
Abstract
Folding and processing newly synthesized proteins are vital functions of the endoplasmic reticulum that are sensitive to a variety of stress conditions. The unfolded protein response is activated to restore endoplasmic reticulum function impaired by stress. While we know that brain ischemia impairs endoplasmic reticulum function, the role of unfolded protein response activation in post-ischemic recovery of neurologic function is only beginning to emerge. Here, we summarize what is known about endoplasmic reticulum stress and unfolded protein response in brain ischemia and discuss recent findings from myocardial ischemia studies that could help to advance research on endoplasmic reticulum stress and unfolded protein response in brain ischemia.
Introduction
The endoplasmic reticulum (ER) is the subcellular compartment in which all newly synthesized membrane and secretory proteins are folded and processed, reactions that require high intraluminal calcium activity.1–3 ER function is sensitive to many stress conditions, and is therefore impaired in a variety of human diseases of major clinical significance including brain ischemia/stroke, degenerative diseases, myocardial ischemia/heart failure, cancer, and diabetes.4–9 Impaired ER function (ER stress) can lead to cell death.
10
To restore ER function, cells activate a battery of adaptive processes via the unfolded protein response (UPR).
11
UPR has three branches (Figure 1) that are controlled by stress sensor proteins in the ER membrane: activating transcription factor 6 (ATF6), inositol-requiring enzyme-1 (IRE1), and the protein kinase RNA-like ER kinase (PERK).
Scheme of the unfolded protein response (UPR). UPR has three response branches controlled by stress sensor proteins in the ER membrane, the ATF6, the inositol-requiring enzyme-1 (IRE1), and the protein kinase RNA-like ER kinase (PERK). Activated ATF6 translocates to the Golgi where it is cleaved by proteases to the short form (sATF6) that translocates to the nucleus and activates expression of sATF6 target genes coding for ER chaperons, and proteins involved in ER-associated degradation (ERAD) and autophagy. ER stress turns IRE1 into an endonuclease that cleaves X-box binding protein-1 (Xbp1) mRNA. IRE1-induced splicing of Xbp1 mRNA triggers a frame-shift of the coding region, and formation of a new 54-kDa protein, XBP1s. XBP1s is a transcription factor that regulates expression of genes coding for ER chaperones, proteins involved in ERAD and in the hexosamine biosynthetic pathway that activates O-linked β-N-acetylglucosamine modification of proteins (O-GlcNAcylation). Activated PERK is turned into a kinase that phosphorylates eIF2α and thereby blocks the initiation process of translation. This promotes translation of Atf4 mRNA into the ATF4 protein, a transcription factor that activates expression of genes coding for pro-apoptosis protein including CHOP.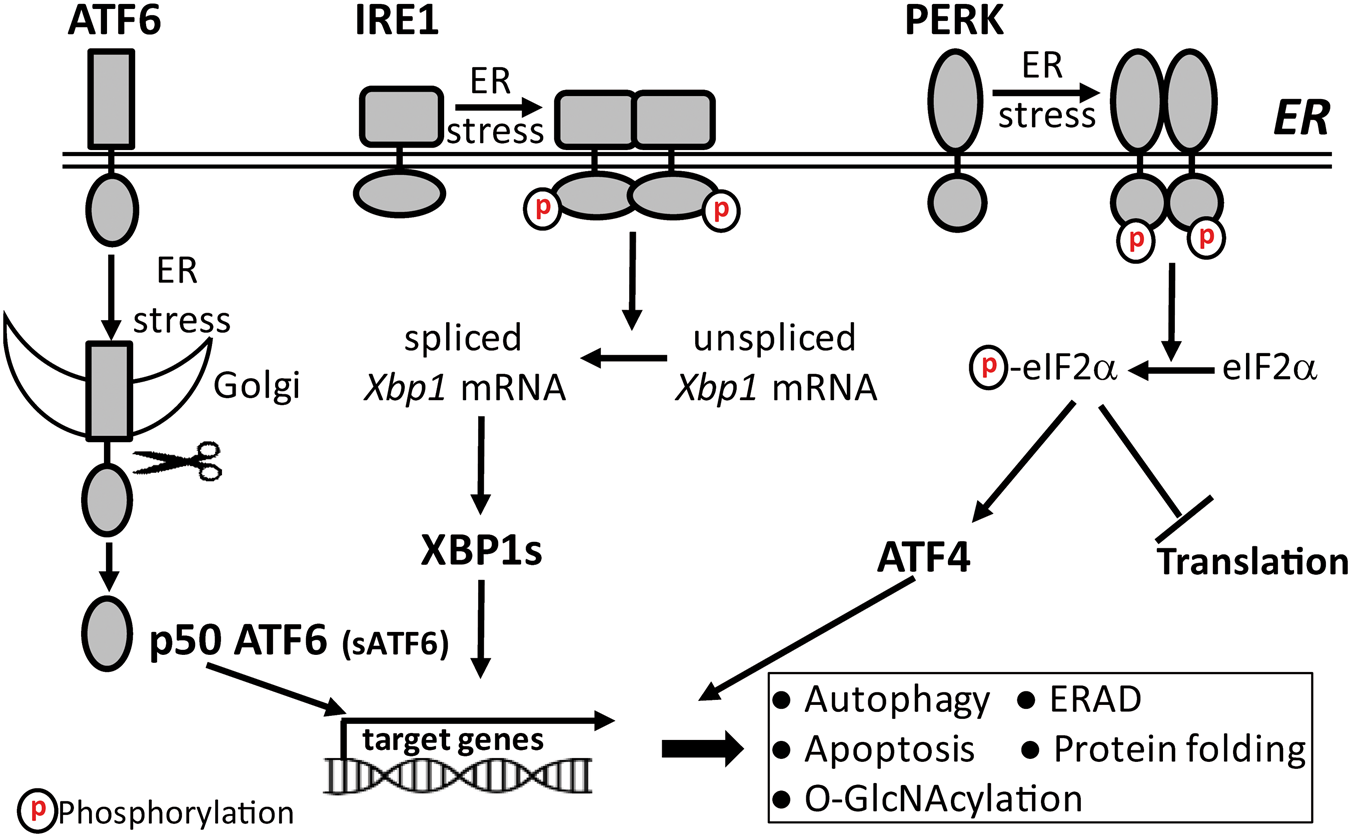
Our knowledge about the mechanisms underlying ER stress-induced activation of UPR, and the role of individual UPR branches in the fate of these cells is based primarily on results from in vitro experiments using pharmacologic tools to disturb ER-resident processes. The traditional, widely accepted view is that glucose-regulated protein 78 (GRP78) is bound to ATF6, IRE1, and PERK in non-stressed cells, thereby keeping these stress sensors in the resting state. 12 However, under stress conditions when unfolded proteins accumulate in the ER, GRP78 dissociates from stress sensor proteins resulting in their activation to increase folding capacity. 12 This view has been challenged because unfolded proteins can bind directly to ER stress sensor proteins to activate UPR. 13 Recently, we have reported that a marked increase in expression of GRP78 does not modify the extent of UPR activation triggered by transient cerebral ischemia, which supports a model of UPR activation independent of GRP78 dissociation from ER stress sensor proteins. 14 Under ER stress conditions, the protein folding demand exceeds folding capacity. Activation of UPR triggers a variety of adaptive responses to restore the folding demand/capacity balance, and to clear unfolded/misfolded proteins from the ER, thereby reestablishing cellular homeostasis. However, if ER stress persists and the protein folding demand/capacity balance cannot be restored, UPR triggers apoptosis.15–17
Each branch of UPR plays a unique role in the recovery of ER function. Upon activation, ATF6 translocates to the Golgi where it is cleaved by proteases to the short form (sATF6), an active transcription factor that translocates to the nucleus, and binds to ER stress elements in the promoter of sATF6 target genes to activate their expression (Figure 1).18,19 Many of the sATF6-dependent gene products protect cells from stress-induced damage. These include the ER chaperons and folding enzymes GRP78, GRP94, and protein disulfide isomerase (PDI), as well as ER-associated degradation (ERAD) proteins such as HMG-CoA reductase degradation protein 1 (Hrd1) and Derlin-3 (Derl3).20–22 ERAD is a protein degradation system that clears accumulated unfolded/misfolded proteins from the ER. 23
ER stress converts IRE1 to an active endonuclease that cleaves X-box binding protein-1 (Xbp1) mRNA (Figure 1). IRE1-mediated splicing of Xbp1 mRNA triggers a frame-shift of the coding region, and formation of a new 54-kDa protein, XBP1s.24,25 XBP1s is a transcription factor that regulates expression of a subset of genes coding for ER-resident chaperons and proteins involved in ERAD. 26 Recently, it was reported that in the heart XBP1s activates expression of the gene coding for glutamine fructose-6-phosphate aminotransferase-1 (GFAT1), the rate-limiting enzyme of the hexosamine biosynthetic pathway (HBP). 27 HBP generates uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), the substrate for O-linked β-N-acetylglucosamine (O-GlcNAc) modification of proteins (O-GlcNAcylation). 28 IRE1 also cleaves other mRNAs through a process known as regulated IRE1-dependent decay (RIDD). 29
When PERK is activated by autophosphorylation, it becomes an active kinase that specifically phosphorylates the alpha subunit of the eukaryotic initiation factor-2 (eIF2α) at Ser 51 (Figure 1). 30 This blocks translation at the initiation step by converting eIF2α to an inhibitor of the guanine nucleotide-exchange factor eIF2B, thereby suppressing a process that is critical for initiating translation. 31 While PERK activation triggers a global suppression of translation, it paradoxically activates translation of certain mRNAs with several short open reading frames upstream of the main coding region (uORF), such as the mRNA that encodes for activating transcription factor 4 (ATF4). This complex structure favors translation of the main open reading frame when phosphorylated eIF2α levels are high and thus eIF2B activity is low.32,33 ATF4 regulates expression of the C/EBP-homologous protein (CHOP, also known as GADD153), a key player in ER stress-induced apoptotic cell death.34–36
Thus, when ER function is impaired by stress, cells activate UPR to restore ER function. UPR recovery processes include a shutdown of translation to decrease the ER load of newly synthesized proteins that must be folded, a genetic program to increase protein folding and processing capacity, and activation of the ERAD pathway to remove unfolded/misfolded proteins from the ER. Additional adaptive processes downstream of UPR activation include autophagy37,38 and O-GlcNAc modification of proteins. 27 If ER stress persists, and cellular homeostasis is not restored, UPR triggers apoptosis, which is mainly mediated through the IRE1 and PERK branches.
Results from in vitro studies using HEK293 cells support the notion that a switch from pro-survival to pro-apoptosis processes is modulated by differences in the duration of ATF6, IRE1, and PERK activation. 39 Specifically, under conditions of persistent ER stress, only the PERK UPR branch shows long-lasting activation, as indicated by eIF2α and PERK phosphorylation, and CHOP expression. 39 However, the pro-apoptosis effect of long-lasting PERK activation is shifted toward pro-survival when IRE1 activation-induced splicing of Xbp1 mRNA is experimentally prolonged. 39
ER stress is implicated in the pathophysiology of a wide range of human diseases (see above). Therefore, UPR has attracted the attention of many biomedical researchers, and small molecules that target UPR for therapeutic purposes have been identified or are under development (for recent reviews, see Hetz et al. 40 and Maly and Papa 41 ). Recently, small molecules that target the ATF6 UPR branch with high specificity have been identified,42,43 and the non-toxic activators of endogenous ATF6 could be of major interest in stroke (see below). In experimental brain ischemia, several strategies have been used to restore ER function impaired by ischemia. These include the activator of GRP78 expression 1-(3,4-dihydroxy-phenyl)-2-thiocyanato-ethanone, 44 the chemical chaperon 4-phenylbutyrate, 45 and Salubrinal,46,47 a specific inhibitor of eIF2α dephosphorylation. 48 Together, these results suggest that restoration of ER function is a promising target for therapeutic intervention in brain ischemia.
Brain ischemia
About 20 years ago, we hypothesized that disturbances of ER calcium homeostasis may contribute to ischemic cell damage, because depletion of ER calcium stores triggers stress responses that resembles, in many respects, those induced by transient cerebral ischemia. 49 This was particularly evident when comparing the effect of transient ischemia to the effect of a depletion of ER calcium stores on protein synthesis. It is well established that transient brain ischemia triggers a shutdown of translation at the initiation step that is associated with a decrease in the activity of the initiation factor eIF2 resulting from phosphorylation of the alpha subunit, and a disaggregation of polyribosomes.50–53 However, the molecular mechanisms responsible for the post-ischemic shutdown of translation at the initiation step were not known. Since an isolated depletion of ER calcium stores also triggers a shutdown of translation at the initiation step that is associated with phosphorylation of eIF2α and disaggregation of polyribosomes,54–56 it was proposed that brain ischemia impairs ER function.4,49 This suggests that the post-ischemic shutdown of translation is not per se a pathologic process but rather part of a stress response that is widely known today as UPR.
The ER stress hypothesis triggered many experimental studies to determine whether brain ischemia impairs ER function and activates UPR, assess the role of ER stress in the fate of post-ischemic neurons, and develop strategies that modulate ER stress pathways to improve outcomes. Specifically, expression of target genes of the three UPR branches, including GRP78, GADD153/CHOP, ER stress protein 72 (ERp72), and PDI, is up-regulated after brain ischemia.57–63 Further, splicing of Xbp1 mRNA is activated in post-ischemic brains,14,64 and eIF2α is phosphorylated in a PERK-specific manner.65,66 It should be noted that post-ischemic activation of UPR is unequivocal proof that transient ischemia impairs ER function, and is also an indicator that cells activate adaptive responses to cope with this pathologic state. However, is post-ischemic activation of UPR sufficient to restore ER function impaired by ischemia? Kumar et al. have proposed that dysfunction of UPR after brain ischemia contributes to neuronal death. 67
Many additional observations support the notion that brain ischemia/stroke impairs ER function, and that UPR is a potential target for therapeutic intervention. For example, preconditioning activates GRP78 expression, and thereby reduces ER stress triggered by brain ischemia. 62 Ischemia-induced apoptosis is reduced in Chop-/- mice, implying that CHOP has a role in ischemia-induced neuronal cell death. 16 Inducing UPR by low levels of tunicamycin, an inhibitor of ER protein glycosylation, or thapsigargin, an irreversible inhibitor of the ER calcium pump, confers protection in experimental stroke. 68 And salubrinal, a highly selective small molecule inhibitor of eIF2α dephosphorylation, 48 is neuroprotective in experimental forebrain ischemia and stroke.47,69 Collectively, these reports support a role for ER stress in brain ischemia/stroke, but they do not provide information on the cells involved because global approaches were used in each case to analyze or manipulate ER pathways.
Despite the wealth of information on ER stress in brain ischemia, we still know very little about the role of individual UPR branches in the fate and function of post-ischemic neurons and in brain ischemia/stroke outcome. This knowledge is, however, critical to the development of therapeutic strategies that modulate UPR pathways to improve post-ischemic recovery of neurologic function impaired by ischemic stress. Our group is particularly interested in clarifying the role of each UPR branch in the fate of neurons exposed to transient ischemia because recovery of neurologic function ultimately defines quality of life for patients suffering from an ischemic attack. Recently, a mouse model of global deletion of Atf6 was used to evaluate the role of the ATF6 UPR branch in stroke. 70 Notably, Atf6-/- mice had larger infarcts than wild-type mice, and reduced astroglial activation; however, a direct effect of Atf6 deletion on post-ischemic neurologic function was not reported. To assess the role of the ATF6 UPR branch in recovery of neurologic function after stroke, we generated a conditional and inducible sATF6 knock-in mouse to express sATF6 in forebrain neurons. 14 Notably, forced activation of sATF6 expression reduces infarct volume and improves neurologic function after stroke. This suggests that modulating UPR branches predominantly in neurons is sufficient to improve outcome after stroke.
Myocardial ischemia
In 2006, Dr. Glembotski and his group began to generate and work with UPR-specific transgenic mice to investigate the role of ER stress in ischemia-induced damage to the heart. 8 Earlier observations linked aberrant ER protein quality control to dilated cardiomyopathy, and aortic constriction-induced heart failure to prolonged ER stress and apoptosis.71,72 Several of the ATF6-inducible gene products were reported to protect cardiomyocytes from a variety of stress conditions.73,74 Therefore, Dr. Glembotski’s group generated a new mouse model with tamoxifen-inducible activation of the ATF6 UPR branch specifically in cardiomyocytes. 8 Notably, forced ATF6 expression markedly protects hearts from ischemia/reperfusion injury. Microarray analysis on hearts of ATF6 transgenic mice identified many ATF6-dependent genes including Derl3 and Hrd1, which encode for ER proteins involved in ERAD. 75 Derl3 is upregulated in the myocardial infarct border zone and protects cardiomyocytes from ischemic stress-induced damage, while Hrd1 decreases cardiac pathology associated with pressure overload.22,76 Thus, cell-specific genetic modulation of UPR branches provides important information about the role of individual UPR pathways in the disease under investigation, and helps to identify novel targets for therapeutic intervention.
Recent studies showed that XBP1s couples the IRE1 UPR branch to the HBP, and thereby activates O-GlcNAcylation, a protein modification that protects cells under various stress conditions including robust cardioprotection from ischemia/reperfusion-induced damage.27,36 Specifically, using cardiomyocyte-specific XBP1s transgenic and Xbp1 knockout mice, the authors provided strong evidence that the post-ischemic activation of O-GlcNAc modification is XBP1-dependent, and that XBP1s induction in cardiomyocytes is sufficient to protect hearts from ischemia/reperfusion injury. 27 Considering the wealth of important information derived from cardiomyocytes-specific XBP1s transgenic and Xbp1 knockout mice, it would be of major interest to generate the respective neuron-specific mouse models to investigate the role of the IRE1/XBP1/O-GlcNAc modification axis in stroke outcome.
Conclusion
After 20 years of experimental studies on ER stress and UPR in brain ischemia, a new level of research is beginning to emerge that advances from a more descriptive approach and global manipulations of ER stress pathways to genetic modulation of individual UPR branches in a cell/organ-specific manner. 14 The successful work on the cardioprotective effects of individual UPR branches has informed new studies on the specific role of individual UPR branches in the fate and function of post-ischemic neurons. Indeed, we know today that forced activation of the ATF6 UPR branch in neurons improves outcome after stroke in a similar fashion to the protective effect of forced ATF6 activation in cardiomyocytes at risk for ischemia/reperfusion-induced damage.8,14 Further, both myocardial ischemia and transient brain ischemia activate O-GlcNAc modification of proteins,27,77 but we have not yet verified that activation of O-GlcNAcylation after brain ischemia is also XBP1s-dependent as in myocardial ischemia. However, since O-GlcNAc modification of proteins is activated in a variety of stress conditions, 27 we expect to find that the IRE/XBP1/HBP/O-GlcNAc axis is also activated after brain ischemia/stroke. Finally, our group has found that O-GlcNAcylation is activated after brain ischemia in young but not in aged mice. 77 This knowledge could have a tremendous impact on the development of new strategies to protect neurons/brains from stress-induced damage. It is, indeed, a perplexing fact that age is a key risk factor for a variety of brain pathologies that are also associated with ER dysfunction. This suggests that boosting UPR and downstream pro-survival pathways must be considered as a neuroprotective strategy to improve outcome in aged patients suffering from an episode of ischemic stress.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Department of Anesthesiology (to WP) by National Institutes of Health R01 grant NS099590 and American Heart Association grants 12SDG11950003 and 16GRNT30270003 (to WY), and by National Institutes of Health R01 grants HL095552, NS081299, and NS097554, R21 grant NS057375, and R03 grant NS078590 (to WP).
Acknowledgments
We thank Kathy Gage for her excellent editorial contributions.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.