
Abstract
The endoplasmic reticulum (ER), which plays a role in apoptosis, is susceptible to oxidative stress. Because superoxide is produced in the brain after ischemia/reperfusion, oxidative injury to this organelle may be implicated in ischemic neuronal cell death. Activating transcription factor-4 (ATF-4) and C/EBP-homologous protein (CHOP), both of which are involved in apoptosis, are induced by severe ER stress. Using wild-type and human copper/zinc superoxide dismutase transgenic rats, we observed induction of these molecules in the brain after global cerebral ischemia and compared them with neuronal degeneration. In ischemic, wild-type brains, expression of ATF-4 and CHOP was increased in the hippocampal CA1 neurons that would later undergo apoptosis. Transgenic rats had a mild increase in ATF-4 and CHOP and minimal neuronal degeneration, indicating that superoxide was involved in ER stress-induced cell death. We further confirmed attenuation on induction of these molecules in transgenic mouse brains after focal ischemia. When superoxide was visualized with ethidium, signals for ATF-4 and superoxide overlapped in the same cells. Moreover, lipids in the ER were robustly peroxidized by ischemia but were attenuated in transgenic animals. This indicates that superoxide attacked and damaged the ER, and that oxidative ER damage is implicated in ischemic neuronal cell death.
Keywords
Introduction
Activation of apoptosis is part of the pathogenesis of neuronal injury in the ischemic brain. The appearance of cells positive for terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (States et al, 1996) and DNA fragmentation (MacManus et al, 1993; Nitatori et al, 1995; Fujimura et al, 1999) has been confirmed in various types of brain ischemia models. The molecular mechanisms underlying apoptosis are diverse and are sometimes the cause of controversy, since many subcellular organelles are likely to be involved (Ferri and Kroemer, 2001). Of note is the involvement of the endoplasmic reticulum (ER) in ischemic neuronal cell death (Paschen and Doutheil, 1999). Unfolded protein accumulation in the ER lumen (Hu et al, 2000), eukaryotic initiation factor 2α (eIF2α) phosphorylation (Althausen et al, 2001), protein synthesis inhibition (Nowak et al, 1985; DeGracia et al, 2002), ER lumen calcium depletion (Paschen, 1996; Paschen and Doutheil, 1999), and caspase-12 induction (Mouw et al, 2003) all suggest an active role for the ER in neuronal cell death after ischemia.
The molecular mechanisms of ER damage in ischemic neurons are not clear (Berridge, 2002), but a number of in vitro studies has demonstrated that ER function is sensitive to oxidative stress (Dreher et al, 1995; Racay et al, 1995; Viner et al, 1996). The ER is one of the organelles that produces reactive oxygen species (ROS), which are also robustly produced in the ischemic brain (Chan, 1996, 2001; Lewén et al, 2000). We therefore speculated that ROS are involved in ischemic neuronal cell death because they damage the ER.
Cells show various responses when stress is imposed on the ER. Protein synthesis inhibition and ER resident molecular chaperone induction are known as the ‘unfolded protein response’ (Ma and Hendershot, 2001). These reactions are attempts to reduce the load on the ER and prevent consequent cellular catastrophe. When ER stress is very severe, however, the cells activate the apoptotic machinery. Proteins on the ER membrane are activated and dispatch cell death signals to other parts of the cell (Nakagawa et al, 2000; Calfon et al, 2002). In the ischemic brain, RNA-dependent protein kinase-like ER eIF2α kinase (PERK) is activated (Kumar et al, 2001). This in turn phosphorylates eIF2α, which induces activating transcription factor-4 (ATF-4) (Harding et al, 2002). Activating transcription factor-4 then induces the C/EBP-homologous protein (CHOP), causing apoptosis (Wang et al, 1998; Harding et al, 2000, Harding et al, 2002). Hence, induction of ATF-4 and CHOP is the key step in ER stress-induced neuronal cell death after ischemia.
We have reported that human copper/zinc superoxide dismutase (SOD1) transgenic (Tg) rats showed attenuated phosphorylation of PERK and eIF2α (Hayashi et al, 2003b). If oxidative ER damage is involved in ischemic neuronal cell death, we should observe neuronal induction of ATF-4 and CHOP, and SOD1 overexpression should attenuate such neuronal induction. In this study, we investigated changes in ATF-4 and CHOP expression after brain ischemia and compared the results between wild-type (Wt) and Tg animals. We also carried out double staining for ATF-4 and oxidized ethidium, a marker for superoxide, and measured ER lipid peroxidation as possible mechanisms of oxidative ER damage.
MATERIALS AND METHODS
Experimental Paradigms
To investigate the possible role of superoxide in ER stress-induced neuronal cell death, we used two different in vivo ischemia models. Earlier studies found that hippocampal CA1 pyramidal cells are selectively vulnerable to ischemia and undergo apoptosis after transient ischemia (Kirino, 1982; Sugawara et al, 1999). Ample evidence that the ER plays an active role in ischemic neuronal cell death has been obtained with this model. Therefore, we investigated the effect of Tg SOD1 overexpression on ATF-4 and CHOP expression using a rat global ischemia model in the first series of experiments. We investigated the relationship between superoxide production and ATF-4 induction by visualizing the ethidium signal in this model (described below). We also investigated thiobarbituric acid reactive substances (TBARS) with this model (described below). Because most cases of human ischemic stroke are caused by occlusion of cerebral arteries, the middle cerebral artery (MCA) occlusion model is more akin to human stroke than other models. We therefore used a mouse MCA occlusion model in a second series of experiments to investigate the effect of SOD1 overexpression on ER stress-induced cell death signal activation. All efforts were made to minimize the number of animals used and their suffering. All procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by Stanford's Administrative Panel on Laboratory Animal Care.
Superoxide Dismutase Transgenic Rats
Heterozygous SOD1 Tg rats with a Sprague-Dawley background, carrying human SOD1 genes with a four- to sixfold increase in copper/zinc SOD, were derived from the founder stock described previously (Chan et al, 1998). They were further bred with Wt Sprague-Dawley rats to generate heterozygous rats. The SOD1 Tg rats were identified by isoelectric focusing gel electrophoresis as described (Chan et al, 1998). There were no observable phenotypic differences, including in the cerebral vasculature, between the Tg rats and Wt littermates, as reported previously (Chan et al, 1998).
Superoxide Dismutase Transgenic Mice
Heterozygous SOD1 Tg mice of the TGHS/SF218–3 strain with a CD-1 background, carrying human SOD1 genes with a threefold increase in SOD1 activity, were derived from the founder stock described previously (Epstein et al, 1987). There were no differences in phenotypes, including cerebrovascular anatomy, between the Tg mice and their Wt littermates (Yang et al, 1994). Our previous study revealed that there were no differences in the regional cerebral blood flow between these two groups before or after focal ischemia (Chan et al, 1993).
Transient Global Ischemia in Rats
Transient global brain ischemia was induced as previously reported, with slight modification (Smith et al, 1984). In brief, the rats were anesthetized with 1.5% isoflurane, 68.5% nitrous oxide, and 30% oxygen using a face mask, and the femoral artery was exposed and catheterized with a PE-50 catheter (427410; Becton Dickinson, San Diego, CA, USA) for continuous recording of arterial blood pressure. A midline neck skin incision was made and the right jugular vein and both common carotid arteries were exposed. After intravenous injection of 150 IU/kg heparin, blood was quickly withdrawn via the jugular vein. When the mean arterial blood pressure became 30 mm Hg, both common carotid arteries were clamped with surgical clips. Blood pressure was maintained at 30 to 35 mm Hg by withdrawing or infusing blood through the jugular vein during the ischemic period. After 5 mins of ischemia, the clips were removed and the blood was reinfused. Body temperature was monitored with a rectal probe and controlled at 37°C with a homeothermic blanket. Sham-operated animals underwent exposure of vessels without withdrawal of blood or clamping of carotid arteries. At 1, 4, 24, and 48 hours after transient ischemia, as well as after the sham operation, samples for histological examination (n = 3 at each time point) and Western blot analysis (n = 3 at each time point) were prepared as described below. For the TBARS measurement, the animals (including sham-operated) were decapitated 1 hour after ischemia (n = 3 for each group) and the ER fraction was extracted as described below.
Focal Cerebral Ischemia in Mice
Using adult SOD1 Tg mice and their littermates (3-month-old males, 35 to 40 mg), we induced focal cerebral ischemia by intraluminal occlusion of the MCA using a nylon suture, as previously described (Murakami et al, 1998). In brief, the mice were anesthetized with 1.5% isoflurane, 68.5% nitrous oxide, and 30% oxygen using a face mask. The body temperature was monitored with a rectal probe and controlled at 37°C with a homeothermic blanket. A midline neck incision was made and the left external carotid artery was exposed. After electrocoagulation of its branches, an 11-mm 5–0 monofilament nylon suture with a blunted tip was introduced into the left internal carotid artery through the external carotid artery stump. After 30 mins of ischemia, cerebral blood flow was restored by removal of the nylon suture. At 1, 4, and 24 hours after reperfusion, as well as after the sham operation, samples for histological study (n = 3 at each time point) and Western blot analysis (n = 3 at each time point) were prepared as described below.
Assessment of Neuronal Degeneration
For assessment of neuronal damage, the animals were deeply anesthetized with isoflurane and perfused with 5 U/mL heparinized saline through the left cardiac ventricle until colorless fluid was obtained. Subsequently, 4% paraformaldehyde in 0.1 mol/L phosphate buffer was perfused and the brains were removed. After postfixation in 4% paraformaldehyde, the brains were washed with water, dehydrated with ethanol, immersed in xylene, and embedded in paraffin. Coronal brain sections 5 μm thick at the hippocampal level (rat) or striatal level (mouse) were prepared and placed on glass slides. The sections were then stained with cresyl violet and neuronal degeneration was quantified as described previously (Kondo et al, 1997; Sugawara et al, 1999). In the study with the rat transient global ischemia model, the number of intact pyramidal cells with a distinct nucleus was counted and divided by the total number of neurons in a 1-mm length of the middle portion of the CA1 subfield (Sugawara et al, 1999). In the study with the mouse focal ischemia model, neuronal degeneration was expressed as infarct area/ischemic hemisphere area × 100% at the striatal level section (Kondo et al, 1997).
Immunohistochemistry
We performed immunohistochemical analysis for ATF-4 and CHOP. The sections were the same as those used for the histological study. After deparaffinization, endogenous peroxidase activity was quenched with 30% methanol and 0.3% hydrogen peroxide in phosphate-buffered saline and the slides were boiled in citrate buffer with microwaves. After blocking nonspecific binding with 1% to 3% bovine serum albumin, the slides were incubated with primary antibodies at room temperature for 2 hours. The primary antibodies used and the dilutions for each were rabbit polyclonal anti-ATF-4 antibody (sc-200; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 1:100 and mouse monoclonal anti-CHOP antibody (sc-7351; Santa Cruz Biotechnology) at 1:100. The slides were washed and then incubated with biotinylated anti-rabbit immunoglobulin G (IgG) (B3275; Sigma, St. Louis, MO, USA) or anti-mouse IgG (BA-2000; Vector Laboratories, Burlingame, CA, USA) at 1:200 dilution. They were subsequently incubated with avidin-biotin-peroxidase complex (PK-6100; Vector Laboratories) for 30 mins and then developed using vector VIP peroxidase substrate (SK-4600; Vector Laboratories). Methyl green was used for counterstaining when necessary. Sets of sections were treated without the first antibody to confirm the specificity of the primary antibodies.
Double Fluorescent Study of Superoxide Production and Activating Transcription Factor-4 Expression
Production of superoxide anions in the rat brains after transient global ischemia was investigated by in situ detection of oxidized hydroethidine as described previously (Sugawara et al, 2002). Hydroethidine (D-11347; Molecular Probes, Eugene, OR, USA) is taken up by living cells and oxidized by superoxide to a red fluorescent dye, ethidium (Bindokas et al, 1996). Hydroethidine solution (1 mL; 1 mg/mL in 1% dimethylsulfoxide with saline) was administered intravenously 15 mins before induction of ischemia. Twenty-four hours after ischemia, the animals were deeply anesthetized and the brains were perfused with heparinized saline and 4% paraformaldehyde in 0.1 mol/L phosphate buffer (n = 3). After postfixation in 4% paraformaldehyde, the brains were cut on a vibratome into slices 50 μm thick at the level of the hippocampus and placed on glass slides. Using these brain slices, we performed fluorescent immunohistochemistry for ATF-4. In brief, after incubation with primary and then secondary antibodies as described above, they were incubated with avidin-conjugated fluorescein isothiocyanate at a dilution of 1:40 (A-2001; Vector Laboratories) and covered with VECTASHIELD mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (H-1200; Vector Laboratories). Fluorescence was observed at excitation 495 nm and emission >515 nm for fluorescein isothiocyanate, excitation 510 nm and emission >580nm for ethidium, and excitation 360 nm and emission >460 nm for DAPI.
Double Fluorescent Study of Activating Transcription Factor-4 and C/EBP Homologous Protein
To confirm that ATF-4 was involved in the induction of CHOP, we performed double staining for ATF-4 and CHOP using rat brain sections. The sections used were the same as those prepared for the histologic study. After deparaffinization and microwave treatment, the sections were incubated with an anti-ATF-4 antibody and then with a biotinylated anti-rabbit IgG antibody, as described above. After washing in phosphate-buffered saline, they were incubated with avidin-conjugated fluorescein isothiocyanate. The sections were subsequently treated with the excess avidin followed by the excess biotin (SP2001; Vector Laboratories) to prevent nonspecific double labeling. The sections were then incubated with an anti-CHOP antibody and a biotinylated anti-mouse IgG antibody, and then reacted with avidin-conjugated Texas Red (A-2006; Vector Laboratories). They were then covered with VECTASHIELD mounting medium with DAPI. Fluorescence was observed at excitation 495 nm and emission >515 nm for fluorescein isothiocyanate, excitation 510 nm and emission >580 nm for Texas Red, and excitation 360 nm and emission >460 nm for DAPI.
Western Blot Analysis
We performed a Western blot analysis for ATF-4 and CHOP using rat and mouse brain samples. After decapitation, the brains were removed and cut into coronal slices 2-mm thick. The hippocampal CA1 subregion was carefully separated from the rat brains and quickly frozen in dry ice. The cerebral cortex and caudate of the left (ischemic side) MCA territory were separately removed from the mouse brains and quickly frozen. For protein extraction, the tissue was sonicated with about seven volumes of protein extraction buffer (20 mmol/L HEPES potassium hydroxide (pH 7.5), 250 mmol/L sucrose, 10 mmol/L potassium chloride, 1.5 mmol/L magnesium chloride, 1 mmol/L EDTA, 1 mmol/L EGTA, 0.7% protease inhibitor cocktail (P8340; Sigma), and 1% phosphatase inhibitor cocktails (P2850 and P5726; Sigma)). The homogenate was centrifuged at 10,000g for 15 mins and the supernatant was used for this study. Assays to determine the protein concentration were performed by comparison with a known concentration of bovine serum albumin using a kit (23227; Pierce, Rockford, IL, USA).
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed according to a previous report (Noshita et al, 2001). In brief, the lysate equivalent of 5 μg of protein from each brain was run on the gel at 120 V together with a size marker (RPN800; Amersham, Piscataway, NJ, USA). The protein on the gel was subsequently transferred to a polyvinylidene fluoride transfer membrane (LC2002; Invitrogen, Carlsbad, CA, USA) in a buffer containing methanol, glycine, Tris base, and sodium dodecyl sulfate. After the transfer, the membrane was placed in 10% powdered milk in phosphate-buffered saline with 0.1% Tween 20 to block nonspecific binding and was then incubated with primary antibodies for 12 hours at 4°C. The primary antibodies were the same as those used for immunohistochemistry, and were diluted at 1:1,000 in 1% powdered milk in phosphate-buffered saline with Tween 20. After washing, the membrane was incubated with horseradish peroxidase-conjugated anti-rabbit IgG (7074; Cell Signaling Technology, Beverly, MA, USA) and/or anti-mouse IgG (PI-2000; Vector Laboratories), and then the signal was detected with a chemiluminescent kit (RPN2132; Amersham). The same membranes were subsequently used for β-actin immunodetection, and equal protein loading was ensured.
Thiobarbituric Acid Reactive Substances Measurement
Using the ER fraction, we investigated ER membrane lipid peroxidation by measuring TBARS. This test estimates the amount of malondialdehyde precursors, including hydroperoxides and endoperoxides (Keller et al, 1998). The ER fraction was extracted as described previously with slight modification (Ghribi et al, 2001). In brief, brain tissue from the hippocampal CA1 subregion was gently homogenized with a glass homogenizer in seven volumes of the above-mentioned protein extraction buffer. The homogenate was first centrifuged at 750g for 10 mins and then at 10,000g for 20 mins at 4°C. The supernatant was further centrifuged at 100,000g for 1 hour at 4°C to separate the cytosolic fraction from the ER fraction. We performed Western blot analysis and confirmed that only the ER fraction contained immunoreactive calnexin, showing that our fractionation was successful. Aliquots of the ER fraction with 10 μg of protein were added to the sodium dodecyl sulfate solution, and were then incubated with TBARS reaction buffer at 95°C for 1 hour (#0801192; ZeptoMetrix, Buffalo, NY, USA). Absorbance at a 532-nm wavelength was measured and the amount of TBARS was quantified using a standard curve of malondialdehyde from the kit. The results were expressed as nanomoles per 10 μg of ER protein.
Quantification and Statistical Analysis
For the histologic assessments, the results were expressed as mean ± s.d. To evaluate the results of the Western blot studies, the film was scanned with an imaging densitometer (GS-700; Bio-Rad, Hercules, CA, USA) and the optical density was quantified using Multi-Analyst software (Bio-Rad) as previously reported (Noshita et al, 2001). In both studies, statistical significance between the two groups was established with an F test followed by an unpaired Student's t-test. A P-value smaller than 0.05 was considered statistically significant.
RESULTS
Rat Global Ischemia Model
Histologic Assessment of Rat Brains After Transient Ischemia: With cresyl violet staining, no neuronal degeneration in the hippocampal CA1 region was confirmed 1 or 4 hours or 1 day after 5 mins of ischemia in either the Wt or SOD1 Tg rat brains. At 2 days, however, 78.1% ± 11.0% of the CA1 pyramidal neurons had degenerated in the Wt brains, which is known as delayed neuronal cell death (Kirino, 1982). In the SOD1 Tg rats, in contrast, only 40.8% ± 12.1% of those neurons had degenerated and most of the cells remained morphologically intact as previously demonstrated (Chan et al, 1998; Sugawara et al, 2002). The difference was statistically significant (P<0.01).
Induction of Activating Transcription Factor-4 in the Hippocampal CA1 Region was Attenuated by Superoxide Dismutase Overexpression: Endoplasmic reticulum stress causes phosphorylation of eIF2α through activation of PERK, which results in inhibition of new protein synthesis (Kumar et al, 2001; Harding et al, 2002). Nevertheless, the translation into proteins is accelerated in some genes through a ‘bypass scanning’ mechanism (Kaufman, 1999; Kumar et al, 2003). Activating transcription factor-4 translation is facilitated through this mechanism. It activates other gene expressions that are implicated in apoptosis (Harding et al, 2002); therefore, ATF-4 plays an important role in ER stress-induced cell death. We investigated changes in ATF-4 expression with Western blotting and immunohistochemical analysis using an anti-ATF-4 antibody (Figure 1). A faint band with a molecular weight of 26 kDa was observed in the sham-operated Wt and Tg brains by Western blot analysis (Figure 1A). In the Wt rats, the density of the band gradually increased from 1 hour, peaked at 1 day, and decreased 2 days after ischemia. In the Tg rat brains also, there was a gradual increase from 1 hour and a peak at 1 day, but the degree of increase was less prominent than in the Wt animals. With quantitative analysis (Figure 1B), we found that SOD1 overexpression significantly attenuated induction of ATF-4 1 day after ischemia (*P<0.05 compared with Wt animals at the same time point). Immunohistochemical analysis showed that hippocampal CA1 neurons possessed almost no immunoreactivity for ATF-4 in the Wt or Tg control brains (Figure 1C). The immunoreactivity became slightly strong at 1 hour and stronger at 4 hours in both the Wt and Tg groups. Some neurons showed a very strong immunoreactivity at this time point. At 1 day, many neurons were quite densely stained in both the Wt and Tg animals, but the number of strongly stained neurons and the degree were more prominent in the Wt animals, which is compatible with the results of the Western blot study. At 2 days, most of the neurons had degenerated in the Wt animals, and the remaining neurons showed positive immunoreactivity. The SOD1 Tg rats, however, had only a few degenerated neurons, and the immunoreactivity became very weak. Of note is that most neurons showed immunoreactive ATF-4 in the cytoplasm as well as in the nuclei. This finding does not conflict with the fact that ATF-4 is a transcription factor and exerts its effect in the nuclei, because proteins are synthesized at the cytoplasm and thereafter translocated to the nuclei. Previous reports also demonstrated expression of ATF-4 at the cytoplasm (Nehring et al, 2000). Sections without the first antibody showed no immunostaining (data not shown).
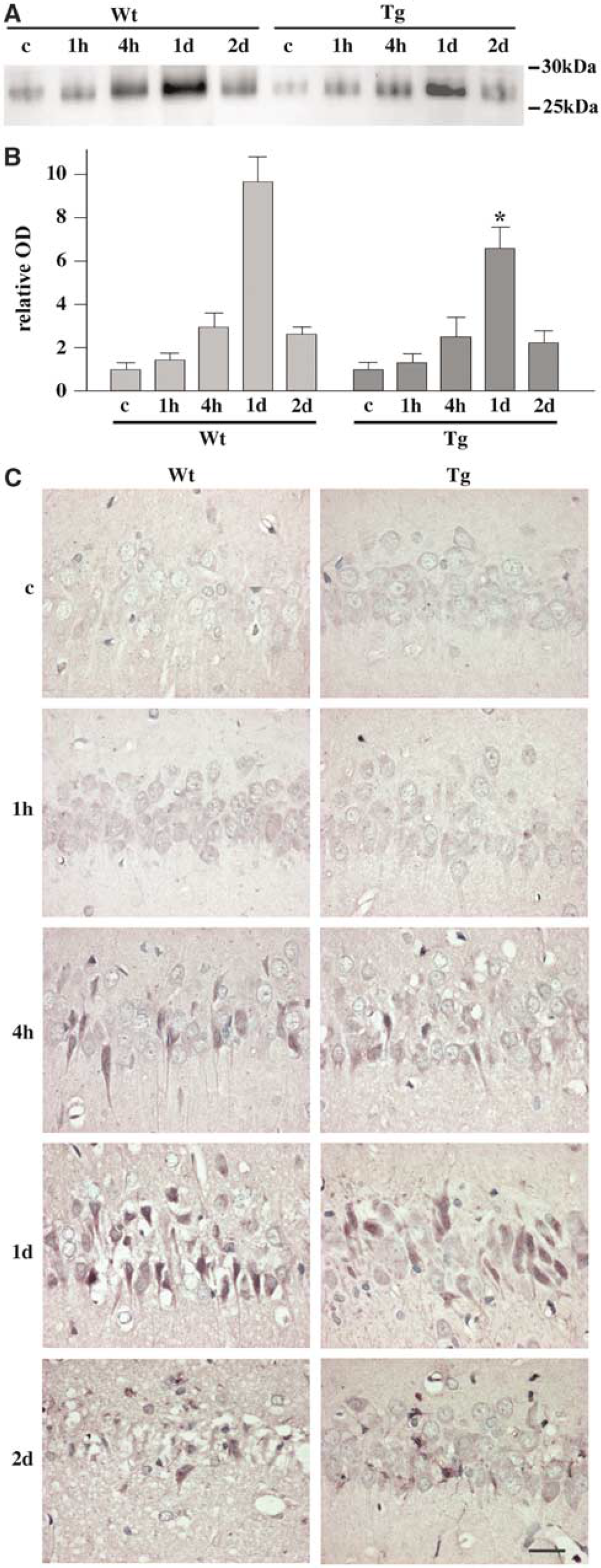
Change in level of ATF-4 expression in rat hippocampal CA1 neurons after transient ischemia. (
Attenuated Induction of Activating Transcription Factor-4 was Accompanied by Decreased Superoxide Production: Superoxide production can be visualized by injecting hydroethidine into the animals and then detecting the oxidized ethidium signals (Bindokas et al, 1996; Sugawara et al, 2002). To confirm that superoxide production is involved in ATF-4 induction, we carried out a double fluorescent study of ethidium and ATF-4. Hippocampal CA1 pyramidal cells showed a strong signal for ethidium in the Wt rats 1 day after transient ischemia, indicating that superoxide was robustly produced in these neurons (Figure 2). The signal for ATF-4 was also strong in these cells and overlapped with the ethidium signal. In the SOD1 Tg brains, signals for both ethidium and ATF-4 were less strong than in the Wt animals, although they again overlapped. These results suggest that superoxide may be involved in ATF-4 induction and that SOD1 overexpression attenuates ATF-4 induction by reducing superoxide accumulation in cells.
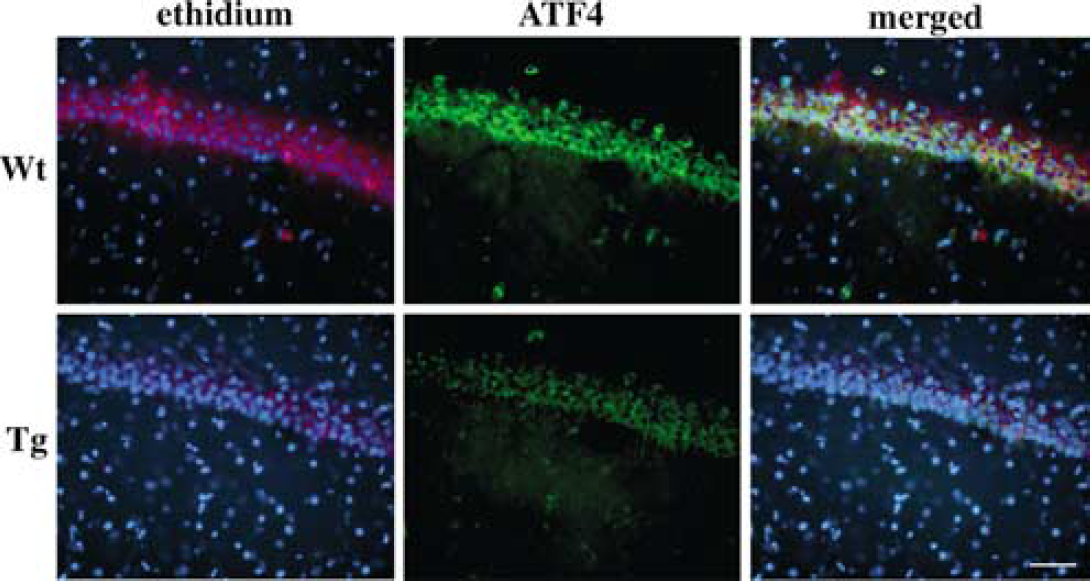
Fluorescent double staining for ethidium (red) and ATF-4 (green) signals in the rat hippocampal CA1 region 1 day after ischemia. Nuclei were counterstained with DAPI (blue). In the Wt animals, a strong ethidium signal was observed in the neurons at the pyramidal cell layer, in which strong immunoreactivity for ATF-4 was also confirmed. In the Tg animals, in contrast, signals for both ethidium and ATF-4 were milder than in the Wt animals, indicating that production of superoxide is implicated in ATF-4 induction. Bar = 50 μm.
Activation of C/EBP Homologous Protein Expression in the Hippocampal CA1 Region was Attenuated by Superoxide Dismutase Overexpression: C/EBP homologous protein expression is increased by ER stress, and ATF-4 plays an active role in the induction of this molecule (Oyadomari et al, 2001; Harding et al, 2002). C/EBP homologous protein binds with a certain group of transcription factors and induces proapoptotic molecules (Fawcett et al, 1996; Wang et al, 1998; Cortés-Canteli et al, 2001). It is, therefore, a key molecule that connects ER stress to apoptotic cell death. We investigated changes in CHOP expression with Western blotting and immunohistochemical analysis (Figure 3). A barely detectable band with a molecular weight of 50 kDa was observed by Western blot analysis in the sham-operated Wt and SOD1 Tg brains (Figure 3A). This is compatible with the fact that the level of CHOP expression is very low under normal conditions (Paschen et al, 1998; Jousse et al, 1999; Gotoh et al, 2002). In the Wt rat brains, the density of the band gradually increased from 1 hour, peaked at 1 day, and decreased 2 days after ischemia. In the brains of the SOD1 Tg rats also, there was a gradual increase from 1 hour and a peak at 1 day, but the degree of increase was less prominent than in the Wt animals. With quantitative analysis (Figure 3B), we found that SOD1 overexpression significantly attenuated activation of CHOP expression 4 hours and 1 day after ischemia (*P<0.05 compared with the Wt animals at the same time point). Immunohistochemical analysis showed that hippocampal CA1 neurons possessed almost no immunoreactivity for CHOP in the Wt and Tg control brains (Figure 3C). Some neurons became positively stained at 1 hour, and the number of such cells increased at 4 hours in both the Wt and Tg groups. Immunoreactivity was localized in the cytoplasm until this time point. At 1 day, many neurons became strongly stained in both the Wt and Tg rats. Of note is that nuclei were also positively stained at 1 day, indicating that this transcription factor actually exerted its effect. The number of strongly stained cells was smaller in the Tg rats than in the Wt rats, which is compatible with the results of the Western blot study. At 2 days, most of the neurons had degenerated in the Wt animals and the remaining neurons showed positive immunoreactivity. There were only a few degenerated neurons in the Tg rats, however. Sections without the first antibody showed no immunostaining (not shown).
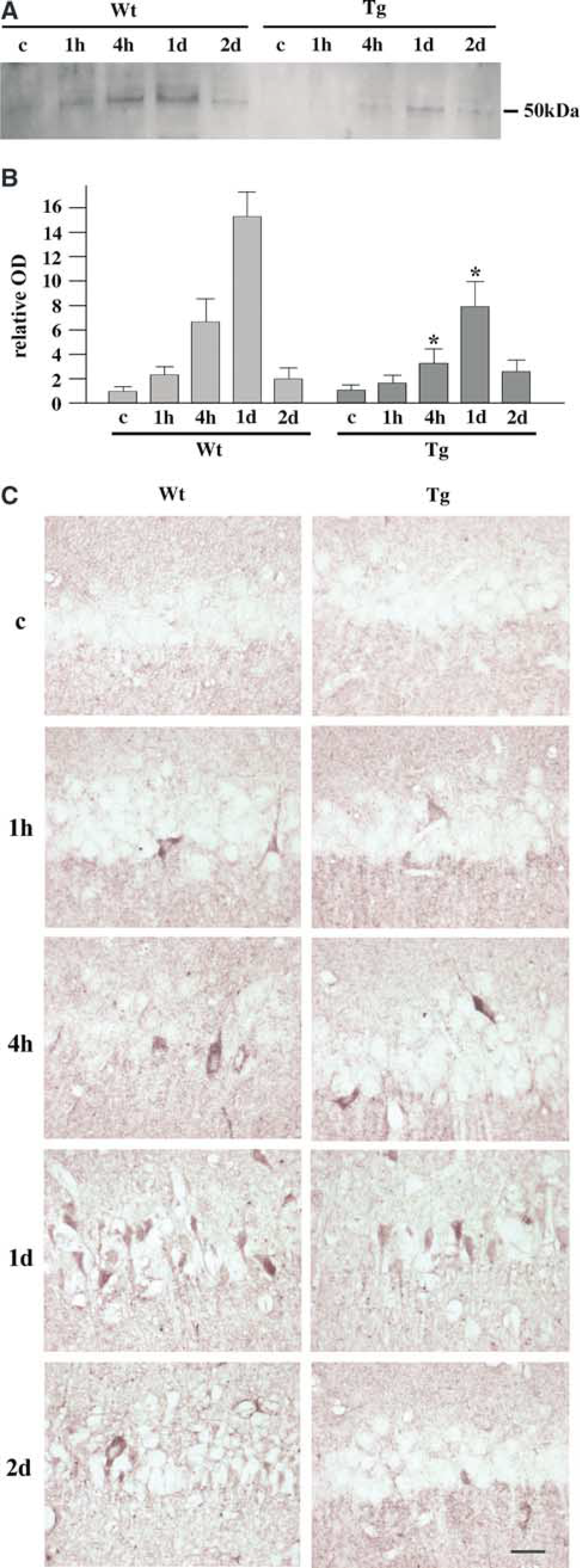
Change in the level of CHOP expression in rat hippocampal CA1 neurons after transient ischemia. (
C/EBP Homologous Protein was Colocalized with Activating Transcription Factor-4: Although CHOP expression is controlled by ATF-4, other molecules may also be implicated (Yoshida et al, 2001; Gotoh et al, 2002). To confirm that ATF-4 plays an active role in CHOP induction, therefore, we carried out a double fluorescent study of CHOP and ATF-4. Neurons immunoreactive for CHOP gradually increased from 1 hour and peaked at 1 day in both the Wt and Tg animals, which is compatible with the results of the above-mentioned immunohistochemistry study. The number of strongly stained cells was larger in the Wt than in the Tg rats. The signal for ATF-4 was also increased from 1 hour, as demonstrated above. Of note was that the signal for CHOP was mostly colocalized with that for ATF-4 in both the Wt and Tg groups, but both were stronger in the Wt animals (Figure 4). This finding indicates that although the role of other molecules cannot be excluded, ATF-4 is implicated in the activation of CHOP expression in this model.
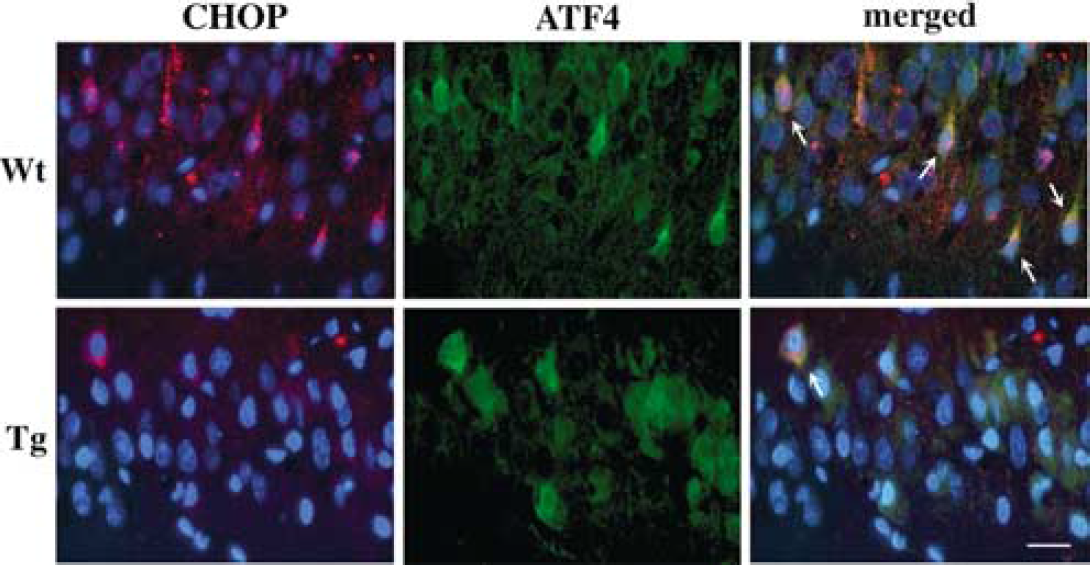
Fluorescent double staining for CHOP (red) and ATF-4 (green) in the rat hippocampal CA1 region 1 day after transient ischemia. Nuclei were counterstained with DAPI (blue). In the Wt brains, signals for both CHOP and ATF-4 were observed and overlapped (arrows). These two signals also overlapped in the Tg brains (arrow), although they were substantially milder than in the Wt animals. Bar = 20 μm.
Endoplasmic Reticulum Lipid Peroxidation was Partially Prevented by Superoxide Dismutase Overexpression: The ER is a highly membranous organelle and lipid peroxidation should easily occur when superoxide attacks its membrane (Halliwell and Gutteridge, 1999). To provide evidence of oxidative damage to the ER, we observed TBARS formation in this organelle using ER fraction samples (Figure 5). The TBARS method is a reliable index of lipid peroxidation (Ohtsuki et al, 1992; Keller et al, 1998). Massive production of superoxide occurs at an early stage after reperfusion (Murakami et al, 1998). Therefore, we carried out this study using sham-control samples 1 hour after reperfusion. In the Wt control brains, 5.8 nmol of TBARS were measured in the ER fraction of 10 μg of protein, which means that there was a small amount of peroxidized lipids under normal conditions. At 1 hour after 5 mins of ischemia, it increased to 24.1 nmol. Transient ischemia and reperfusion caused massive lipid peroxidation in the ER. In the SOD1 Tg rats, in contrast, TBARS were 3.9 nmol per 10 μg of ER protein, which was smaller than in the Wt control animals (P<0.05), indicating that SOD1 overexpression reduced peroxidized lipids under normal conditions. TBARS were increased to 13.4 nmol 1 hour after transient ischemia, but this increase was far smaller than in the Wt rats (P<0.01), which suggests that Tg SOD1 overexpression attenuated oxidative damage to the ER. This finding indicates that ROS may directly cause ER damage, and that it can be partially prevented by SOD1.
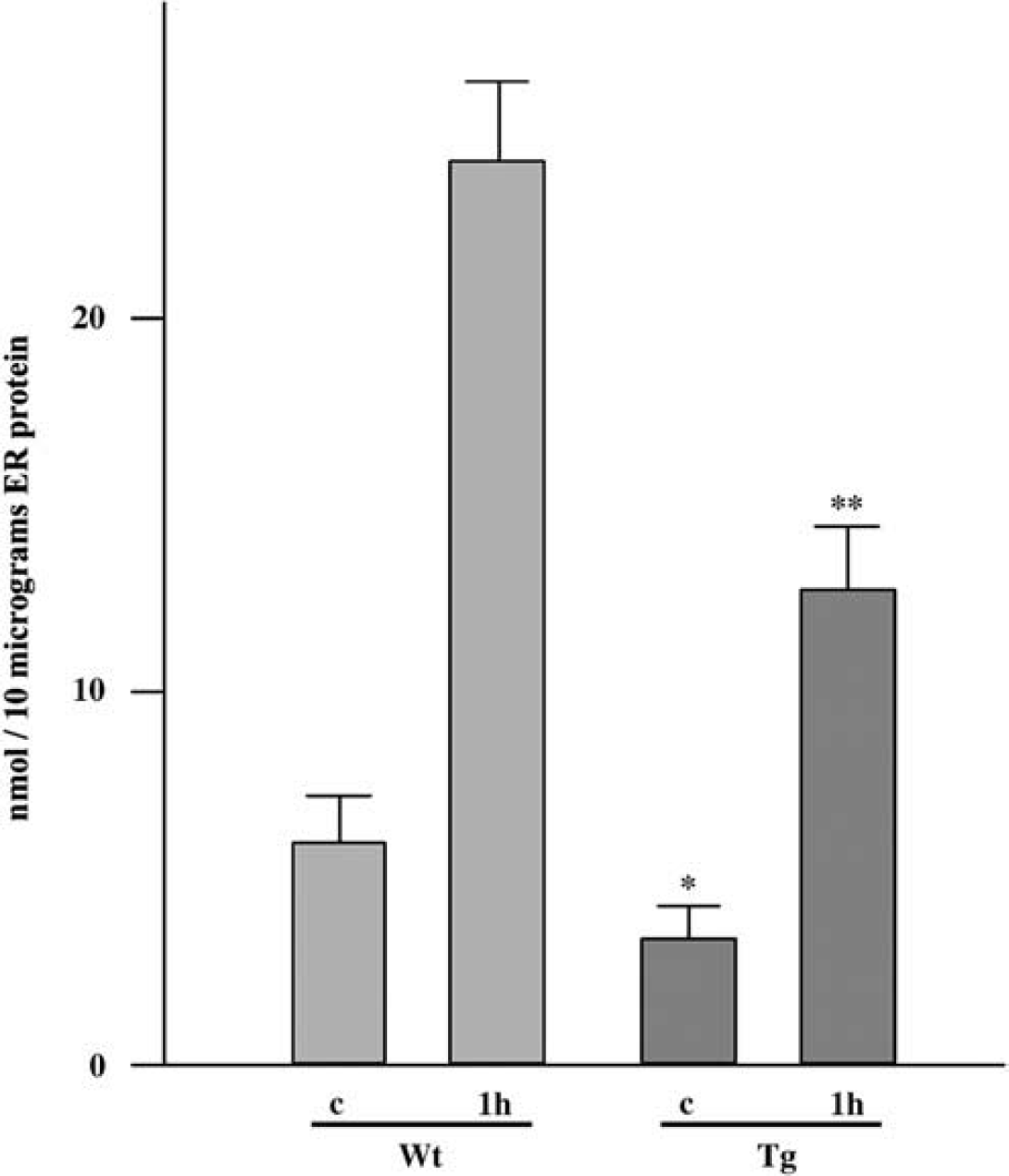
Amount of TBARS in the ER fraction. In the Wt rats, 5.8 nmol of TBARS were contained in the ER fraction with 10 μg of protein, which increased about four times 1 hour after ischemia. In the SOD1 Tg rats, TBARS were normally 3.9 and 13.4 nmol 1 hour after ischemia. In both the control (c) and ischemic groups, the amount of TBARS was significantly smaller in the SOD1 Tg rats (*P<0.05, **P<0.01) compared with the Wt animals at the same time point.
Mouse Middle Cerebral Artery Occlusion Model
Histologic Assessment of Mouse Brains After Middle Cerebral Artery Occlusion: As a second step, we used the mouse MCA occlusion model to investigate the possible involvement of ROS in ER stress-induced cell death. Most human ischemic strokes are caused by occlusion of the cerebral artery and this model is more akin to human stroke than the transient global ischemia model. A cresyl violet staining study showed no neuronal degeneration until 4 hours, but 22.4% ± 4.6% of the ischemic-side hemisphere became infarcted at 1 day in the Wt animals. The infarction was restricted in the striatum and the area of massive neuronal degeneration extended very close to the lateral ventricular wall. In the Tg animals, however, the size of the infarct area at 1 day was smaller than in the Wt animals (12.0% ± 3.9% of the ischemic-side hemisphere); neurons at the innermost part of the striatum were spared. By statistical analysis, the difference reached statistical significance (P<0.01). With this ischemic model, there were no histologic changes in the cerebral cortex in either the Wt or the Tg animals (Hayashi et al, 2004).
Induction of Activating Transcription Factor-4 in the Striatum was Attenuated by Superoxide Dismutase Overexpression: Induction of ATF-4 was investigated in the mouse striatum after transient MCA occlusion. Western blot analysis using samples from the left striatum (ischemic side) showed a faint band at 26 kDa in the control brains of the Wt and SOD1 Tg mice (Figure 6A). In the Wt samples, it became slightly dense at 1 hour and dramatically denser at 4 hours, and peaked at 1 day. In the SOD1 Tg mice, in contrast, it became slightly dense at 1 hour, with increased density at 4 hours, but then the signal decreased 1 day after ischemia. With quantitative analysis (Figure 6B), we found that Tg overexpression of SOD1 significantly prevented induction of ATF-4 in the ischemic striatum 4 hours and 1 day after reperfusion (*P<0.05 compared with the Wt animals at the same time point). Immunohistochemical analysis (Figure 6C) showed that neurons at the medial part of the left striatum possessed almost no immunoreactivity under normal conditions in either the Wt or Tg groups. A few neurons became positively stained at 1 hour, and the number of positively stained cells dramatically increased at 4 hours. The number of these cells was larger in the Wt animals than in the Tg animals, which is compatible with the Western blot study. The fact that the cytoplasm as well as nuclei were stained indicates that ATF-4 is synthesized within the cytoplasm and translocated to nuclei. At 1 day, neurons in the Wt animals within, as well as outside, the infarct area showed strong immunoreactivity for ATF-4 (Figure 6C, arrowheads). In the striatum of the Tg mice, however, only a small number of cells within the infarct area, but none outside the infarct area, were positively stained. Sections without the first antibody showed no immunostaining (not shown). This may suggest that robust production of ROS, superoxide in particular, causes ER stress-induced neuronal cell death outside the pan-necrotic infarct area.
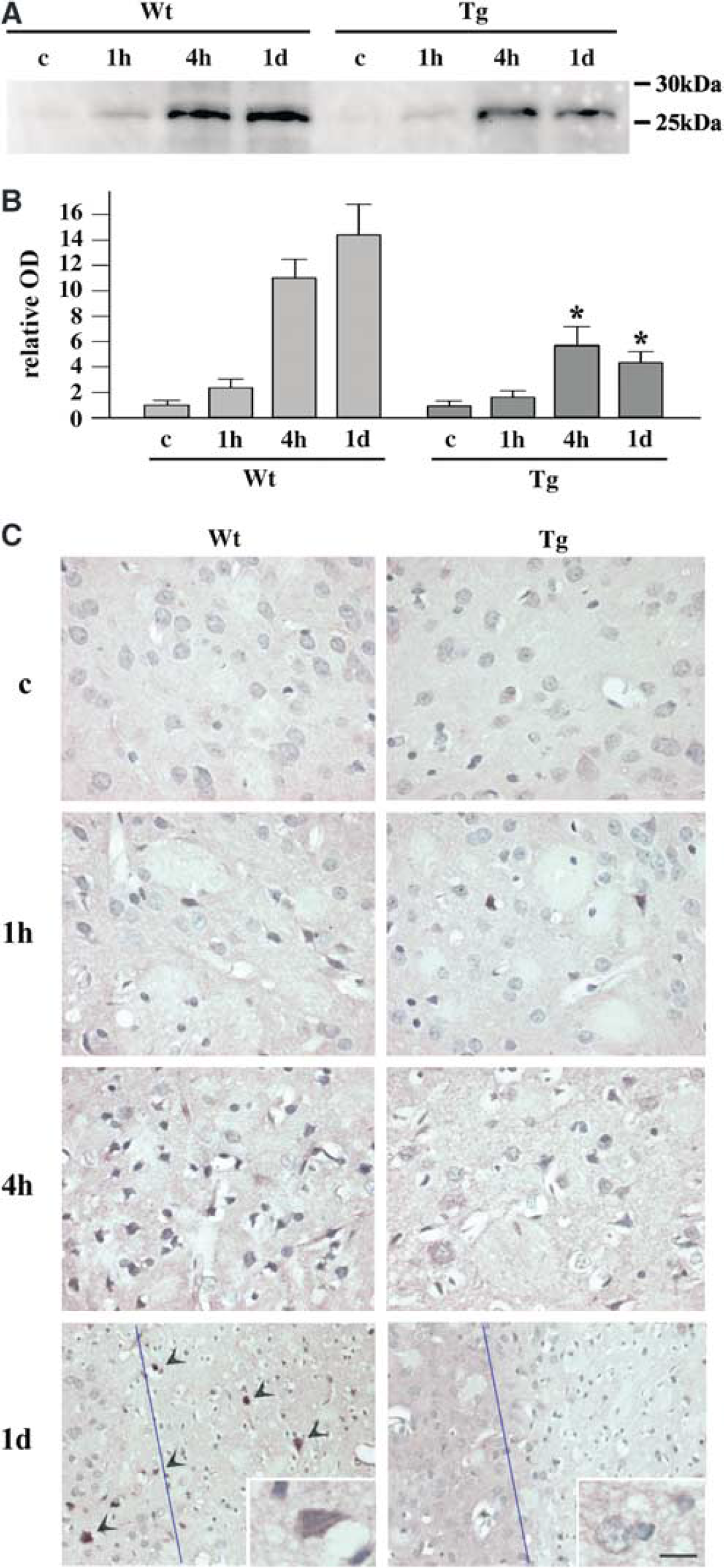
Change in the level of ATF-4 expression in mouse brains after transient focal ischemia. (
Activation of C/EBP Homologous Protein Expression in the Striatum was Attenuated by Superoxide Dismutase Overexpression: Expression of CHOP, which is important for ER stress-induced cell death, was investigated in the mouse striatum after MCA occlusion. Western blot analysis of samples from the left striatum showed a barely detectable band at 50 kDa in the control brains of the Wt and Tg animals (Figure 7A). In the Wt samples, it became slightly dense at 1 hour and dramatically dense at 4 hours, but was partially decreased at 1 day. In the SOD1 Tg mice, it also became slightly dense at 1 hour, denser at 4 hours, and was partially decreased at 1 day. However, the degree of increase in expression was less in the Tg than in the Wt animals. With quantitative analysis (Figure 7B), we found that Tg overexpression of SOD1 significantly prevented activation of CHOP expression in the ischemic striatum 4 hours and 1 day after reperfusion (*P<0.05 compared with the Wt animals at the same time point). Immunohistochemical analysis (Figure 7C) showed that neurons in the medial section of the left striatum possessed no immunoreactivity under normal conditions in either the Wt or Tg groups. Some neurons became positively stained at 1 hour, and the number of positively stained cells and the degree of staining was further increased at 4 hours. Compared with the Wt animals, the number of immunoreactive cells was smaller and the signal intensity was weaker in the Tg animals. At 1 day, neurons within, as well as outside, the infarct area showed strong immunoreactivity to CHOP in the Wt animals (Figure 7C, arrowheads). In the Tg striatum, in contrast, some neurons within, but not outside, the infarct area were mildly stained. Sections without the first antibody showed no immunostaining (not shown). These findings are compatible with those for ATF-4 expression and also support our suggestion that superoxide causes ER stress-induced neuronal cell death outside the pan-necrotic infarct area.
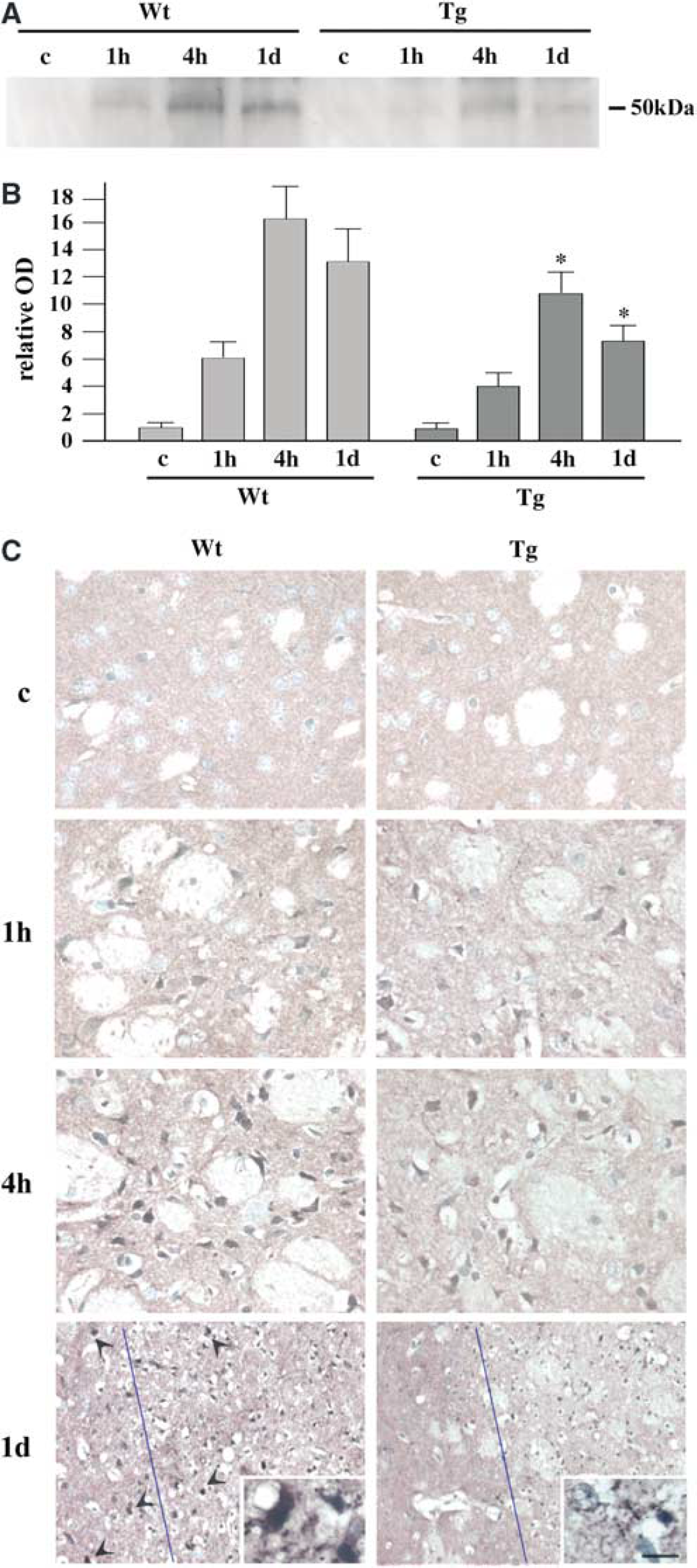
Change in the level of CHOP expression in mouse brains after transient focal ischemia. (
DISCUSSION
In these studies, we demonstrated that expression of ATF-4 and CHOP was increased in the brain after ischemia. This result seems to contradict a previous study, in which ATF-4 and CHOP induction after ischemia did not reach statistical significance (Kumar et al, 2003). This might be due to the difference in animal models, but is more likely because of the difference in investigated time points. In the previous report, the authors investigated ATF-4 and CHOP expression until 4 hours; we investigated it until 2 days. Activating transcription factor-4 and CHOP expression peaked at 1 day (Figures 1 and 3). Induction might have been successfully detected in the previous report if the authors had extended their studies.
We found that Tg overexpression of SOD1 attenuated ATF-4 and CHOP induction. CHOP expression is a representative feature of ER stress-induced cell death (Harding et al, 2000; Oyadomari et al, 2001); thus, our results strongly indicate that oxidative ER stress is involved in ischemic neuronal cell death. This is a novel finding because it is known that the upper stream of the ER stress response occurs after ischemia, but whether this is actually relevant to neuronal cell death is uncertain (Althausen et al, 2001; Kumar et al, 2001; Hayashi et al, 2003a, b). In both the ischemia models used in this study, neurons with a prominent induction of ATF-4 and CHOP underwent apoptosis, but those without it survived. This fact further supports the idea that ER stress not only occurs, but also triggers the cell death program in ischemic neurons.
We suggest several possible mechanisms of oxidative ER damage. Since adenosine triphosphate (ATP) is necessary for regular ER functions such as protein folding and calcium homeostasis (Berridge, 2002), a lack of ATP might be responsible for ER stress. The results from the SOD1 Tg animals that showed attenuated ER stress response could be compatible with a previous report demonstrating that superoxide inhibits restoration of ATP (Siems et al, 1991). However, a lack of ATP should first disturb the plasma membrane ion gradient, which causes cell rupture. Activation of the apoptotic machinery observed in this study cannot be fully explained by the ATP theory. Rather, our TBARS study provides some insights into the mechanisms of oxidative ER damage. We demonstrated that ER lipid peroxidation occurs after ischemia. This may result in a disturbance in ER function and culminate in cellular catastrophe because denatured lipids can influence protein function (Parola et al, 1999). In contrast, superoxide is known to instantly react with nitric oxide to form peroxynitrite, which can directly nitrosylate protein and impair protein function (Beckman and Crow, 1993). We recently demonstrated nitrosylated protein in ER fractions of rat brain proteins obtained from transient global cerebral ischemia (Hayashi et al, 2003b), which supports the role of ROS in ER damage.
Protein synthesis is generally inhibited in cells under ER stress. Protein kinase-like ER eIF2α kinase is phosphorylated by ER stress, which in turn phosphorylates eIF2α (Althausen et al, 2001; Kumar et al, 2001; DeGracia et al, 2002). Phospho-eIF2α prevents removal of the guanine nucleotide from the eukaryotic initiation factor, thus inhibiting translation initiation (Rowlands et al, 1988). Induction of ATF-4 and CHOP in neurons after ischemia seems, therefore, to contradict this. However, translation is accelerated in some genes under ER stress. The ER resident molecular chaperones, for example, are also increased in cells under ER stress (Yu et al, 1999; Kitao et al, 2001). This apparently strange phenomenon is brought about through ‘bypass scanning,’ in which ribosomes with a ternary complex have an increased probability of reaching the protein coding region of the mRNA (Degracia et al, 2002). Induction of ATF-4 in our study might have been brought about through this bypass scanning. C/EBP homologous protein expression is, at least in part, controlled by ATF-4 (Harding et al, 2000, 2002), and therefore the increase in CHOP was also the result of bypass scanning.
The exact mechanisms of CHOP-induced apoptotic cell death are not fully elucidated. It binds with a transcription factor, CEBPβ, to make a heterodimer and to activate several target genes, and this is one of the possible mechanisms (Fawcett et al, 1996; Wang et al, 1998; Cortés-Canteli et al, 2001) because there is a high amount of CEBPβ in the brain (Sterneck and Johnson, 1998). Indeed, our preliminary study revealed that CHOP was coimmunoprecipitated with CEBPβ and that this was increased by ischemia. However, CEBPβ may not be the sole partner of CHOP (Ron and Habener, 1992) and we cannot rule out other possibilities for CHOP-induced neuronal cell death.
There is ample evidence to indicate that ROS exacerbate ischemic brain injury (Lewén et al, 2000); however, the way in which this happens is not clear. Many types of processes can be implicated (Sugawara et al, 2002; Saito et al, 2003). Meanwhile, we are aware that the ER is especially vulnerable to oxidative stress (Dreher et al, 1995; Racay et al, 1995; Viner et al, 1996; Yu et al, 1999; Oyadomari et al, 2001). The fact that the ER is a membranous organelle and contains a large amount of lipids may make it very susceptible to oxidative stress. The ER is one of the organelles that produces ROS (Halliwell and Gutteridge, 1999), and this may be one of the reasons for its vulnerability to oxidative stress. Because oxidative stress causes various kinds of cellular damage, we cannot conclude that ER stress is the sole event that leads to neuronal cell death. In this study, however, we found that Tg overexpression of SOD1 reduces the induction of ATF-4 and CHOP. Double staining for an ethidium signal and ATF-4 also suggests that ROS play an active role in ATF-4 induction. These results are in accord with the vulnerability of the ER to oxidative stress and its involvement in ischemic neuronal cell death. We strongly suggest, from the results of this study, that ROS cause ER damage and activate the ATF-4 and CHOP death signal pathways, which play important roles in subsequent neuronal cell death after cerebral ischemia and reperfusion.
Footnotes
Acknowledgements
The authors thank Liza Reola, Bernard Calagui and Ghezal Omar for technical assistance, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for figure preparation.