
Abstract
Although the endoplasmic reticulum (ER) is implicated in neuronal degeneration in some situations, its role in delayed neuronal cell death (DND) after ischemia remains uncertain. The authors speculated that ER stress is involved in DND, that it is reduced by ischemic preconditioning, and that ER stress reduction by preconditioning is due to ER molecular chaperone induction. The phosphorylation status of eukaryotic initiation factor 2α (eIF2α) and RNA-dependent protein kinase–like ER eIF2α kinase (PERK) was investigated in the rat hippocampus after ischemia with and without preconditioning. PERK is phosphorylated by ER stress, which phosphorylates eIF2α. To investigate the role of ER molecular chaperones in preconditioning, the authors examined GRP78 and GRP94 expression, both of which are ER chaperones that inhibit PERK phosphorylation, and compared their induction and ischemic tolerance time windows. Phosphorylation of eIF2α and PERK was confirmed after severe ischemia but was inhibited by preconditioning. After preconditioning, GRP78 was increased in the brain with a peak at 2 days, which corresponded with the ischemic tolerance time window. Immunoprecipitation and double staining demonstrated involvement of GRP78 in prevention of PERK phosphorylation. These results suggest that GRP78 induced by preconditioning may reduce ER stress and eventual DND after ischemia.
Neurons in the hippocampal CA1 pyramidal cell layer are selectively vulnerable to ischemia (Pulsinelli et al., 1982). They degenerate several days after transient ischemia, a process known as delayed neuronal cell death (DND) (Kirino, 1982). Molecular mechanisms underlying DND are not fully understood, but evidence suggests that activation of apoptotic machinery plays an important role in this process. Appearance of terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL)-positive cells, internucleosomal fragmentation of genomic DNA, and activation of executioner caspase, all of which are features of apoptosis, have been confirmed in these neurons after transient ischemia (Asahi et al., 1997; Endres et al., 1997; Héron et al., 1993; Petito et al., 1997).
There are two major pathways for apoptosis induction. One is the extrinsic pathway, which is activated by plasma membrane death receptor ligation, and the other is the intrinsic pathway in which release of cytochrome c from mitochondria and activation of caspase-9 are implicated (Yuan and Yankner, 2000). In DND, the intrinsic pathway plays an important role, and the mitochondrial injury mechanism has been extensively investigated in the ischemic model used in this study (Eldadah and Faden, 2000; Fujimura et al., 1998). However, it has become clear that not only mitochondria but also other organelles, such as the endoplasmic reticulum (ER), Golgi apparatus, and lysosomes, are involved in the intrinsic pathway of apoptosis (Deiss et al., 1996; Ferri and Kroemer, 2001; Mancini et al., 2000; Nakamura et al., 2000). In brain ischemia, there is ample evidence to indicate the possible involvement of the ER in neuronal injury. Accumulation of unfolded protein in the ER lumen, phosphorylation of eukaryotic initiation factor 2α (eIF2α), inhibition of protein synthesis, and calcium depletion from the ER lumen all suggest an active role for the ER in neuronal cell death after ischemia (Althausen et al.; 2001; DeGracia et al., 2002; Hu et al., 2000; Nowak et al., 1985; Paschen and Frandsen, 2001). Induction of ER molecular chaperones after brain ischemia also suggests ER stress (Kitao et al., 2001). Therefore, we can speculate that the ER plays a pivotal role in DND.
A brief period of nonlethal ischemia, known as ischemic preconditioning (PC), prevents DND caused by subsequent severe ischemia (Pérez-Pinzón et al., 1997). If the ER is involved in DND, ischemic tolerance produced by PC may be associated with ER stress reduction. Glucose-regulated proteins (GRP), which are molecular chaperones in the ER, play important roles in regulation of ER stress (Bertolotti et al., 2000). GRP may thus be implicated in the establishment of ischemic tolerance in the brain after PC. In the present study, we speculated that (1) ER stress is implicated in DND, (2) PC reduces ER stress and eventual DND, and (3) ER stress reduction by PC is due to induction of GRP. We investigated the phosphorylation status of eIF2α and RNA-dependent protein kinase–like ER eIF2α kinase (PERK) and expression of GRP78 and GRP94, and provide evidence to confirm that these molecular pathways are involved in DND and PC.
MATERIALS AND METHODS
Experimental paradigms
Using male Sprague-Dawley rats (300–350 g), 3 minutes of global brain ischemia was induced for PC and 5 minutes of global ischemia was induced for hippocampal CA1 neuronal lethality as in our previous report (Sugawara et al., 2001). To investigate the effect of PC on ER stress and cell injury caused by lethal ischemia, the time interval between PC and lethal ischemia was fixed at 2 days. These animals were decapitated 1 and 4 hours and 1 and 2 days after lethal ischemia and used for histologic, TUNEL, and immunohistochemical studies and for Western blotting and immunoprecipitation studies (n = 3 or 4). Sham-operated control animals were also used (n = 3). To investigate changes in GRP expression, we used only PC, and the animals were decapitated 1 and 4 hours and 1, 2, and 7 days after PC and used for immunohistochemical and Western blot studies (n = 3). Sham-operated control animals were also used (n = 3). Some of these samples were also used for immunoprecipitation studies. For the studies to investigate the time window of ischemic tolerance, the time interval between PC and lethal ischemia was varied from 4 hours to 7 days. The animals were decapitated 2 days after lethal ischemia and used for histologic and TUNEL studies (n = 3). All procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by Stanford's Administrative Panel on Laboratory Animal Care.
Transient global ischemia
Transient global brain ischemia was induced as previously reported with slight modification (Smith et al., 1984). In brief, the animals were anesthetized with 1.5% isoflurane, 68.5% nitrous oxide, and 30% oxygen using a facemask, and the femoral artery was exposed and catheterized with a PE-50 catheter (427410; Becton Dickinson, San Diego, CA, U.S.A.) for continuous recording of arterial blood pressure. A midline neck skin incision was made and the right jugular vein and both common carotid arteries were exposed. After intravenous injection of 150-IU/kg heparin, blood was quickly withdrawn via the jugular vein. When the mean arterial blood pressure became 30 mm Hg, both common carotid arteries were clamped with surgical clips. Blood pressure was maintained at 30 to 35 mm Hg by withdrawing or infusing blood through the jugular vein during the ischemic period, at the end of which the clips were removed and the blood was reinfused. The body temperature was monitored with a rectal probe and controlled at 37°C with a homeothermic blanket. Sham-operated animals underwent exposure of vessels without blood withdrawal or clamping of carotid arteries.
Histologic assessment and TUNEL study
Deeply anesthetized animals were perfused with 5-U/mL heparinized saline through the left cardiac ventricle until colorless fluid was obtained. Subsequently, 4% paraformaldehyde in 0.1-mol/L phosphate buffer was perfused and the brain was removed. After postfixation in 4% paraformaldehyde, the brains were washed with water, dehydrated with ethanol, immersed in xylene, and embedded in paraffin. Coronal brain sections 5-μm thick were prepared and placed on glass slides. The neuronal cell damage was histologically evaluated by cresyl violet staining as previously reported (Murakami et al., 1998). To investigate DNA fragmentation, we performed a TUNEL study using a commercial kit (S7100; Intergen, Norcross, GA, U.S.A.). Briefly, after pretreatment with 0.2% pepsin, the sections were incubated with terminal deoxynucleotidyl transferase and digoxigenin-conjugated nucleotides at 37°C for 1 hour. The sections were then incubated with peroxidase-conjugated antidigoxigenin antibody, and cells undergoing apoptosis were visualized using diaminobenzidine as a color substrate. Methyl green was used for counterstaining.
Immunohistochemical analysis
PERK becomes phosphorylated when stress is imposed on the ER, which in turn phosphorylates eIF2α (Harding et al., 1999; Rao et al., 2002a). The ER molecular chaperones, GRP78 and GRP94, are involved in cellular defense against ER injury by inhibiting PERK phosphorylation as well as preventing ER resident caspase and mediating peptide translocation across the ER membrane (Bertolotti et al., 2000; Liu et al., 1998; Rao et al., 2002b). We performed immunohistochemical analysis for the phosphorylated forms of eIF2α and PERK and for the expression of GRP78 and GRP94. The sections were prepared the same as those for histological and TUNEL studies. After deparaffinization, endogenous peroxidase activity was quenched with 30% methanol and 0.3% hydrogen peroxide in phosphate-buffered saline (PBS). The slides were boiled in citrate buffer with microwaves when necessary. After blocking nonspecific binding with 1% to 3% bovine serum albumin, the slides were incubated with primary antibodies at room temperature for 2 hours. The primary antibodies used and the dilutions for each were as follows: rabbit polyclonal anti–phospho-eIF2α antibody (#9721; Cell Signaling, Beverley, MA, U.S.A.) at 1:100, rabbit polyclonal anti–phospho-PERK antibody (#3191; Cell Signaling) at 1:100, rabbit polyclonal anti-GRP78 antibody (SPA-826; Stressgen, Victoria, Canada) at 1:250, and rabbit polyclonal anti-GRP94 antibody (SPA-851; Stressgen) at 1:250. The slides were washed and then incubated with biotinylated mouse monoclonal anti–rabbit immunoglobulin G (IgG) (B3275; Sigma, St. Louis, MO, U.S.A.) at 1:200 dilution. They were subsequently incubated with avidin-biotin-peroxidase complex (PK-6100; Vector Laboratories, Burlingame, CA, U.S.A.) for 30 minutes and then developed using vector VIP peroxidase substrate (SK-4600; Vector Laboratories). Methyl green was used for counterstaining.
Fluorescent double staining
We performed double staining for phospho-PERK and GRP78 to investigate the role of GRP78 overexpression on PERK phosphorylation. After microwave pretreatment and blocking of nonspecific binding, the sections were incubated with anti-GRP78 antibody and then with biotinylated anti–rabbit IgG antibody. The primary and secondary antibodies and each dilution were the same as those described previously. After washing in PBS, they were incubated with avidin-conjugated fluorescein isothiocyanate (FITC) at a dilution of 1:40 (A-2001; Vector Laboratories). The sections were subsequently reacted with the Fab fragment of anti–rabbit IgG (111-007-003; Jackson Immunoresearch Laboratories, West Grove, MA, U.S.A.) and treated with the excess avidin followed by the excess biotin (SP2001; Vector Laboratories) to prevent nonspecific double labeling. The sections were then incubated with anti–phospho-PERK antibody, and biotinylated anti–rabbit IgG antibody, and reacted with avidin-conjugated Texas Red (A-2006; Vector Laboratories) at a dilution of 1:40. They were covered with VECTASHIELD mounting medium with 4′,6 diamidino-2-phenylindole (DAPI) (H-1200; Vector Laboratories). Fluorescence was observed at excitation of 495 nm and emission of more than 515 nm for FITC, excitation of 510 nm and emission of more than 580 nm for Texas Red, and excitation of 360 nm and emission of more than 460 nm for DAPI.
Western blot analysis
We performed Western blot analysis for the phosphorylated forms of eIF2α and PERK and for the expression of GRP78 and GRP94. The animals were deeply anesthetized and decapitated, and the CA1 subregion of the hippocampus was quickly removed. For protein extraction, the tissue was sonicated with about seven volumes of protein extraction buffer containing (in mmol/L) 20 HEPES potassium hydroxide (pH 7.5), 250 sucrose, 10 potassium chloride, 1.5 magnesium chloride, 1 ethylenediaminetetraacetic acid, 1 EGTA, 0.7% protease inhibitor cocktail (P8340; Sigma), and 1% phosphatase inhibitor cocktails (P2850 and P5726; Sigma) as previously reported (Kumar et al., 2001). The sonicated sample was centrifuged at 10,000 g for 15 minutes at 4°C and the supernatant was used for this study. Assays to determine the protein concentration were performed with comparison to a known concentration of bovine serum albumin with the use of a kit (23227; Pierce, Rockford, IL, U.S.A.). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed according to previous reports (Hayashi et al., 1997; Noshita et al., 2001). In brief, the lysate equivalent of 5 μg of protein from each brain was run on the gel at 120 V together with a biotinylated protein ladder size marker (#7727, Cell Signaling). The protein on the gel was subsequently transferred to a polyvinylidene fluoride transfer membrane (LC2002; Invitrogen, Carlsbad, CA, U.S.A.) in a buffer containing methanol, glycine, Tris base, and sodium dodecyl sulfate. After the transfer, the membrane was placed in 3% to 10% powdered milk in PBS with 0.1% Tween 20 for 1 to 4 hours to block nonspecific binding and was then incubated with primary antibodies for 12 hours at 4°C. The primary antibodies used were the same as those for immunohistochemistry, and the dilution was 1:2500 for phospho-eIF2α, 1:2000 for phospho-PERK, 1:5000 for GRP78 and 1:10000 for GRP94. After washing, the membrane was incubated with horseradish peroxidase conjugated anti-rabbit IgG (7074; Cell Signaling) together with horseradish peroxidase conjugated antibiotin antibody (#7727, Cell Signaling), and then the signal was detected with a chemiluminescent kit (RPN2132; Amersham).
Immunoprecipitation study
A coimmunoprecipitation study was carried out to investigate the change in GRP78 binding with PERK. GRP78 is one of the molecules that bind with PERK and inhibits its phosphorylation, which then inhibits ER stress-induced cell death (Bertolotti et al., 2000; Yu et al., 1999). The protein extract was prepared in the same manner as for the Western blot analysis. To prevent nonspecific precipitation, 300 μg of protein from each brain sample was incubated with protein G Sepharose (71-7083-00; Pharmacia Biotech, Wikstroms, Sweden) and centrifuged, and the supernatant was used for the subsequent immunoprecipitation study. The supernatant sample was incubated with goat polyclonal anti–total-PERK antibody (sc-9477; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) and then protein G Sepharose was added. After 2 hours of incubation at 4°C, they were centrifuged and the supernatant was discarded. The precipitates were washed five times with protein-extraction buffer and used for the Western blot analysis of GRP78. Electrophoresis, blotting and detection were the same as described previously. The membrane was then used for PERK immunoblotting to confirm that the change in PERK-bound GRP78 was not caused by induction of PERK. It was placed in 10% powdered milk in PBS with 0.1% Tween 20 to block nonspecific binding, and was subsequently incubated with anti-PERK antibody (sc-9477; Santa Cruz Biotechnology) for 12 hours at 4°C. It was incubated with horseradish peroxidase-conjugated anti–goat IgG (PI-9500, Vector Laboratories) and then the signal was detected with a chemiluminescent kit (RPN2132; Amersham).
Quantification and statistical analysis
For evaluation of the results of the Western blot studies, the film was scanned with an imaging densitometer (GS-700; Bio-Rad, Hercules, CA, U.S.A.) and the optical density (OD) was quantified using Multi-Analyst software (Bio-Rad) as previously reported (Noshita et al., 2001). In the coimmunoprecipitation study, the OD obtained by immunoblotting for GRP78 was divided by that for PERK, and this ratio was used for data. For quantification of histologic or TUNEL studies, the number of intact pyramidal cells with a distinct nucleus or TUNEL-positive cells was counted in a 1-mm length of the middle portion of the CA1 subfield, and the values are expressed as mean ± SD (Sugawara et al., 2002). Statistical significance between the two groups was established with an F test followed by an unpaired t-test. A P value smaller than 0.05 was considered statistically significant.
RESULTS
Delayed neuronal death occurred 2 days after lethal ischemia and was prevented by ischemic preconditioning
One and 4 hours and 1 day after lethal ischemia, no neuronal degeneration in the hippocampal CA1 region was confirmed, and no TUNEL-positive cells were observed at these time points. Two days after lethal ischemia, however, more than 90% of the CA1 neurons were degenerated and became TUNEL positive. In the brains with PC and lethal ischemia, the tissue was intact until 1 day, the same as those without PC. At 2 days, about 20% of the neurons had condensed nuclei and morphological features of degeneration and became TUNEL positive, but the number of these cells was far smaller than those in the no-PC brains (data not shown).
eIF2α phosphorylation after lethal ischemia was prevented with ischemic preconditioning
eIF2α plays a critical role in the initiation of protein synthesis (Pain, 1996). It is phosphorylated and inactivated by stimuli such as ER stress, which is known as the unfolded protein response (DeGracia et al., 1997, 2002; Paschen and Doutheil, 1999; Paschen and Frandsen, 2001). We investigated changes in the phosphorylation of eIF2α with Western blot and immunohistochemical analysis using an anti–phospho-eIF2α antibody (Fig. 1). In the control brains, a single band with a molecular weight (MW) of 40 kd was barely detected by Western blot analysis (Fig. 1A). The MW of this band is compatible with phospho-eIF2α. The band became about six times stronger 1 hour after 5 minutes of ischemia, and became less than three times intense at 4 hours and 1 day. At 2 days, the band became three-quarters more intense than in the control brain. In the brains with PC, the band became strong 1 hour after 5 minutes of ischemia and then gradually became weak, but the degree of increase was smaller than that in the no-PC brains. With quantitative analysis, we found that PC significantly inhibited phosphorylation of eIF2α 1 and 4 hours and 1 day after 5 minutes of ischemia (P <0.01) (Fig. 1B). Immunohistochemical analysis showed that hippocampal CA1 neurons were only faintly stained in the control brains, but the immunoreactivity became more intense 1 hour after 5 minutes of ischemia without PC (Fig. 1C). At 4 hours, the immunoreactivity became less strong, and only some neurons retained dense immunoreactivity at 1 day (arrows in Fig. 1C). The neurons degenerated at 2 days and immunoreactivity could not be evaluated. In contrast, in the brains with PC, only a few neurons showed strong immunoreactivity 1 hour after 5 minutes of ischemia (arrows in Fig. 1C). The number of strongly stained cells gradually decreased and was almost undetectable 2 days after 5 minutes of ischemia.
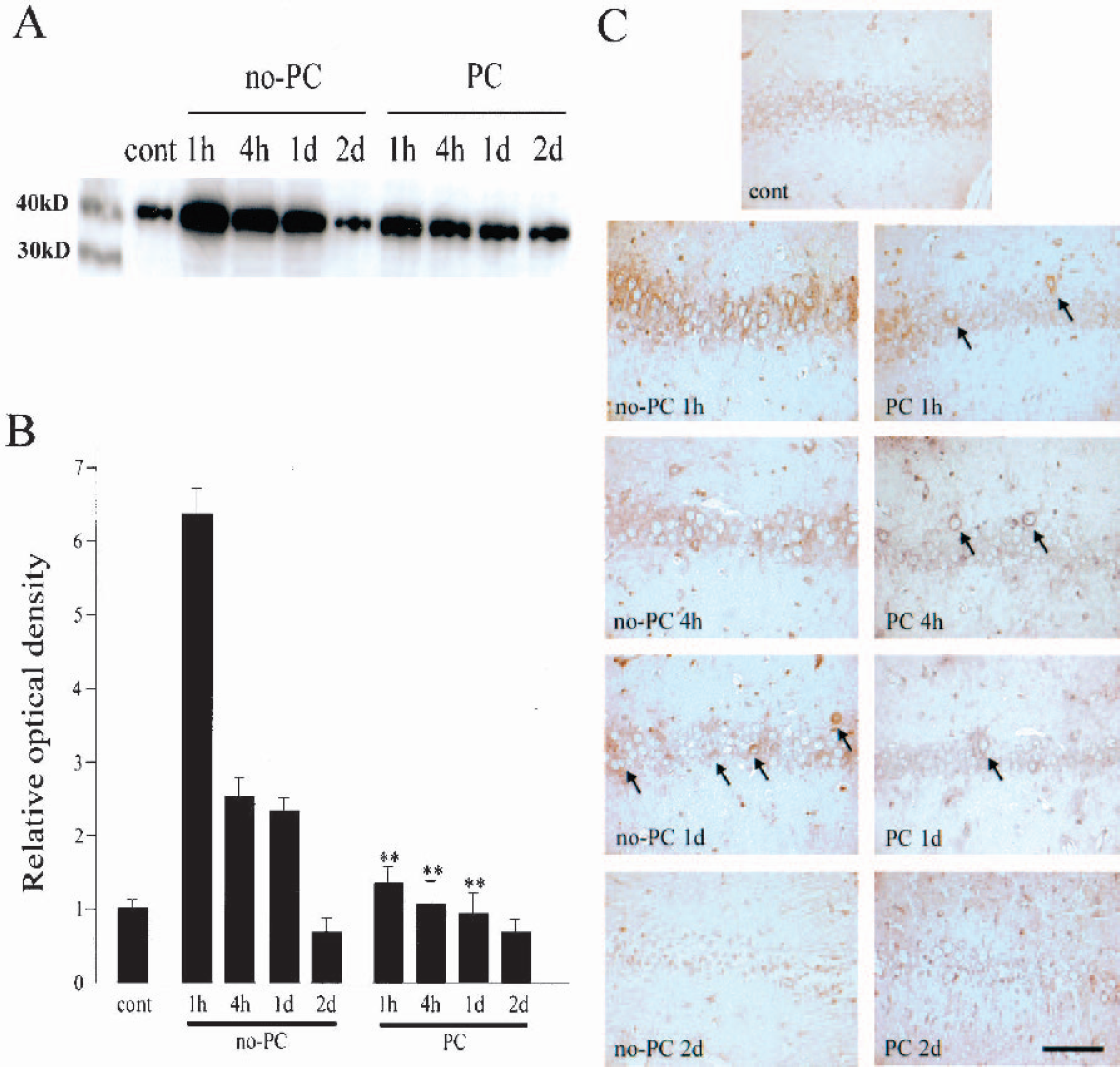
Change in phosphorylation of eIF2α at the hippocampal CA1 pyramidal cell layer.
PERK phosphorylation after lethal ischemia was prevented with ischemic preconditioning
The phosphorylated form of PERK is responsible for phosphorylation of eIF2α (Bertolotti et al., 2000; Kumar et al., 2001). PERK is localized on the ER membrane, and is phosphorylated when stress is imposed on the ER (unfolded protein response) (Harding et al., 1999). Phospho-PERK can thus be used as a molecular marker for ER stress. We investigated changes in phosphorylation of PERK with Western blot and immunohistochemical analysis using an anti–phospho-PERK antibody (Fig. 2). In the control brains, a faint band with a MW of 170 kd was observed by Western blot analysis (Fig. 2A). The MW of this band is compatible with phospho-PERK. The band became about four times denser 1 hour after 5 minutes of ischemia, and became only three times denser at 4 hours and weaker at 1 day. At 2 days, the band for phospho-PERK became less than three-quarters dense and was almost undetectable. In the brains with PC, the band became 1.3 times stronger 1 hour after 5 minutes of ischemia and then gradually weakened, but the degree of increase was smaller than in the no-PC brains. With quantitative analysis, we found that PC significantly inhibited phosphorylation of PERK 1 and 4 hours and 1 day after 5 minutes of ischemia (P <0.05) (Fig. 2B). Immunohistochemical analysis showed that hippocampal CA1 neurons were only faintly stained in the control brains, but the immunoreactivity became more intense 1 hour after 5 minutes of ischemia without PC (Fig. 2C). The immunoreactivity was strongest in the perinuclear region, which is compatible with the distribution in the ER (Nakagawa et al., 2000; Peters et al., 1991). At 4 hours, only some neurons retained a strong immunoreactivity. The number of strongly stained neurons was further decreased at 1 day (arrows in Fig. 2C), and the neurons degenerated and the immunoreactivity could not be evaluated at 2 days. In contrast, in the brains with PC, some neurons showed strong immunoreactivity 1 hour after 5 minutes of ischemia. The number of strongly stained cells gradually decreased (arrows in Fig. 2C), and was almost undetectable 2 days after 5 minutes of ischemia.
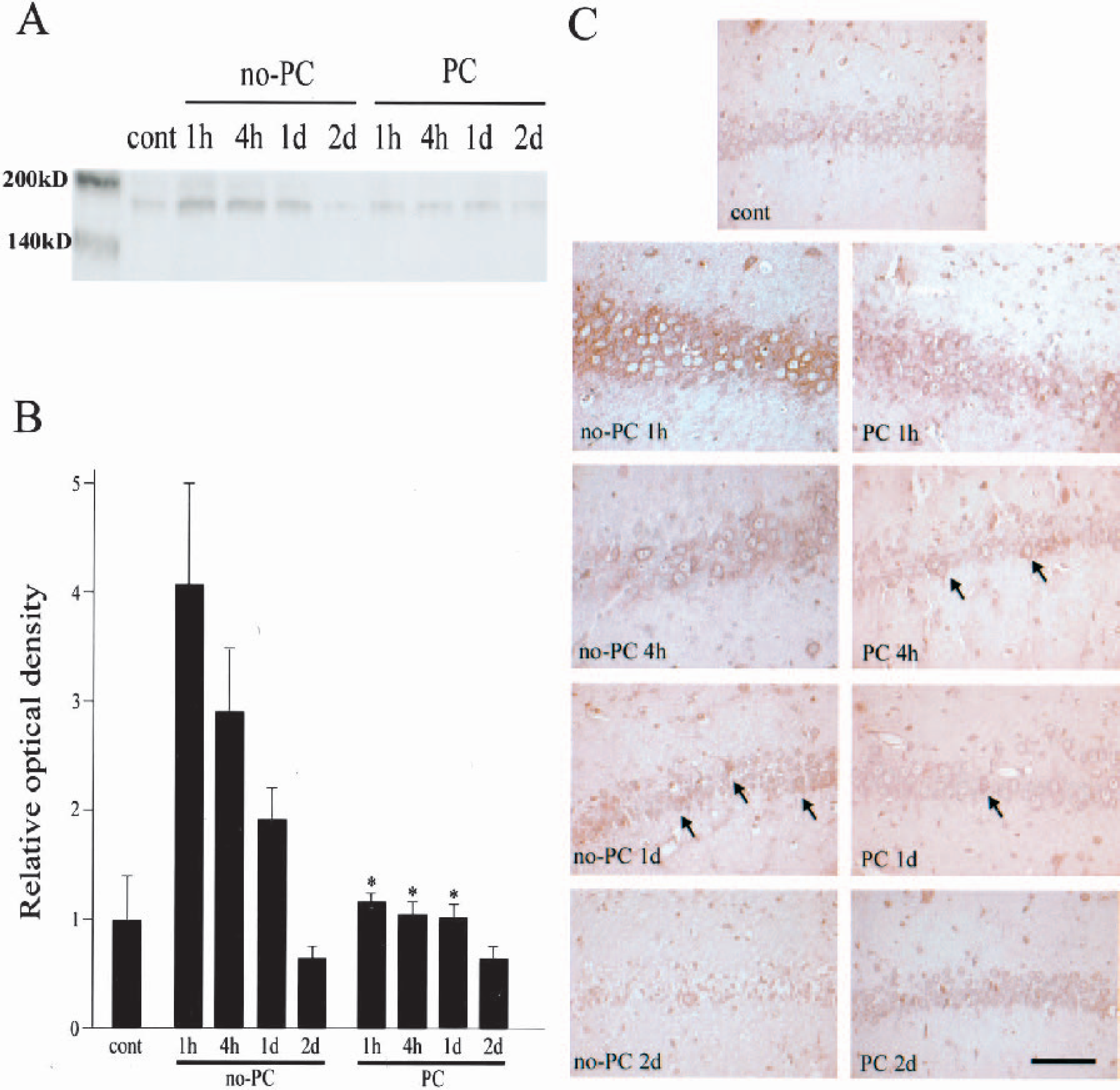
Change in phosphorylation of PERK at the hippocampal CA1 pyramidal cell layer.
Induction of GRP78, but not GRP94, corresponded to the time window of ischemic tolerance
GRP78 and GRP94 are ER resident molecular chaperones. They bind with PERK as well as mediate the translocation of polypeptides across the ER membrane and complex with caspase-7 and caspase-12 (Rao et al., 2002b). They are induced when the ER is subjected to stress, and act to reduce ER injury (Bertolotti et al., 2000; Liu et al., 1998). As mentioned previously, studies have demonstrated that PC reduces ER stress, and we speculated that GRP plays an important role in the establishment of ischemic tolerance. Therefore, we investigated changes in GRP78 and GRP94 expression with Western blot and immunohistochemical analysis in the brains after PC, and compared the results with those of the ischemic tolerance time window. The results of the GRP expression investigation are summarized in Fig. 3. In the sham-control brains, the band for GRP78 was weak, but it gradually became dense and peaked 2 days after PC. At 7 days, the band returned almost to the control level. Quantitative analysis revealed that the density of the band increased with statistical significance from 1 hour to 2 days after PC (P <0.05 at 1 hour, P <0.01 at 4 hours and 1 and 2 days). With immunohistochemical analysis, mild immunoreactivity for GRP78 was confirmed in the control brains. The perinuclear staining pattern was compatible with the fact that this is an ER resident protein (Nakagawa et al., 2000; Peters et al., 1991). The immunoreactivity gradually became dense, and peaked 2 days after PC. At 7 days, however, the immunoreactivity became mild. Western blot analysis of GRP94 revealed no definite change in its level of expression. Quantitative analysis showed a slight but statistically nonsignificant increase after PC. Immunohistochemical analysis for GRP94 also revealed that the induction of GRP94 was mild.
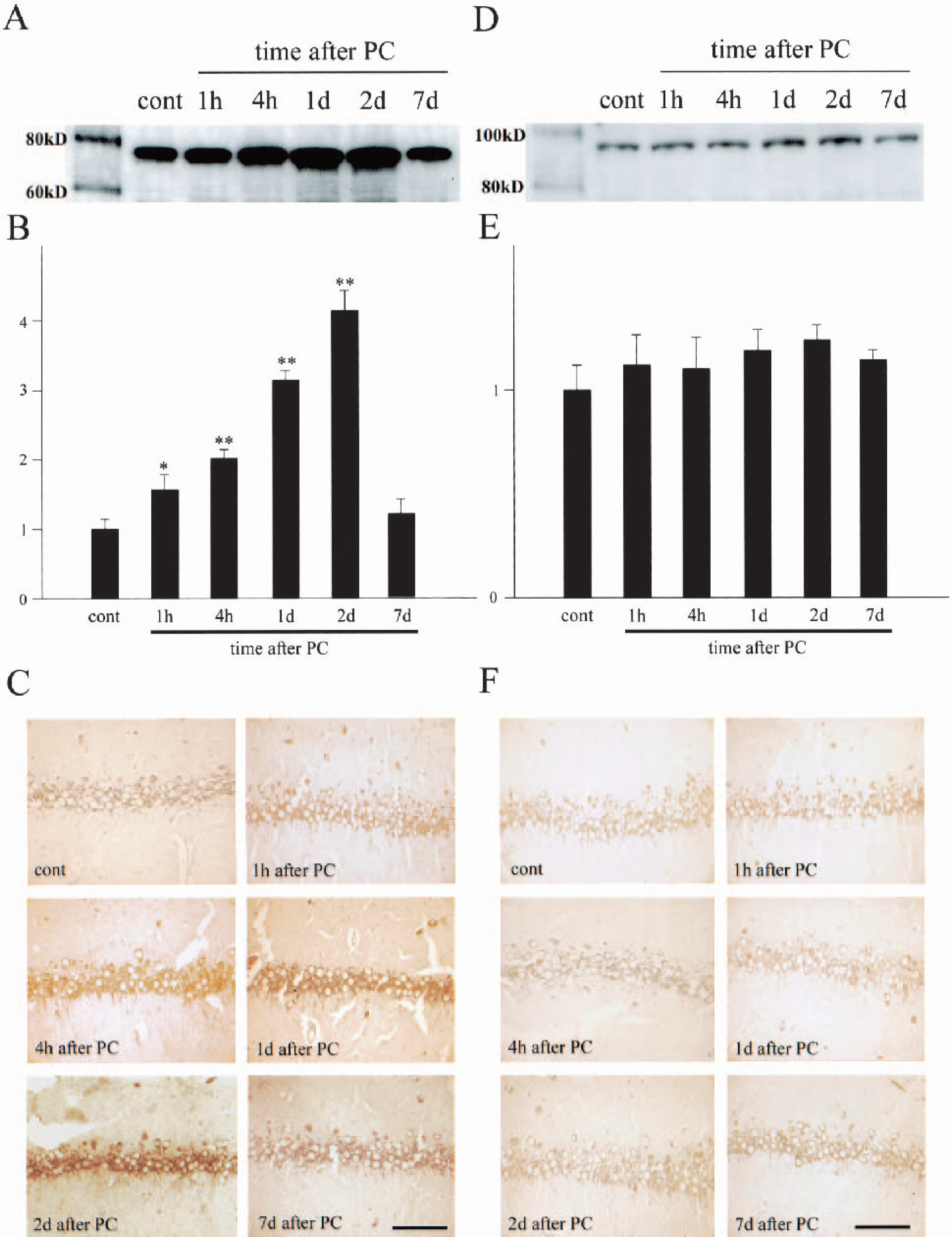
Change in expression of GRP78 and GRP94 at the hippocampal CA1 pyramidal cell layer.
The results of the investigation of the ischemic tolerance time window are summarized in Fig. 4. Without PC, more than 90% of the hippocampal CA1 neurons degenerated and became TUNEL positive 2 days after lethal ischemia. When the time interval between PC and 5 minutes of ischemia was 4 hours, the results from cresyl violet staining and a TUNEL study were essentially the same as those without PC; more than 90% of the neurons degenerated and became TUNEL positive. When 5 minutes of ischemia were induced 1 or 2 days after PC, about 80% of the neurons remained viable and only 20% of the neurons became TUNEL positive. When the interval was 7 days, however, the neuroprotective effect of PC was not observed; more than 90% of the neurons degenerated and became TUNEL positive. The results of quantitative analysis for cresyl violet staining and the TUNEL study are compatible with a previous report (Sugawara et al., 2001). In the present study, we found that GRP78 overexpression was correlated to the establishment of ischemic tolerance in a temporal manner.
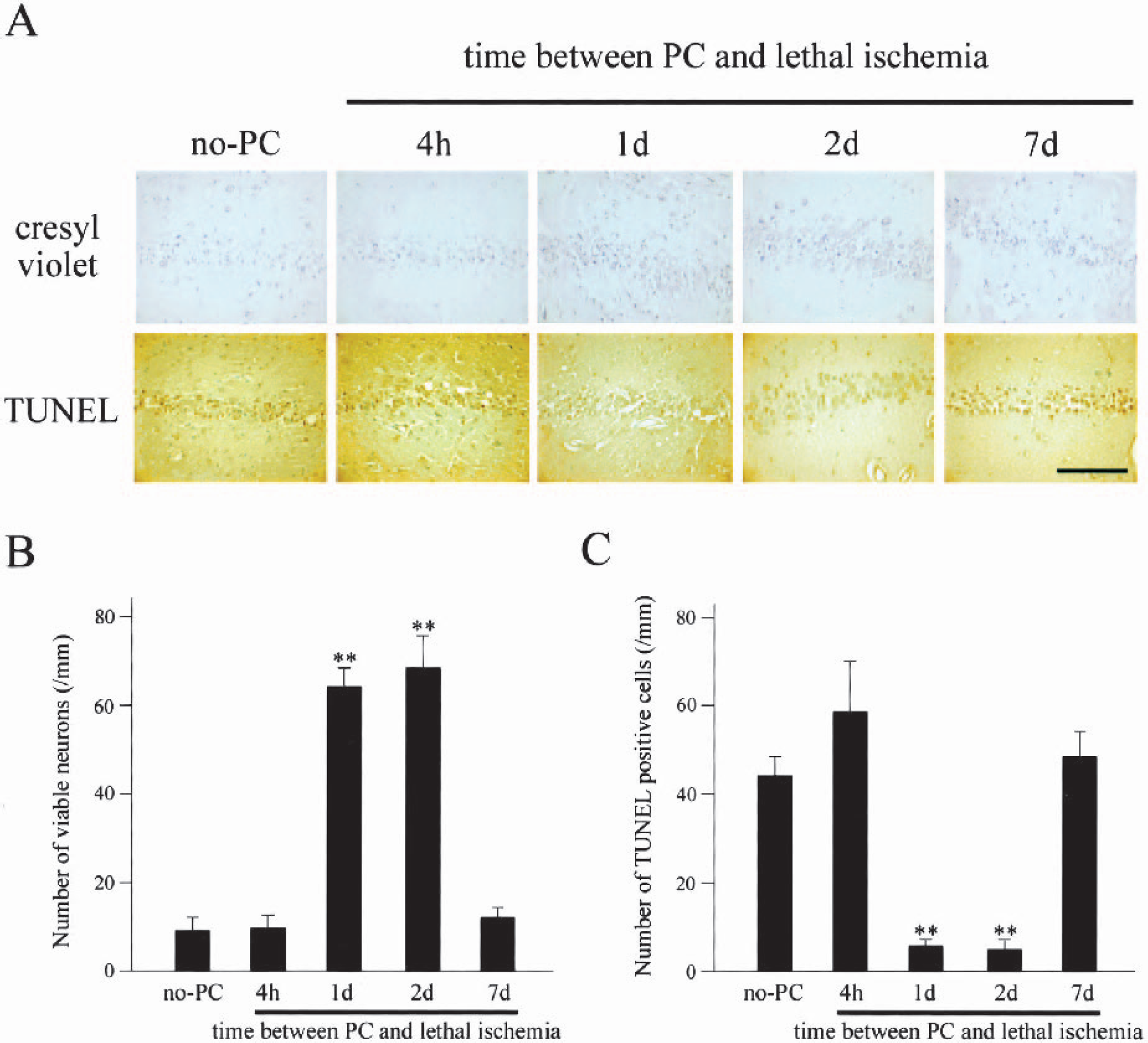
Interval between ischemic preconditioning (PC) and 5 minutes of ischemia and the degree of ischemic tolerance in the hippocampal CA1 pyramidal cell layer.
Overexpressed GRP78 interacted with PERK
To provide more direct evidence that overexpressed GRP78 reduces ER stress and induces neuronal tolerance to ischemia, we carried out a coimmunoprecipitation study. GRP78 is one of the molecules that bind with PERK and inhibit its phosphorylation, thus preventing cell death (Bertolotti et al., 2000; Yu et al., 1999). Protein lysates were immunoprecipitated with an anti-PERK antibody and then analyzed with Western blotting for GRP78. With this experiment, we detected the amount of GRP78 bound with PERK. The same membrane was subsequently used for PERK immunoblotting to confirm that the change in GRP78 band density was not caused by a change in PERK expression. The results are shown in Fig. 5A for GRP78 and PERK detection, and the relative OD analysis is shown in Fig. 5B. In the control brains, a substantial amount of GRP78 and PERK binding was confirmed (arrow in panel A). In the no-PC brains, the band density was about half that in the control brains 1 hour after 5 minutes of ischemia, which indicates that about half of the GRP78 was detached from PERK. In the PC brains without 5 minutes of ischemia, the band was about six times denser, indicating that overexpressed GRP78 bound with PERK. In the PC brains 1 hour after 5 minutes of ischemia, the band became milder than in those without 5 minutes of ischemia. However, the density of the band was still about four times stronger than in the no-PC brains after lethal ischemia. This indicates that a surplus amount of GRP78 prevented PERK from phosphorylating. The membrane was also used for PERK detection, but there were no changes in total PERK expression levels (Fig. 5A, lower panel). Of note is that the phosphorylated form of PERK (lighter arrowhead in Fig. 5A) showed slower mobility in the gel than the unphosphorylated form (darker arrowhead), which is compatible with a previous report (Kumar et al., 2001). Five minutes of ischemia increased PERK phosphorylation and PC partially prevented it. Quantitative analysis revealed that PC increased GRP and PERK binding, whereas 5 minutes of ischemia significantly decreased binding.
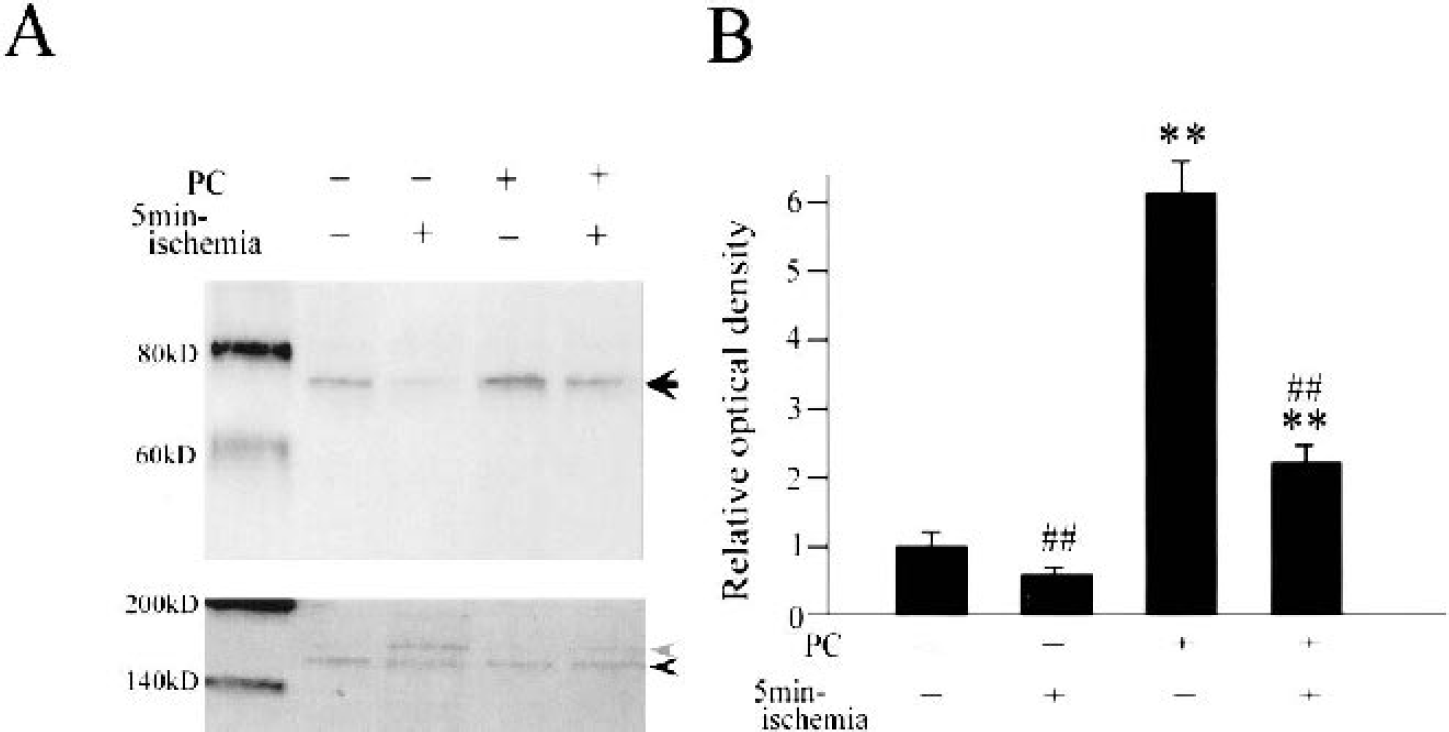
Effects of ischemic preconditioning (PC) and 5 minutes of ischemia on the interaction between PERK and GRP78.
Phosphorylated form of PERK was decolocalized with a strong immunoreactivity for GRP78
To further confirm that GRP78 overexpression prevents PERK phosphorylation, we performed fluorescent double staining for GRP78 and phospho-PERK. As seen in Fig. 6, the control brain showed mild immunoreactivity for GRP78 (FITC, green) and an almost undetectable immunoreactivity for phospho-PERK (Texas Red, red). In the no-PC brains 1 hour after 5 minutes of ischemia, the signal for phospho-PERK was substantially increased, but that for GRP78 showed only a slight change. In contrast, the PC brains without 5 minutes of ischemia showed a strong signal for GRP78, with an almost undetectable level of phospho-PERK. In the PC brains 1 hour after 5 minutes of ischemia, a strong signal for GRP78 was retained and that for phospho-PERK also became stronger. However, the degree of increase in the phospho-PERK signal was milder in the PC brains. Of importance is that neurons with a strong signal for GRP78 showed almost no signal for phospho-PERK (arrowheads in Fig. 6), and neurons with a strong signal for phospho-PERK showed almost no signal for GRP78 (arrows in Fig. 6). This finding suggests that overexpression of GRP78 inhibits phosphorylation of PERK in hippocampal CA1 neurons.
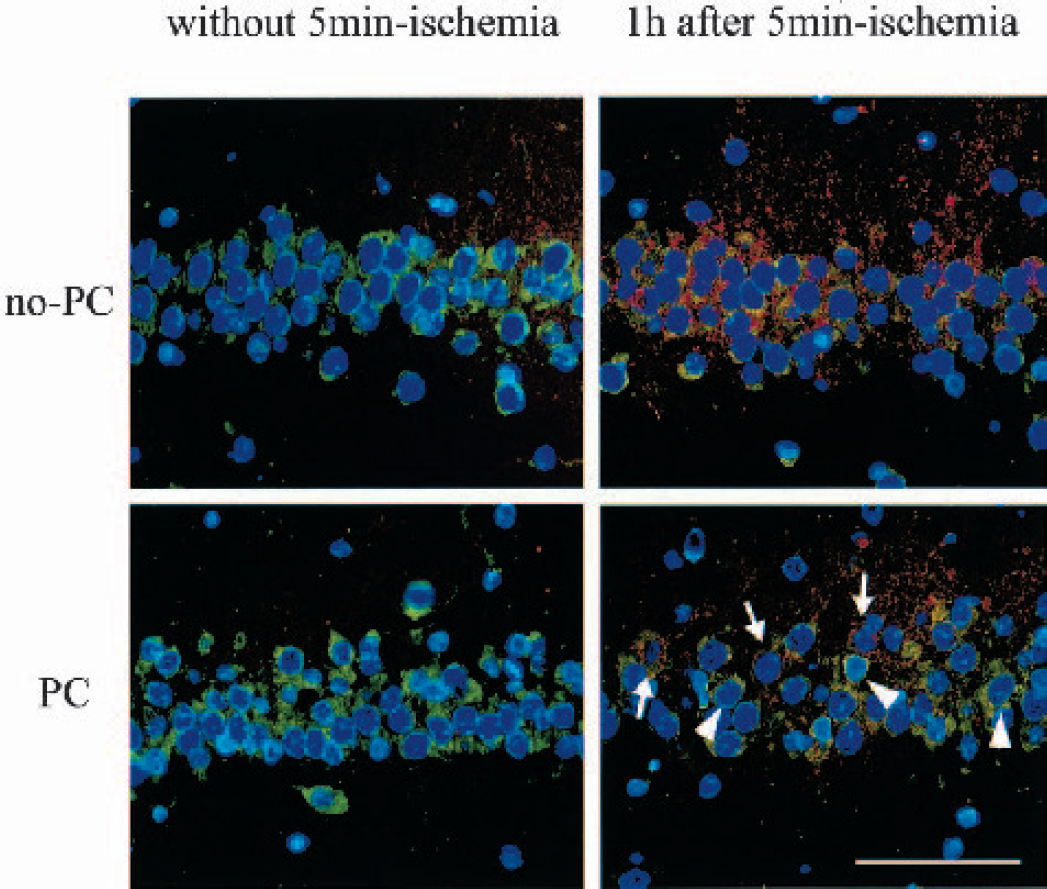
Fluorescent double staining for GRP78 (FITC, green) and the phosphorylated form of PERK (Texas Red, red). Nuclei were counterstained with DAPI (blue). In the brains without treatment, weak immunoreactivity for GRP78 was confirmed, although that for the phosphorylated form of PERK was barely detectable. One hour after 5 minutes of ischemia without ischemic preconditioning (no-PC), immunoreactivity for the phosphorylated form of PERK became more intense, but that for GRP78 showed almost no change. In the brains 2 days after PC without 5 minutes of ischemia, immunoreactivity for GRP78 became intense, but that for the phosphorylated form of PERK showed no change. In the brains with PC and 5 minutes of ischemia, both GRP78 and the phosphorylated form of PERK were increased. Of note is that neurons with strong immunoreactivity for GRP78 showed almost no signal for the phosphorylated form of PERK (arrowheads), but those with a very weak signal for GRP78 showed quite a strong signal for the phosphorylated form of PERK (arrows), indicating that GRP78 prevents phosphorylation of PERK. Scale bar = 50 μm.
DISCUSSION
Our study provides evidence to support our speculations that (1) ER stress is involved in DND, (2) PC prevents DND by reducing ER stress, and (3) ER stress reduction by PC is due to overexpression of the ER resident molecular chaperones. Phosphorylation of eIF2α and PERK reflects exposure of the hippocampal CA1 neurons to ER stress. PC prevented these phenomena, which are, at least in part, mediated by GRP78 (but not GRP94) overexpression. GRP78 involvement in neuroprotection was confirmed by (1) comparing time windows of its induction and ischemic tolerance, (2) an immunoprecipitation study, and (3) fluorescent double staining. That GRP78, but not GRP94, decided neuronal susceptibility to ischemic injury is compatible with a previous report (Yu et al., 1999). The molecular mechanisms of GRP78 neuroprotection and ER stress-induced neuronal cell death elucidated with this study are schematically summarized in Fig. 7.
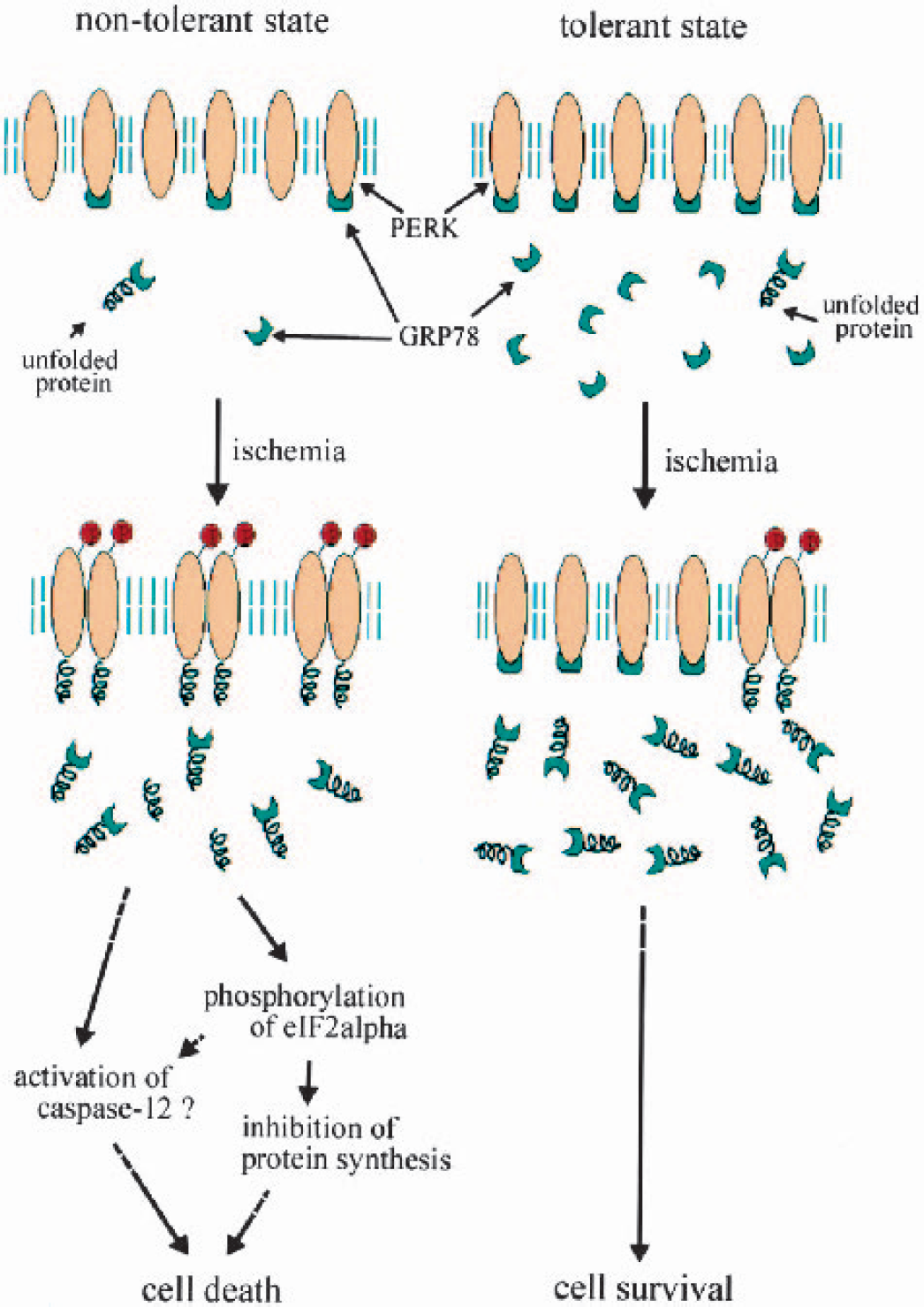
Mechanisms of neuroprotection by GRP78 overexpression. In the nontolerant state, neurons possessed little or no surplus of GRP78, but the cell death signal was not activated if no injury was induced. Once severe injury was induced, such as 5 minutes of ischemia, many unfolded proteins were produced in the ER lumen and they phosphorylated PERK. Because the amount of GRP78 was small, it could not prevent phosphorylation of PERK, and phosphorylation of eIF2α, activation of caspase-12, and neuronal cell death occurred. In the tolerant state, such as 2 days after ischemic PC, there was a large surplus of GRP78. When severe injury was imposed, such as 5 minutes of ischemia, a large amount of unfolded protein was produced, but a large surplus of GRP78 inhibited their interaction with PERK. Therefore, phosphorylation of PERK was prevented, and phosphorylation of eIF2α, activation of caspase-12, and neuronal cell death will seldom occur.
eIF2α phosphorylation blocks the initial step of new protein synthesis (DeGracia et al., 2002; Pain, 1996). This phenomenon is observed in cells under ER stress and is known as the unfolded protein response (Harding et al., 1999; Nakagawa et al., 2000; Pain et al., 1996). Previous reports have demonstrated that neurons that degenerate later show a marked unfolded protein response (Althausen et al., 2001; Kumar et al., 2001). The eIF2α phosphorylation mechanism is controversial, but it was recently reported that PERK phosphorylation plays an important role in the ischemic brain (Kumar et al., 2001). Therefore, phosphorylation of PERK should be a key step in ER stress-induced neuronal cell death in the ischemic brain. In the normal brain, the expression level of GRP78 is not very high, but because the amount of unfolded protein in the ER lumen is small, most of PERK remains nonphosphorylated (Fig. 7, nontolerant state before ischemia). When severe ischemia is induced, however, unfolded protein increases in the ER lumen (Hu et al., 2000). Some of the protein detaches GRP78 from PERK and binds with PERK, which causes dimerization and phosphorylation of PERK and then phosphorylation of eIF2α (Fig. 7, nontolerant state after ischemia) (DeGracia et al., 2002; Kumar et al., 2001). Whether eIF2α phosphorylation causes cell death remains uncertain, but ample evidence suggests that it causes apoptotic neuronal cell death. In the brain after PC, in contrast, there is a surplus of GRP78 (Fig. 7, tolerant state before ischemia). Even after severe ischemia, most of the unfolded protein binds with GRP78 in the ER lumen and does not cause phosphorylation of PERK (Fig. 7, tolerant state after ischemia). Based on the findings of the current study, we postulate that eIF2α phosphorylation takes place to only a slight degree, and ER stress-induced neuronal cell death seldom occurs.
Many reports indicate an important role for ER stress in neuronal cell death. Among these, much attention has been focused on depletion of ER calcium stores (Parsons et al., 1997; Paschen and Doutheil, 1999; Paschen and Frandsen, 2001). Dantrolene, which inhibits calcium release from the ER, can prevent DND (Wei and Perry, 1996). When the calcium store is depleted from the ER, the amount of unfolded protein increases in the ER lumen (Verkhratsky and Petersen, 2002). Therefore, depletion of ER calcium stores might be the initial common step in ER stress-induced neuronal cell death. However, neurons in the ischemic brain are deficient in ATP, which is necessary for the folding of proteins (Nguyen and Bensaude, 1994). In addition, reactive oxygen species may play an active role in ER stress (Paschen et al., 2001). Therefore, the mechanisms of unfolded protein production in the ER lumen may be diverse, and further study is required to elucidate how protein in the ER became unfolded in this model.
In the present study, the degree of eIF2α phosphorylation at 1 hour was more prominent than that of PERK. As reviewed by Clemens (2001), there are several mechanisms in eIF2α phosphorylation. Martín de la Vega et al. (2001) reported that eIF2α phosphatase plays an active role, and we cannot exclude the possibility that enzymes other than PERK are also involved in eIF2α phosphorylation. eIF2α phosphorylation is often associated with neuronal cell death in vivo, but inhibition of new protein synthesis could be a protective reaction of the cells; it preserves the cellular ATP level and reduces the load on the ER (Sherman and Goldberg, 2001). Whether phosphorylation of eIF2α triggers the cell death program or is only an epiphenomenon is not clear at present; however, we propose that it is implicated in the apoptotic cell death program. Other studies support the finding in our model that eIF2α phosphorylation causes DND (Srivastava et al., 1998; Zinszner et al., 1998). For example, the phosphorylated form of eIF2α increases some proapoptotic proteins such as activating transcription factor-4 and the C/EBP-homologous protein (Jousse et al., 1999). To elucidate the mechanisms of ER stress-induced cell death more precisely, further studies are required.
The role of mitochondria in DND has been extensively investigated. Release of cytochrome c from mitochondria is a pivotal step in the process of ischemic neuronal cell death (Fujimura et al., 1998; Eldadah and Faden, 2000; Sugawara et al., 2002), though the mechanism of this release is not fully understood. Superoxide is involved in cytochrome c release, but its energy in vivo is not high enough to directly break lipid bilayers (Halliwell and Gutteridge, 1999). Calcium also causes cytochrome c release from mitochondria (Zamzami and Kroemer, 2001). One study showed that the ER and mitochondria have numerous close contacts and that calcium released from the ER can influence the function of mitochondria (Rizzuto et al., 1998). Therefore, damage in the ER may be related to cytochrome c release from mitochondria. We believe that the damaged ER and mitochondria are intimately linked, and that “mitochondrial cell death” and “ER-induced cell death” cannot be distinctly separated in this model.
The molecular mechanism of PC on neuroprotection is not fully elucidated. Previous reports showed that bcl-2 induction, inhibition of cytochrome c release, and activation of nuclear factor-κB (NF-κB) play pivotal roles (Barone et al., 1998; Blondeau et al., 2001; Shimizu et al., 2001). Although these pathways seem so diverse, we note that all of them could be related to ER stress. The bcl-2 protein is localized on the ER as well as on mitochondrial and nuclear membranes, and is implicated in regulation of ER calcium homeostasis (Rudner et al., 2002). Overexpression of the bcl-2 protein may thus reduce ER stress after ischemia. Inhibition of cytochrome c release from mitochondria could be the result of ER stress reduction, as calcium release from the ER may be the cause of this process. NF-κB is activated by ER stress, and the fact that this activation is necessary for PC indicates that the ER plays a pivotal role in PC (Pahl and Baeuerle, 1997).
The results obtained from this study suggest the crucial role of the ER in DND and PC. One cannot explain all the phenomena of neuronal cell injury by focusing on the ER. For example, DNA repair enzymes such as Ku-70 and APE/Ref-1, or cytosolic molecular chaperones such as heat shock protein 72 (which may by implicated in reduction of ischemic neuronal injury), do not seem to be related to ER stress (Chen et al., 1996; Fujimura et al., 1999; Sugawara et al., 2001). However, with the scenario we postulate here, we can reasonably explain many findings of molecular events in ischemic neuronal cell death and PC. Not only protein synthesis inhibition and ER calcium store depletion, but also other cellular processes related to DND and PC, such as caspase activation, cytochrome c release, and change in gene expression, can be attributed to ischemic disturbance or damage to the ER.
Footnotes
Acknowledgments
The authors thank Liza Reola, Bernard Calagui, and Ghezal Omar for technical assistance, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for figure preparation.