bouzier et al., 2000; Hassel and Bråthe, 2000) have been cited as providing convincing evidence that lactate is specifically a neuronal substrate in vivo (Bouzier-Sore et al., 2002). In these studies, a similar labeling of the glutamine C2 and C3 positions was found after infusion of rats with 3−13C-lactate. Bouzier et al. (2000) suggested that the similar labeling of the glutamine C2 and C3 positions meant that 3−13C-lactate was metabolized in a compartment devoid of pyruvate carboxylase (PC), specifically neurons. However, whereas a greater labeling of glutamine C2 than glutamine C3 may indicate higher glial PC activity (Bouzier et al., 2000; Merle et al., 2002), it does not necessarily follow that similar labeling of glutamine C2 and C3 means that astrocytes were not metabolizing 3−13C-lactate. For example, 3−13C-pyruvate derived from 3−13C-lactate could have been metabolized in astrocytes via the pyruvate dehydrogenase (PDH)-mediated pathway. Zwingman et al. (2000) found little difference in the labeling of the glutamine C2 and C3 positions in cultured astrocytes after they were incubated in 3−13C-alanine, which, like 3−13C-lactate, leads to the production of 3−13C-pyruvate. Martin et al. (1993) reported that labeling of the glutamine C2 position was 1.4 times greater than that of the glutamine C3 position in astrocyte cultures exposed to 1-13C-glucose, which is also converted metabolically to 3−13C-pyruvate. The inconsistency of the glutamine C2/C3 labeling ratio in astrocytes between different studies indicates that the labeling pattern from 3−13C-pyruvate may not reflect the site(s) of substrate use.
Moreover, the glutamine C2/C3 ratio can be affected by other factors such as time (Hassel et al., 1995; Merle et al., 2002) and anesthesia (Shank et al., 1993). Several investigators have reported on the effects of time (e.g., Hassel et al., 1995; Merle et al., 2002; Shank et al., 1993). For example, Hassel et al. (1995) found that the glutamine C2/C3 ratio in the mouse brain decreased from 1.96 at 15 minutes to 1.12 at 30 minutes after they injected 1-13C-glucose intravenously into conscious mice. Similarly, Shank et al. (1993) reported a decline in the glutamine C2/C3 ratio of the rat forebrain from around 1.8 at 30 minutes to around 1.2 at approximately 45 minutes (estimated from Shank et al., 1993) after they administered 1-13C-glucose intraperitoneally to conscious rats. The glutamine C2/C3 ratios obtained at the later time points in these studies were similar to the ratios found by Hassel and Bråthe (2000) (1.3 at 15 minutes) and by Bouzier et al. (2000) (1.1 at 20 minutes), all of whom used 3−13C-lactate as the labeling substrate.
The time dependence of glutamine C2/C3 labeling may be caused in part by the relative contributions of neuronal and astrocytic pools of glutamate to glutamine synthesis changing with time (Merle et al., 2002). It could also be caused by the equilibration of oxaloacetate with fumarate, which scrambles the label between the glutamine C2 and C3 positions (Hassel et al., 1995). It has been suggested that the glutamine C2/C3 ratio may be most accurate in the early phase after the infusion of labeled substrates (Merle et al., 2002; Shank et al., 1993). However, accurate determination of the amino acid enrichment in the initial phase is hindered by many technical difficulties, including low signal-to-noise ratios in 13C-NMR spectra (Merle et al., 2002). These factors further increase the uncertainty of predicting the sites of substrate use based on the labeling of the glutamine C2 and C3 positions.
In addition, results from a study by Sonnewald and coworkers (Qu et al., 2000) conflict with the studies of Hassel and Bråthe (2000) and Bouzier et al. (2000). In the Qu et al. study (2000), the relative contributions of PC and PDH to glutamine labeling in the rat brain were approximately 0.31 after rats were injected with [U-13C]lactate, and approximately 0.41 after rats were injected with [U-13C]glucose. Their results suggested that lactate, like glucose, is used by astrocytes in vivo.
Evidence for an astrocyte-neuron lactate shuttle during normal neural activity
Substrate use in an isolated retinal preparation
Major support for the existence of an astrocyte-neuron lactate shuttle has been drawn from a study by Poitry-Yamate et al. (1995). They concluded that photoreceptors used lactate released from retinal astrocytes (Müller cells) even in the presence of glucose. The principal finding was that the radioactivity of bath 14C-lactate released from Müller cells was reduced 70% when photoreceptors were included in the cell suspension. However, as discussed in detail elsewhere (Chih et al., 2001a, 2001b; Roberts and Chih, in press), most of the decrease in 14C-lactate radioactivity was due to dilution caused by adding photoreceptors to Müller cell cultures. Thus, it is difficult to conclude from the Poitry-Yamate et al. study whether a shuttle exists between Müller cells and photoreceptors.
Exchange of lactate between astrocytes and neurons in coculture systems
Two other studies of astrocyte-neuron cocultures (Waagepetersen et al., 2000; Zwingmann et al., 2000) have been used as direct evidence for the existence of an astrocyte-neuron lactate shuttle (Bouzier-Sore et al., 2002). In one study, monocultures of neurons and astrocytes, and astrocyte-neuron cocultures, were incubated in 1 mM [U-13C]lactate and 2.5 mM glucose (Waagepetersen et al., 2000). After 4 hours of incubation, unlabeled lactate formed in all cultures. The consumption of [U-13C]lactate in each group was calculated based on the difference between bath [U-13C]lactate at the beginning and end of incubation. Neurons in monoculture apparently consumed 0.7 μmol/mg protein of [U-13C]lactate in 4 h, whereas consumption of [U-13C]lactate in both astrocyte cultures and astrocyte-neuron cocultures was listed as “nonquantifiable”. Waagepetersen et al. postulated that because less [U-13C]lactate was consumed by neurons in cocultures than by the neurons in monocultures, the neurons in the cocultures must have used the unlabeled lactate produced by astrocytes. However, when estimated from the data in Table 1 of Waagepetersen et al. (2000), the calculated averaged value of bath [U-13C]lactate in both astrocyte cultures and astrocyte-neuron cocultures apparently increased from the beginning to the end of the incubation period. This calculated increase in [U-13C]lactate would reflect experimental error and may not be equated to zero (nonquantifiable) [U-13C]lactate consumption. For statistical comparisons of the presumed consumption of [U-13C]lactate among the three groups, the mean values and variations of [U-13C]lactate concentration in each culture must be evaluated. Also, the proportional content of neurons in the cocultures was not determined. Without the preceding information, comparisons of [U-13C]lactate consumption between neurons in monoculture and coculture are not possible. Moreover, even if bath [U-13C]lactate decreased less in cocultures than it did in neuronal monocultures, the result does not necessarily mean that neurons used the unlabeled lactate released from astrocytes instead of [U-13C]lactate. Other plausible explanations for the results exist, such as increased glucose consumption by neurons in the coculture. Thus, the data from the Waagepetersen et al. study provide little evidence that a lactate shuttle exists between astrocytes and neurons.
Also in the Waagepetersen et al. study, the formation of unlabeled lactate exceeds the calculated decrease in bath [U-13C]lactate in neuronal monocultures by a factor of two. A net formation of lactate suggests that the thermodynamics of the LDH reaction in neurons favors lactate instead of pyruvate production for the given experimental conditions. Thus, it is unclear whether the calculated decrease in bath [U-13C]lactate in the media really represents neuronal use of [U-13C]lactate. Even if it does, the calculated [U-13C]lactate consumption in neuronal monocultures only represents approximately 38% of energy production from glucose use, taking into account the facts that glucose produces two pyruvates and lactate produces only one pyruvate and that approximately 47% of glucose was metabolized anaerobically in this study, which does not support the idea of the ANLSH that neurons prefer lactate as a substrate over glucose.
In another study, monocultures of neurons and astrocytes, and astrocyte-neuron cocultures, were incubated in 1.5 mM [3−13C]alanine and 2.5 mM glucose (Zwingmann et al., 2000). In this study, astrocytes released more lactate than neurons, and lactate produced from alanine was higher in astrocyte-neuron cocultures than in astrocyte cultures. The authors of this study speculated that a lactate-alanine shuttle between astrocytes and neurons existed, but no evidence was presented showing that neurons consumed lactate released by astrocytes. Thus, the existence of an astrocyte-neuron lactate shuttle cannot be substantiated based on the results of either the Zwingmann et al. study or the Waagepetersen et al. study.
Breakdown of glycogen in astrocytes as a source of lactate for neighboring cells
Two studies (Dringen et al., 1993a; Wender et al., 2000) are thought by some investigators (e.g., Bouzier-Sore et al., 2002) to provide evidence that lactate produced by astrocytes as a result of glycogen breakdown is transferred to neurons. In the first study, cultured astrocytes deprived of glucose depleted their glycogen stores and released lactate instead of glucose (Dringen et al., 1993a). However, in the absence of glucose, astrocytes may have to metabolize glucose produced through glycogen breakdown to meet their own energy needs. Also, glucose derived from glycogen may be metabolized anaerobically to lactate in cultured astrocytes because cultured astrocytes typically produce large amounts of lactate even under normoxic conditions (Schousboe et al., 1997; Waagepetersen et al., 2000; Walz and Mukerji, 1988). The breakdown of glycogen to lactate in the absence of glucose does not imply that a similar breakdown of glycogen to lactate will occur in situ when glucose is present. In fact, Fillenz and colleagues (Forsyth et al., 1996; Fray et al., 1996) found evidence that astrocytes may export glucose derived from glycogen degradation to the extracellular space of the brain in vivo when the brain is stimulated. Whether glucose is released by astrocytes and used by active neurons during normal neural activity in vivo is as yet a question needing further exploration.
In a second study, Wender et al. (2000) showed that the lactate transport blockers α-cyano-4-hydroxycinnamate (4-CIN) and p-chloromercurobenzosulfonate (p CMBS) reduced the time needed for failure of axonal function during glucose deprivation and decreased the recovery of axonal function after glucose replenishment. The study implied that when glucose is unavailable, such as during hypoglycemia, active neurons may use lactate derived from glycogen degradation in astrocytes as a fuel. However, the results can only be applied to pathologic conditions when glucose is scarce. As mentioned earlier, lactate must compete with, instead of supplement, glucose as a substrate for active neurons under normal physiological conditions (see Part I, Suppression of lactate oxidation by increased glycolysis). The results of Wender et al. do not imply that, when glucose is readily available, lactate released by astrocytes will be shuttled to and used by active neurons during normal neural activity, as proposed by the ANLSH. Also, the results of this study do not conflict with the conventional view of glucose metabolism, which contends that glucose is the predominant substrate during normal neural activity in the mature brain but that alternative substrates such as lactate may be used when glucose is unavailable.
Distributions of carbonic anhydrase and the sodium-bicarbonate transporter
The distributions of carbonic anhydrase (CA) and the Na+-HCO3 transporter (NBC) also have been considered in some reviews (e.g., Ames, 2000; Bouzier-Sore et al., 2002) as reinforcing lactate shuttling between astrocytes and neurons. In these reviews, CA is said to be present only in astrocytes, and the NBC is said to be found only in astrocytes. However, CA and the NBC occur in both neurons and astrocytes. Indeed, CA has been found in neurons and nerve fibers, particularly those associated with sensory functions in different regions of the brain (Ridderståle and Wistrand, 1998; Willoughby and Schwiening, 2002). The NBC also has been found in rat brain neurons (Schmitt et al., 2000). Thus, speculation that the distribution of CA and of the NBC plays a role in promoting lactate shuttling in the brain is not supported by the existing evidence.
Comparison of glucose and lactate as substrates in the brain and brain tissue
Changes in extracellular glucose and lactate levels in vivo
Several studies have investigated changes in extracellular glucose or lactate levels during and after neural activation in the brain in vivo (Fellows and Boutelle, 1993; Fellows et al., 1993; Fillenz and Lowry, 1998; Fray et al., 1996; Hu and Wilson, 1997). In one study, glucose and lactate use was inferred from changes in extracellular lactate (Hu and Wilson, 1997). However, changes in extracellular glucose or lactate levels are complex because they represent not simply metabolite use alone but also a balance between supply and use. For example, increases in extracellular glucose resulting from neural activation may reflect increases in the glucose supplied from the bloodstream. This is because (1) the plasma glucose concentration (approximately 7 to 8 mM) (Hu and Wilson, 1997; Silver and Erecinska, 1994) is higher than glucose levels in brain tissue, and (2) cerebral blood flow goes up sharply (50% to 100%) in stimulated regions (Fellows and Boutelle, 1993). As a result, extracellular glucose during induced neural activity has been shown to increase in some studies (Fellows and Boutelle, 1993) and decrease in other studies (Fillenz and Lowry, 1998; Fray et al., 1996; Hu and Wilson, 1997). Thus, drawing conclusions regarding glucose or lactate use based on changes in extracellular glucose or lactate levels is difficult.
In the study by Hu and Wilson (1997) mentioned above, transient changes in extracellular lactate and glucose levels measured with lactate- and glucose-sensitive microsensors were used as evidence for glucose and lactate use. As has been discussed in detail elsewhere (Chih et al., 2001a, 2001b; Roberts and Chih, in press), the results of this study were inconclusive because of several reasons, including a lack of statistical analysis.
Changes in brain lactate levels in vivo, as measured with magnetic resonance spectroscopy
In vivo magnetic resonance spectroscopy has been used in several studies to measure changes in lactate levels during stimulated neural activity (e.g., Chhina et al., 2001; Prichard et al., 1991; Ueki et al., 1988). Whereas one study found that intense visual stimulation did not produce significant changes in lactate levels (Chhina et al., 2001), other studies have shown that brain lactate levels were elevated after physiological stimulation (Prichard et al., 1991; Ueki et al., 1988). In a study by Prichard et al. (1991), lactate transiently increased 0.3 to 0.9 mM during the first 3 minutes of stimulation, followed by a gradual decline (approximately 20% to 60% in 10 minutes) during the stimulation period and a continuous decline after termination of stimulation. It is unclear whether this slow decline in lactate levels represented lactate use in brain cells. If it did represent this, then the rate of lactate use was probably declining as brain lactate levels went down because the rate of lactate dehydrogenation depends primarily on lactate levels (see Part I, Responses of glycolytic enzymes and LDH to increases in energy demand during neural activity). Also, the type (neurons or astrocytes) and status (active or inactive) of brain cells contributing to this decline remain unidentified.
Inhibition of glucose consumption by lactate in brain cells in vitro
One study frequently used to support the ANLSH shows that, in unstimulated sympathetic ganglia isolated from chicken embryos, lactate carbons can displace 50% to 70% of the glucose carbons used for CO2 production (Larrabee, 1995). It should be noted that the concentration of lactate used in this study (5 mM) was higher than the extracellular lactate concentration estimated in situ (0.6 to 1.37 mM) (see Part I, Suppression of lactate oxidation by increased glycolysis) and that the cell types using lactate were not identified. The results from this study have been taken as evidence that brain tissue prefers lactate to glucose as an energy substrate (Magistretti et al., 1999). However, as mentioned earlier, the glycolytic rate is tightly coupled to energy demand and so is greatly depressed at rest (section Part I, Responses of glycolytic enzymes and LDH to increases in energy demand during neural activity). In contrast, the rate of lactate dehydrogenation is mainly determined by the concentrations of its substrates and products (Part I, Responses of glycolytic enzymes and LDH to increases in energy demand during neural activity). Thus, when extracellular lactate levels rise through experimental manipulation, lactate oxidation will increase and may suppress glucose use in unstimulated neural tissue. For example, increasing lactate levels inhibit glucose oxidation in both neuronal and astrocyte cultures (Itoh et al., 2003) and reduce 2-DG uptake in vivo (Bliss and Sapolsky, 2001). High lactate levels (10 mM) also suppress lipogenesis from glucose in cultured neurons and astrocytes (Tabernero et al., 1996). The removal of lactate in the brain after neural activity (Fellows et al., 1993; Fray et al., 1996; Hu and Wilson, 1997) may be a protective mechanism allowing the brain to avoid the potentially detrimental effects of a lactate buildup on both neuronal and glial glycolysis.
Inhibition of glycolysis by high lactate levels also occurs during resting conditions in other tissues, such as red skeletal muscle fibers. When unstimulated rat soleus muscles were incubated with 8 mM lactate, lactate metabolism accounted for 70% of the oxygen consumption, and both glucose use and glycerol use were suppressed (Pearce and Connett, 1980). This result does not mean that lactate is the preferred substrate for red muscle fibers during exercise. Likewise, the results from unstimulated nerve tissue in Larabee's study do not necessarily apply to active nerve tissue in vivo.
Comparison of the abilities of glucose and lactate to support neural function in brain slices
The ability of glucose and lactate to support neural activity has been studied in several brain slice preparations. Some studies have shown that lactate supports basal neural activity in vitro as well as glucose (Izumi et al., 1997; Schurr et al., 1988). However, other studies have reported that lactate cannot support neural activity as well as glucose (Cox and Bachelard, 1988; Kanatani et al., 1995; Takata and Okada, 1995; Wada et al., 1997). In two of these studies, population spikes maintained in brain slices exposed to glucose-containing artificial cerebrospinal fluid (ACSF) disappeared in slices provided only with lactate (Kanatani et al., 1995; Wada et al., 1997). In addition, lactate is less able than glucose to aid the recovery of ion homeostasis in brain tissue that is nonpathologically activated (Roberts, 1993): When CA1 pyramidal cells of rat hippocampal slices are activated transsynaptically at high frequency (40 Hz), extracellular K+ recovers more slowly in ACSF containing lactate than in ACSF containing glucose (Roberts, 1993). Thus, although lactate may be used to support activity in brain tissue in the absence of glucose, the issue remains whether lactate can successfully compete with glucose when energy demand increases.
A series of studies in hippocampal slices by Schurr and coworkers (1997, 1999) has been considered as strong support for the ANLSH (Magistretti et al., 1999) because these studies showed that 4-CIN, which inhibits the cell membrane monocarboxylate transporter, prevented the recovery of synaptic transmission even in the presence of adequate glucose. However, 4-CIN blocks not only lactate transport across the plasmalemma but also pyruvate entry into isolated mitochondria (Cox et al., 1985; Halestrap and Denton, 1974). Several studies have shown that 4-CIN by itself compromises normal oxidative metabolism and blocks neural activity (e.g., Amos and Richards, 1996; Chih et al., 2001a; McKenna et al., 2001). The effect of 4-CIN on oxidative metabolism, rather than inhibition of the lactate (monocarboxylate) transporter, could account for the lack of recovery seen in the studies of Schurr and coworkers.
Summary
The above analysis shows that current experimental evidence provides little support for the ANLSH because both neurons and astrocytes may take up glucose, release lactate, and use lactate. Also, the existence of an astrocyte-neuron lactate shuttle has yet to be demonstrated under normal physiological conditions. Although lactate may support basic brain function in the absence of glucose, lactate may be unable to compete successfully with glucose during neural activity.
PART III: ROLE OF LACTATE AS AN ENERGY SUBSTRATE
Brain tissue produces small amounts of lactate even when it is at rest and well oxygenated (Clarke et al., 1989). The reason for increased lactate production during neuronal activity, as deduced from measurements of oxygen and glucose consumption (Fox and Raichle, 1986; Fox et al., 1988; Raichle, 1991; Sappeymarinier et al., 1992) and as demonstrated by increases in extracellular lactate described previously (Fray et al., 1996; Hu and Wilson, 1997; Prichard et al., 1991) is not entirely clear. Lactate production may occur in those areas of neurons and astrocytes where mitochondria are absent and glycolytic enzymes are present, such as cerebrocortical dendritic spine heads (Wu et al., 1997) or distal astrocytic processes (Dienel and Cruz, 2003) (also see Part I, Glycolysis as an essential energy source for localized areas of brain cells). Lactate production may be seen when glycolytic activity transiently exceeds ongoing oxidative metabolism, when NADH shuttles are not activated as quickly as needed to meet increased energy demands (Clarke et al., 1989), or when cellular supplies of oxygen are temporarily limited. An analogous situation is also found in red muscle fibers, where lactate production occurs during moderate exercise although these muscle fibers are fully oxygenated (Connett et al., 1985). Whatever the basis is for lactate production, increases in lactate in the brain during neural activity are moderate and slow.
Buildups of high levels of lactate must be avoided because of its potential inhibitory effects on both neuronal and glial glycolysis. Because the energy demands of brain cells fluctuate rapidly as cells switch between their active and inactive states, the brain needs to remove excess lactate at rest so that the glycolytic pathway in both neurons and astrocytes can be easily activated when brain cells become active (also see Part I, Responses of glycolytic enzymes and LDH to increases in energy demand during neural activity). Several studies have demonstrated that high lactate levels (5 to 10 mM) may significantly suppress glucose use in both neurons and astrocytes at rest (Itoh et al., 2003; Larrabee, 1995; Tabernero et al., 1996). Similar inhibition of glycolysis by high lactate levels (8 mM) occurs in red muscle fibers (Pearce and Connett, 1980) (also see Part II, Comparison of glucose and lactate as substrates in the brain and brain tissue, Inhibition of glucose consumption by lactate in brain cells in vitro). Rapid removal of lactate has already been demonstrated in skeletal muscle, where rapid fluctuations in energy demand and glycolytic rates often takes place. For example, lactate produced by active skeletal muscle fibers is shuttled to and used by inactive skeletal muscle fibers during exercise (Bangsbo et al., 1995).
Because of the relatively slow transport of lactate across the blood-brain barrier (McIlwain and Bachelard, 1985), some lactate produced during and after neural activity probably is used by inactive brain cells. Several possible pathways exist for the metabolism of lactate built up during neural activity. Neurons and astrocytes may use lactate for oxidative metabolism following activity, when energy demands are reduced and glycolytic rates are downregulated. Astrocytes may also use lactate for glycogen synthesis (Dringen et al., 1993b). Furthermore, lactate may be used by those cells with little glycolytic capacity, such as Purkinje neurons, microglia, and oligodendrocytes (Dringen et al., 1993a). In addition, removal of lactate from the brain via the bloodstream may occur when lactate levels are elevated, as during acute ammonia challenge (Hawkins et al., 1973), spreading cortical depression (Dienel and Cruz, 2003), and generalized sensory stimulation (Dienel and Cruz, 2003). The rates of lactate removal via the bloodstream for these stimuli are 15%, 22%, and 6%, respectively, of glucose influx (Dienel and Cruz, 2003; Dienel and Hertz, 2001). The observed decline in lactate after neural activity elicited by physiologically relevant stimuli in most in vivo studies (e.g., Fellows et al., 1993; Fillenz and Lowry, 1998; Fray et al., 1996; Hu and Wilson, 1997) supports the notion that use or removal of lactate occurs mainly after neural activity.
CONCLUSION
The major difference between the conventional hypothesis and the ANLSH concerns which substrate (glucose or lactate) is used by neurons during neuronal activation. The analysis in Part I has provided evidence that, from a theoretical perspective, glucose is probably the major substrate for active neurons. The analysis in Part II suggests that current experimental evidence provides little unequivocal support for the ANLSH. Most importantly, no convincing evidence has yet been offered to affirm the declaration of the ANLSH that shuttling of lactate from astrocytes to active neurons occurs during normal (physiological) neural activity.
The analyses in this review do not rule out the possibility that some brain cells use lactate to meet their energetic needs. Brain cells are heterogeneous and show great structural and biochemical variability. This includes variability in the cellular levels of glycolytic enzymes and in the oxidative capacity of brain cells. Brain cells with low levels of glycolytic enzymes, such as oligodendrocytes, may preferentially metabolize lactate or other substrates. Lactate may also be used by active brain cells when glucose is temporarily unavailable. The conventional hypothesis contends that glucose is the primary substrate but does not exclude the possibility that lactate can be used as an alternative substrate under certain conditions, such as hypoglycemia. However, the analyses in this review point out (1) that the kinetic characteristics of metabolic enzymes and the observed changes in metabolites lend little support to the basic concept of the ANLSH that lactate is produced by astrocytes and used by neurons in an activity-dependent manner, (2) the often overlooked uncertainty in the evidence used to support the ANLSH, and (3) that the existing evidence and theoretical considerations provide a great deal of credibility to the conventional hypothesis.
Footnotes
1
In this paper, Glycolysis refers to the conversion of glucose to pyruvate under aerobic conditions, and anaerobic glycolysis refers to the conversion of glucose to lactate.
Acknowledgments
The authors thank Dr. Peter Lipton, Department of Physiology, University of Wisconsin School of Medicine, for his invaluable advice and suggestions that aided greatly with the preparation of this manuscript. The authors also thank Dr. Gerald Dienel, Department of Neurology and Physiology & Biophysics, University of Arkansas for Medical Sciences, for the illuminating discussions regarding the manuscript.