
Abstract
A brief period of cerebral ischemia confers transient tolerance to a subsequent ischemic challenge in the brain. This phenomenon of ischemic tolerance has been confirmed in various animal models of forebrain ischemia and focal cerebral ischemia. Since the ischemic tolerance afforded by preceding ischemia can bring about robust protection of the brain, the mechanism of tolerance induction has been extensively studied. It has been elucidated that ischemic tolerance protects neurons, and at the same time, it preserves brain function. Further experiments have shown that metabolic and physical stresses can also induce cross-tolerance to cerebral ischemia, but the protection by cross-tolerance is relatively modest. The underlying mechanism of ischemic tolerance still is not fully understood. Potential mechanisms may be divided into two categories: (1) A cellular defense function against ischemia may be enhanced by the mechanisms inherent to neurons. They may arise by posttranslational modification of proteins or by expression of new proteins via a signal transduction system to the nucleus. These cascades of events may strengthen the influence of survival factors or may inhibit apoptosis. (2) A cellular stress response and synthesis of stress proteins may lead to an increased capacity for health maintenance inside the cell. These proteins work as cellular “chaperones” by unfolding misfolded cellular proteins and helping the cell to dispose of unneeded denatured proteins. Recent experimental data have demonstrated the importance of the processing of unfolded proteins for cell survival and cell death. The brain may be protected from ischemia by using multiple mechanisms that are available for cellular survival. If tolerance induction can be manipulated and accelerated by a drug treatment that is safe and effective enough, it could greatly improve the treatment of stroke.
When exposed to a sufficient but sublethal alteration of environment, most living organisms acquire transient tolerance to otherwise lethal environmental changes. This cellular response is observed in a wide variety of species from bacteria to mammalian cells, indicating that it is an essential function for cells that have to survive various adverse circumstances over a long time. The underlying mechanism of such an important phenomenon is believed to be conserved among species (Feder and Hofmann, 1999).
For cells and organs living under aerobic conditions, a long period of anoxia or ischemia can be fatal. The brain is known to be particularly vulnerable to ischemia. Even a brief ischemic insult of a few minutes can kill a certain population of neurons, such as CA1 neurons of the hippocampus. Since the advent of recombinant thrombolytic agents, recanalizing the occluded vessel and reversing the ischemia in stroke patients has become feasible, and was proven to be beneficial if performed within 3 hours of stroke onset (NINDS rt-PA Stroke Study Group, 1995). We still lack effective measures, however, to protect neurons exposed to ischemia. If neuronal protection becomes possible, it will substantially expand the therapeutic time window for cerebral ischemia.
Neurons share their genetic information with other cells. It is natural, therefore, to postulate that the brain can acquire transient tolerance by a preceding stress as in other cells. Indeed, this possibility has been proven first in the retina, and then in hippocampal neurons. Barbe et al. (1988) showed that retinal photoreceptor cells became resistant to exposure to strong light, after a brief exposure to high temperature. Then Kitagawa et al. (1990) first described that brief ischemia, which is by itself not lethal, renders hippocampal neurons tolerant to subsequent ischemia that was otherwise lethal. This report drew wide attention because the protection of hippocampal CA1 neurons afforded by their method was the most robust among neuroprotective measures ever reported. Following on from this article, a substantial number of studies confirmed the phenomenon of ischemic tolerance in forebrain ischemia models, and then in the penumbral region in focal ischemia models. Further studies revealed that metabolic or physical stresses, which were nonischemic, could also induce cross-tolerance to ischemia. Measurement of cerebral blood flow showed that such tolerance was not accompanied by an improvement of regional tissue perfusion during or after the ischemia that induced tolerance (Matsushima and Hakim, 1995; Chen et al., 1996; Barone et al., 1998). Therefore, the state of ischemic tolerance seems to be based on the alteration of neurons themselves at the cellular level.
The tolerance phenomenon has also been studied in other organs. In the heart, it has become apparent that brief ischemia provides a marked increase in myocardial resistance to a later ischemic insult that would be lethal otherwise (Tomai et al., 1999). Murry et al. (1986) found that four periods of 4-minute coronary occlusion, each separated by 5 minutes of reperfusion, led to a reduction in infarct size produced by a subsequent 40-minute occlusion of the coronary artery. They called this method of enhancing ischemic tolerance in the heart “preconditioning.” Extensive research in experimental animals has provided data on the potential mechanism of ischemic preconditioning in the heart, which includes adenosine receptor activation, opening of mitochondrial ATP-sensitive potassium channels, and production of endogenous protective stress proteins.
There are two temporally distinct types of ischemic tolerance afforded by sublethal pretreatment—immediate and delayed tolerance—which may differ in their mechanisms. Ischemic tolerance found in the brain is usually of the delayed type, but in the heart, the tolerance induced by ischemic preconditioning is almost immediate and wanes quite rapidly within a few hours, representing a marked difference between the heart and the brain. A delayed phase of myocardial protection has since been found, however, and its molecular mechanism is now being extensively studied (Sun et al., 1995).
We have ample evidence that ischemic tolerance is a reproducible phenomenon, but its underlying mechanism is still not fully understood. A possible explanation of tolerance induction is the neuronal stress response. After exposure to a noxious condition, or stress, cells start synthesizing a special group of proteins called stress proteins, an increased amount of which make cells resistant to subsequent stresses (Jäättelä, 1999). This cellular reaction is thus called the stress response. Investigations of the stress response have a long history and date back to the early 1960s. Because the stress response was first studied on the acquisition of tolerance to heat shock, those stress proteins have been called heat-shock proteins (usually designated Hsp). Heat-shock proteins can be divided into six subfamilies, and many molecular species have been documented (Lindquist, 1988; Morimoto and Santoro, 1998). There are numerous reports on the increased expression of Hsp70 in response to cerebral ischemia (Massa et al., 1996; Abe and Nowak, 1996a). The role of Hsps other than Hsp70, however, in the ischemic brain in vivo has only partially been studied.
Although the stress response can be a major component of ischemic tolerance in the brain, neurons seem to be protected by divergent mechanisms of tolerance induction. Even a brief period of ischemia can trigger an enormous complexity of gene expression in neurons (MacManus and Linnik, 1997; Koistinaho and Hokfelt, 1997). Protein kinase signaling, immediate early genes, growth factors, and antioxidant defense enzymes are examples of such a response. It may not be surprising, therefore, that induction of ischemic tolerance is governed by multiple mechanisms. These mechanisms appear in different temporal and spatial patterns as discussed later.
REPETITION OF ISCHEMIC INSULTS
The effect of repeating ischemic insults on the brain varies depending on the duration, the interval, and the number of the episodes. In the Mongolian gerbil, a single episode of 5 minutes of forebrain ischemia damages only a circumscribed region in the hippocampus. If ischemia of 5-minute duration were to be performed repeatedly, however, it would result in cumulative brain damage. For example, three separate 5-minute periods of ischemia in the gerbil resulted in more extensive brain injury than one 15-minute period of ischemia (Tomida et al., 1987; Vass et al., 1988). The cumulative effect was most pronounced when the interval between the insults was 1 hour. Multiple episodes of 2-minute forebrain ischemia in the gerbil model brought about extensive brain injury in the hippocampus, thalamus, and cerebral cortex. The severity of neuronal damage was dependent on the number of and interval between the ischemic insults (Kato et al., 1989, 1991). Survival of animals worsened if the repeating unilateral ischemia was performed with short intervals (3 hours), but it increased gradually up to 100% when the interval was prolonged to 48 hours (Hanyu et al., 1993). A similar cumulative effect was found in the rat (Nakano et al., 1989). These experiments indicated that a preceding ischemic insult makes the brain tissue more susceptible, not tolerant, to further ischemia when the interval between the episodes is short (Ohno and Watanabe, 1996). Immediate induction of tolerance after preconditioning stress, as is seen in the heart, seems unlikely in the brain in vivo (see Time Course of Ischemic Tolerance).
ISCHEMIC TOLERANCE IN THE BRAIN
Although multiple repeating ischemic events bring about cumulative damage to the brain in general, it is quite possible that animals could attain tolerance within a specific time window after a sublethal ischemia. As was stated earlier, Kitagawa et al. (1990) first demonstrated the phenomenon of ischemic tolerance in vivo. After a 2-minute period of ischemia that does not cause neuronal damage by itself, they subjected gerbils to a second ischemia for 5 minutes, which was induced more than 24 hours after the first ischemia. When the second ischemia was induced 1 day or 2 days after the first ischemia, most of the hippocampal CA1 neurons survived (Fig. 1). Double 2-minute periods of preconditioning ischemia, which were induced with a 1-day interval, produced much more protection of CA1 neurons on 5 minutes of forebrain ischemia. Since the majority of CA1 neurons died without pretreatment, the authors concluded that brief, sublethal ischemia could confer ischemic tolerance. Acquisition of tolerance was a transient phenomenon. It took longer than 24 hours to develop, lasted for about 7 days, and faded away within 14 days (Kitagawa et al., 1991a). After this classic report, numerous experiments confirmed and extended this result. The phenomenon of ischemic tolerance was found not only in the gerbil (Kirino et al., 1991; Kato et al., 1991) but also in the rat (Liu et al., 1992; Nishi et al., 1993; Simon et al., 1993) and in the mouse (Wu et al., 2001). Tolerance was inducible even in neonatal animals by preconditioning in utero (Cai et al., 1997). Furthermore, brain regions that can respond to sublethal ischemia and can acquire tolerance were not restricted to the hippocampus: tolerance was also found in the cerebral cortex, basal ganglia and thalamus (Kitagawa et al., 1991a).
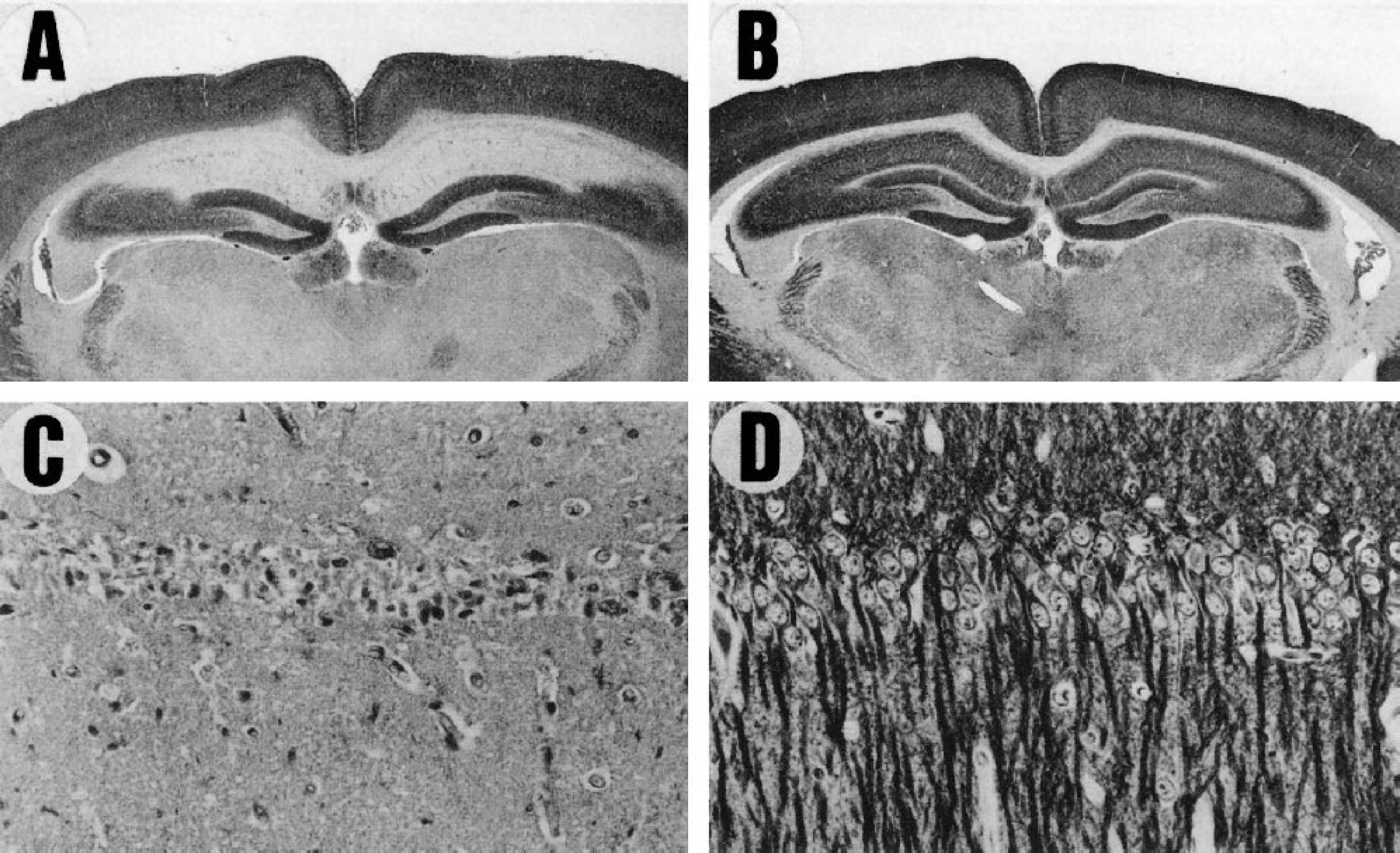
Immunostaining of microtubule-associated protein 2 (MAP2) demonstrates CA1 injury after 5 minutes of forebrain ischemia in the Mongolian gerbil. Extensive loss of neurons is shown in
Ischemic tolerance has also been described in cultured neurons and in brain slices. A neuroprotective effect of sublethal hypoxia on hypoxic neuronal damage in cortical cultures correlated with increased fibroblast growth factor (Sakaki et al., 1995). Sublethal oxygen–glucose deprivation or Na+/K+ ATPase inhibition in cortical culture also showed a neuroprotective effect (Bruer et al., 1997). Cultured mouse cortical neurons preconditioned by sublethal oxygen–glucose deprivation developed resistance to a longer exposure to oxygen–glucose deprivation. The tolerance was blocked by an N-methyl-D-aspartate (NMDA) antagonist (Grabb and Choi, 1999).
Because the majority of ischemic strokes in humans are caused by occlusion of an artery to the brain, induction of ischemic tolerance has also been examined in animal models of focal cerebral ischemia (Kitagawa et al., 1996; Glazier et al., 1994). Indeed, the extent of brain damage in these animals was much smaller on the side of preceding focal ischemia, indicating that tolerance can be induced by sufficient but sublethal ischemic stress, regardless of the type of ischemia. Interestingly, forebrain ischemia as a priming stress can protect the brain from either forebrain ischemia or from focal ischemia. Focal ischemia, vice versa, can confer tolerance to forebrain ischemia or focal ischemia. Small animal magnetic resonance imaging (MRI) has recently become available, and has been applied to study the tolerance phenomenon in focal ischemia. MRI demonstrated the evolution of infarction and its reduction by tolerance induction (Mullins et al., 2001). Once ischemic tolerance is induced, it attenuates not only neuronal injury but also the extent of ischemic brain edema by protecting vascular cells (Masada et al., 2001).
In human studies, the time course of patients presenting with transient ischemic attack (TIA) was analyzed to study whether ischemic tolerance occurs in clinical settings (Moncayo et al., 2000; Weih et al., 1999). It seemed difficult to prove the validity of this hypothesis, but the data suggested that patients who have experienced a number of TIAs showed better recovery than those with a single TIA. These studies suggest the possibility that preceding ischemia protects the human brain by a mechanism similar to ischemic tolerance.
TIME COURSE OF ISCHEMIC TOLERANCE
In general, it is widely accepted that immediate acquisition of tolerance is protein-synthesis independent, is mediated by posttranslational modification, and the effective duration is brief. Conversely, there seems to be a general agreement that delayed induction of ischemic tolerance requires new protein synthesis and is sustained for a few days to weeks. In the brain, the time course of ischemic tolerance apparently follows the delayed pattern, which leads to an assumption that synthesis of active proteins may be necessary for full development of ischemic tolerance. The latency period for ischemic tolerance is usually longer than 1 day, and the protection of brain tissue is induced only when the preconditioning ischemia is performed at least 1 day before the test ischemic challenge (Barone et al., 1998). Once induced, the condition of ischemic tolerance is believed to last for a few days and to wane gradually until it disappears a few weeks after acquisition (Kirino et al., 1991).
In contrast, immediate development of ischemic tolerance has been reported, particularly in vitro. Repeated hypoxic insults in a slice preparation brought about tolerance and adaptation to subsequent hypoxia (Schurr and Rigor, 1987). Amplitudes of evoked potentials recovered significantly better after test anoxic insults in preconditioned hippocampal slices. The latency periods to develop tolerance in the slice preparation ranged from 30 minutes to 2 hours (Pérez-Pinzón et al., 1996). The survival of neurons after a longer observation period, however, is largely unknown in experiments using cultured neurons or brain slices.
In a rat forebrain ischemia experiment, a short (30-minute) reperfusion period between the pretreatment and test ischemic challenge provided significant protection in CA1 neurons in vivo (Pérez-Pinzón et al., 1997). A significant neuronal protection was found at 3 days after ischemia, but neuroprotection had almost disappeared by 7 days after ischemia. This result indicates that immediate protection, if any, may not last long and may only delay the process of neuronal death. In a mouse intraluminal thread model of focal ischemia, a brief, mild ischemia in the territory of the middle cerebral artery can produce ischemic tolerance with a 30-minute interval from the preconditioning ischemia (Stagliano et al., 1999). The protection was not observed when the interval was extended to 2 hours. Since the infarct volume was measured only at 24 hours after ischemia in this experiment, the long-term survival of neurons is not known. These results seem to be contradictory to the previous publications of repetitive ischemia (see Repetition of Ischemic Insults), but the reason for the different outcomes remains unknown. Overall, however, it seems that the protection afforded by immediate tolerance induced by stresses other than ischemia is relatively modest compared with that conferred by an ischemic insult.
ISCHEMIC TOLERANCE AND POSTISCHEMIC FUNCTIONAL RECOVERY
After ischemic loss of CA1 neurons, animals exhibit significant impairment in memory acquisition (Volpe et al., 1984; Kiyota et al., 1991; Zola-Morgan et al., 1992). Ischemic tolerance protects neurons and, at the same time, it also preserves brain function (Ohno and Watanabe, 1996). Memory acquisition was not impaired after preconditioning ischemia that confers ischemic tolerance in the rat. Preconditioned or control rats were subjected to 10 minutes of ischemia that is lethal to CA1 neurons. When the interval between the pretreatment and the second ischemia was short (2 hours), no effect was found, whereas when the interval was long (2 days), the animals displayed maintained memory acquisition.
Contrary to this result, it has been postulated that neuronal protection by ischemic tolerance is only a transient phenomenon. After acquisition of ischemic tolerance, Corbett and Crooks (1997) recognized a gradual decline of CA1 neurons that were initially protected from ischemia. They observed that neuronal loss continued in the CA1 sector with increasing survival time. The decrease depended on the age of the animals. In older animals, the decline in neuronal number was slow and mild, whereas young gerbils showed a faster and greater decrease in number. Ischemic tolerance protected around 85% of CA1 neurons in old and young gerbils. The number decreased down to 53% to 66% in young animals at 60 days after ischemia. In older animals, it declined only down to 75% (Dowden and Corbett, 1999). These experiments have shown that in hippocampal CA1, neurons may continue to decline beyond the initial reduction in neuronal density induced by ischemia. In a rat experiment, ischemic tolerance preserved 88% of CA1 neurons observed at 8 weeks after ischemia, indicating that in this model a gradual loss of neurons is unlikely (Shamloo and Wieloch, 1999). However, insufficient data are currently available on neuronal density changes over prolonged periods.
CROSS-TOLERANCE
Cross-tolerance implies that a noxious stress can confer cellular tolerance to a subsequent stress that is different in nature from the first one (Table 1). A good example of cross-tolerance is acquisition of tolerance against photic stimulation in retinal photoreceptor cells after brief exposure to high temperature (Barbe et al., 1988). Elevation of body temperature (Chopp et al., 1989) in the rat led to acquisition of tolerance to subsequent forebrain ischemia. High body temperature up to 41.5°C for 15 minutes in the rat protected CA1 neurons 24 hours thereafter. Repeated hyperthermia was even more efficient in protecting neurons in the same model (Kitagawa et al., 1991b). The protective effect, however, was less prominent than that evoked by brief ischemia. Elevated body temperature usually causes Hsp70 elevation (Nowak et al., 1990) mainly in glial cells with neuronal expression obvious only in cerebellar granule cells (Marini et al., 1990). Brief forebrain ischemia in the Mongolian gerbil frequently triggers postischemic hyperthermia that rises to 39°C (Kuroiwa et al., 1990; but also see Welsh and Harris, 1991). Hyperthermia itself could further aggravate ischemic brain damage because elevated brain temperature is known to exert cumulative damage on the brain tissue. Preconditioning ischemia in the gerbil model, therefore, could represent a mixture of ischemia and hyperthermia as the preconditioning stress for tolerance induction (Abe and Nowak, 2000; but also see Kitagawa et al., 1997).
Inducers of ischemic tolerance

Anoxia causes brain injury that is similar to ischemic damage and can be used to induce ischemic tolerance in neonatal rats (Gidday et al., 1994). Postnatal rats were exposed to normothermic hypoxia and then subjected to hypoxia–ischemia 24 hours later. This procedure induced almost no brain injury in preconditioned animals, but it resulted in 34% infarction in the control group. Hypoxic preconditioning in neonatal rat induced hypoxia-inducible factor-1, which then modulated the expression of many genes (Jones and Bergeron, 2001). Hypoxic pretreatment also protected hippocampal neurons from kainic acid–induced injury, thus demonstrating that hypoxia can confer another type of cross-tolerance (Pohle and Rauca, 1994). On the other hand, exposure to hypoxia in cultured hippocampal neurons made the cells more vulnerable to glutamate stress (Kohmura et al., 1990). This situation is quite similar to ischemia because repeated stresses with short intervals result in cumulative injury, yet those with longer intervals result in protection or induction of tolerance. Preceding chemical inhibition of oxidative phosphorylation by 3-nitropropionate brought about early-onset long-lasting neuroprotection against in vitro hypoxia (Riepe et al., 1997). This experimental paradigm of “chemical preconditioning” was examined in in vivo ischemia in the gerbil and in the rat. Contrary to in vitro findings, tolerance was only seen 2 to 3 days after the injection of 3-nitropropionate (Sugino et al., 1999; Wiegand et al., 1999).
Spreading depression (SD) is a phenomenon that is characterized by a gradual expansion of the area of electrical suppression in the cerebral cortex. In the area of SD, massive depolarization of neurons and elevation of extracellular K+ occur, but they do not cause irreversible neuronal damage (Nedergaard and Hansen, 1988). Spreading depression is another stress that can confer ischemic tolerance in the rat. After SD, CA1 neurons were protected from neuronal damage after forebrain ischemia (Kawahara et al., 1995; Kobayashi et al., 1995). Protection was also seen in focal ischemia after SD (Matsushima et al., 1996). The mechanism of ischemic tolerance by cortical SD is only poorly understood. Spreading depression in the rat downregulated glial glutamate transporter isoforms. This change was attributed to a decrease in cortical glutamate exposure during the ischemic challenge after SD (Douen et al., 2000). Cortical SD was also shown to increase brain-derived neurotrophic factor (BDNF) levels that were consistent with the induction of ischemic tolerance (Kawahara et al., 1997).
Oxidative stress can confer tolerance in gerbil hippocampal neurons. Diethyldithiocarbamate, an inhibitor of SOD, elicits oxidative stress in the brain when it is injected into the lateral ventricle. Two or 4 days after this procedure in the gerbil, animals were subjected to 5 minutes of forebrain ischemia. When the substance was given 4 days before the ischemic challenge, it showed a protection, but it did not on the second day after drug administration (Ohtsuki et al., 1992). The acquisition of tolerance correlated well with expression of Hsp70 and manganese SOD. Copper-zinc SOD is also known to function as a protectant in cerebral ischemia. Overexpression of CuZn SOD in the mouse has been reported to ameliorate focal ischemic brain injury (Kinouchi et al., 1991). Measurement of focal cerebral blood flow by laser-Doppler flowmetry has revealed that this protective effect is not due to a difference in the cerebral circulation between transgenic and wild-type mice (Kamii et al., 1994). The enzyme is also known to protect brain tissue from ischemia when it is administered intravenously (Liu et al., 1989; Imaizumi et al., 1990; Uyama et al., 1992). Taken together, it is likely that increased expression of SOD by oxidative stress could induce ischemic tolerance.
Small traumatic brain injuries can protect the brain from subsequent damage. Multiple stab wounds in the brain 1 week before ischemia in the mouse improved the survival rate after ischemia (Takahata and Shimoji, 1986). Since the mechanism of brain injury by trauma is at least partially similar to ischemic brain injury or to glutamate neurotoxicity, the protective effect of head trauma may arise through a similar mechanism (Faden et al., 1989). A small surgical wound (Brown et al., 1989) or percussion damage to the brain (Tanno et al., 1993) accelerates expression of many genes including stress proteins in the rat. Furthermore, the extent of ischemic damage increases if the interval between trauma and ischemia is shortened (Jenkins et al., 1989). A short interval of around 1 hour causes aggravated injury, but tolerance is induced when the interval is elongated up to 24 hours.
Stresses such as status epilepticus (Lowenstein et al., 1990), and hypoglycemia (Bergstedt et al., 1993) are also inducers of the stress response and can bring about ischemic tolerance. Prior epileptic seizure can confer tolerance to subsequent seizures (epileptic tolerance). Kindling induced by kainic acid injection protected the brain from epileptic damage resulting from kainic acid–induced status epilepticus (Kelly and McIntyre, 1994). Bicuculline-induced seizure afforded protection of CA3 neurons against subsequent seizure that was otherwise lethal to those neurons (Sasahira et al., 1995). Epileptic preconditioning was also effective in inducing crossischemic tolerance in the rat (Plamondon et al., 1999). In contrast, hypothermia that usually suppresses the stress response (Chopp et al., 1992) was shown to reduce subsequent cerebral infarction (Nishio et al., 1999).
THE MECHANISM OF ISCHEMIC TOLERANCE
Acquisition of tolerance against various stresses requires at least two intracellular components (Table 2). The first component is the stress sensor that can detect various stressful conditions and convert the information into intracellular signals. The second component is the effector of tolerance induction. Since tolerance acquisition is conserved among a wide variety of cells, it is natural to believe that various stress signals are captured by a small number of sensors, and then the signals are gradually converged to a stereotyped final common pathway. This is based on a simplistic assumption that it would be extremely uneconomical for cells to have different sensors to all variety of stresses and to prepare inherent effectors that differ from cell to cell. In other words, such a simplistic view assumes that the mechanism of ischemic tolerance in the brain may be quite similar to that of other organs or other type of cells. On the other hand, it is quite possible to postulate a protective mechanism unique to neurons. Experimental data are not currently sufficient to answer these questions. According to the literature, potential mechanisms may be divided into at least two categories: (1) a cellular defense function against ischemia may be enhanced by the mechanisms inherent to neurons. They may arise by posttranslational modification of proteins or by expression of new proteins via a signal transduction system to the nucleus. These cascades of events may strengthen the influence of survival factors or may inhibit apoptosis. (2) A cellular stress response and synthesis of stress proteins may lead to an increased capacity for health maintenance inside the cell. These proteins work as cellular “chaperones” by unfolding misfolded cellular proteins and helping the cell to dispose of unneeded denatured proteins. Recent experimental data have demonstrated the importance of the processing of unfolded proteins in the cytoplasm and in the endoplasmic reticulum for cell survival and cell death (Ciechanover et al., 2000; Kopito, 2000; Paschen and Doutheil, 1999).
Proposed mechanisms of ischemic tolerance

CELLULAR DEFENSE
Our knowledge on neuronal defense mechanisms against ischemia, other than the stress response, is still fragmentary, and a unified view on this phenomenon is not possible. Sensors that act at the start points of the defense mechanism may be distributed diffusely, and to identify a relatively small number of “universal stress detectors” is difficult. Accordingly, neurons seem to have various effectors for cell protection.
Initial events of tolerance induction may involve the opening of ATP-sensitive K+ channels via the activation of adenosine A1 receptors (Heurteaux et al., 1995). This hypothesis was supported by the fact that adenosine uptake inhibition could potentiate ischemic tolerance (Kawahara et al., 1998) and that adenosine receptor antagonists blocked the ischemic tolerance phenomenon (Hiraide et al., 2001). The alteration of ATP-sensitive K+ channels hyperpolarizes the neuronal cell membrane, thereby protecting the neuron from detrimental depolarization.
Acquisition of tolerance may be linked to an alteration in the electrophysiologic properties of CA1 neurons (Shimazaki et al., 1998). Postanoxic potentiation (anoxic LTP) was inhibited in slices obtained from gerbils that had acquired ischemic tolerance. This observation suggests that ischemic tolerance is mediated by inhibiting postischemic overactivation of CA1 neurons, but the precise mechanism is not known (Kawai et al., 1998). The total amount of mRNA of an isoform of the AMPA receptor is changed by an ischemic insult. Reduction of GluR2, the “Ca2+ ion gatekeeper,” after lethal ischemia is postulated to induce greater Ca2+ influx, resulting in neuronal injury. Preconditioning ischemia prevented the downregulation of GluR2, thereby protecting neurons (Tanaka et al., 2002; Alsbo et al., 2000). Alteration of neuronal circuitry in the hippocampus may therefore be related to tolerance induction. Neurons in the hippocampal hilar region (CA4) that contain somatostatin are exceptionally vulnerable to ischemia. Two minutes of ischemia, used as a tolerance inducer in the gerbil model of forebrain ischemia, does not kill CA1 neurons but it destroys somatostatin neurons in CA4. This observation led to a hypothesis that ischemic tolerance is afforded by the progressive loss of hilar neurons, since the loss of CA4 neurons changes synaptic connections to CA1 pyramidal neurons (Matsuyama et al., 1993). This is unlikely because ischemic tolerance can be inducible repeatedly (Chen et al., 1994), and induction of ischemic tolerance has been shown to be independent of the presence or absence of hilar somatostatin neurons.
Ischemia alters the properties of the neuronal plasma membrane, and this change could suppress extracellular release of glutamate during the second period of ischemia. Measurement of glutamate by microdialysis, however, did not verify this hypothesis (Nakata et al., 1994).
Brief ischemia in the brain triggers a complex pattern of gene expression, which includes the immediate-early genes (IEG) fos, jun, and Krox. Preconditioning ischemia caused postischemic increase in c-JUN protein expression but not in the other IEGs. The enhanced tolerant state after priming ischemia may therefore be associated with the activation of specific transcription factors such as c-JUN (Sommer et al., 1995). Expression of c-fos was implicated in the protection of the contralateral hippocampus after unilateral preconditioning ischemia (Belayev et al., 1996).
Nitric oxide (NO) production may also be linked to ischemic tolerance. In a newborn rat model, hypoxic preconditioning rendered the animals resistant to hypoxic–ischemia. Acquisition of tolerance in this model depended on NO production by endothelial nitric oxide synthase (Gidday et al., 1999). In the rat slice model, neuronal nitric oxide synthase (nNOS)-mediated NO was involved in anoxic preconditioning (Centeno et al., 1999). Nitric oxide production depended on NMDA-receptor activation (Grabb and Choi, 1999), which activated nNOS. Increased NO can activate the Raf/Mek/Erk cascade and can induce new protein synthesis (Nandagopal et al., 2001).
Ischemia activates protein phosphorylation (Shamloo and Wieloch, 1999) and this phosphorylated state persists for a few days, as was confirmed with calcium/calmodulin-dependent protein kinase II (Shamloo et al., 2000) and mitogen-activated protein kinases (Shamloo et al., 1999). This enhanced phosphorylation is blocked by preconditioning ischemia. Since the cascade of phosphorylation may work as an amplifier of neuronal injury, preconditioning could cancel this detrimental cascade and normalize the intracellular signal transduction. Postischemic activation of Akt/protein kinase B may contribute to the induction of ischemic tolerance (Yano et al., 2001). Akt was activated after sublethal ischemia that could induce tolerance and inhibition of Akt activity resulted in attenuation of ischemic tolerance. Possible molecules lying downstream to Akt are BAD, caspase-9, Bcl-2, and trophic factors such as BDNF, and Hsp70.
Gene expression that leads to apoptosis may be related to ischemic tolerance. Expression of p53 has been known to be correlated with neuronal apoptosis after cerebral ischemia. A recent report showed that ischemia activates p53 gene expression along with its downstream genes, and preconditioning ischemia markedly reduces this activation (Tomasevic et al., 1999). Procedures that induce ischemic tolerance enhance Bcl-2 protein expression (Shimazaki et al., 1994), and antisense treatment against Bcl-2 mRNA blocked tolerance. Since Bcl-2 expression is known as a key step that inhibits apoptosis, its increase may well be related to ischemic tolerance (Shimizu et al., 2001).
Inflammatory cytokines, particularly tumor necrosis factor alpha (TNF-α) or interleukin-1 beta (IL-1β), have been implicated in the mechanism of ischemic tolerance (Ohtsuki et al., 1996a; Wang et al., 2000). Tolerance induced by brief priming ischemia can be blocked by an IL-1–receptor antagonist (Barone et al., 1998). Lipopolysaccharide (LPS) injection has been reported to induce ischemic tolerance against subsequent focal cerebral ischemia (Tasaki et al., 1997) and LPS-induced tolerance can be blocked by a TNF-α–binding protein. Intravenous administration of LPS conferred tolerance in the rat and led to elevation of ceramide in the brain, suggesting that ceramide may play a role in LPS-induced protection (Zimmermann et al., 2001).
Nuclear factor (NF)-κ is activated by various signals, such as cytokines, neurotrophic factors, and neurotransmitters. Oxidative stress and intracellular Ca2+ elevation can also induce NF-κ. Nkuclear factor-κ, in turn, plays a pivotal role in the induction of neuroprotective gene products, such as MnSOD and Bcl-2. All of these products are known to be related to tolerance induction. Nuclear factor-κ increases with three different paradigms of ischemic tolerance induced by sublethal ischemia, epilepsy, or polyunsaturated fatty acid (Blondeau et al., 2001). All of these procedures led to a rapid increase in NF-κ activity. Pretreatment with NF-κ inhibitor blocked NF-κ activity and eventually abolished the neuroprotective effect of preconditioning. Erythropoietin-mediated neuroprotection against ischemia was reported to involve cross-talk between the Jak2 and the NF-κ cascades in cultured neurons (Digicaylioglu and Lipton, 2001).
Glial cell proliferation has been implicated in the induction of ischemic tolerance in cortical neurons (Kitagawa et al., 2000). Bromodeoxyuridine-labeled cells, however, did not increase in the hippocampus during the period when ischemic tolerance was acquired (Liu et al., 2001).
STRESS PROTEINS AND ISCHEMIC TOLERANCE
Acquisition of ischemic tolerance could be explained by the stress response of the brain tissue (Table 3). When exposed to a noxious environment, the stress signal in the cell is transferred to heat-shock factors to convey information to the heat-shock element that exist upstream to stress-inducible hsp genes. Then, hsp gene expression is greatly enhanced and the cell acquires transient tolerance to further stresses (Fig. 2). These characteristics are common to a wide variety of cells, and neurons may not be the exception. The stress response is essential for cell survival because cells cannot cope with a sudden accumulation of denatured proteins or protein aggregation that arises from various stresses such as thermal, anoxic or oxidative stresses. Protein aggregates do not accumulate in unstressed cells because they are continuously degraded by cellular health control machinery. This mechanism consists of chaperoning unfolded proteins and subsequent proteolysis by the ubiquitin–proteasome pathway (Fig. 3) (Ciechanover et al., 2000). Approximately one third of newly synthesized proteins, in fact, are degraded within minutes of their synthesis (Yewdell, 2001). Once this cellular health care system fails, apoptosis occurs. This pathomechanism has been implicated in neurodegenerative disorders such as Alzheimer disease, Parkinson disease, and Huntington disease (Kopito, 2000). Recent studies have also revealed that processing of denatured proteins is disturbed in postischemic hippocampal neurons (Ide et al., 1999; Asai et al., 2002).
Stress proteins
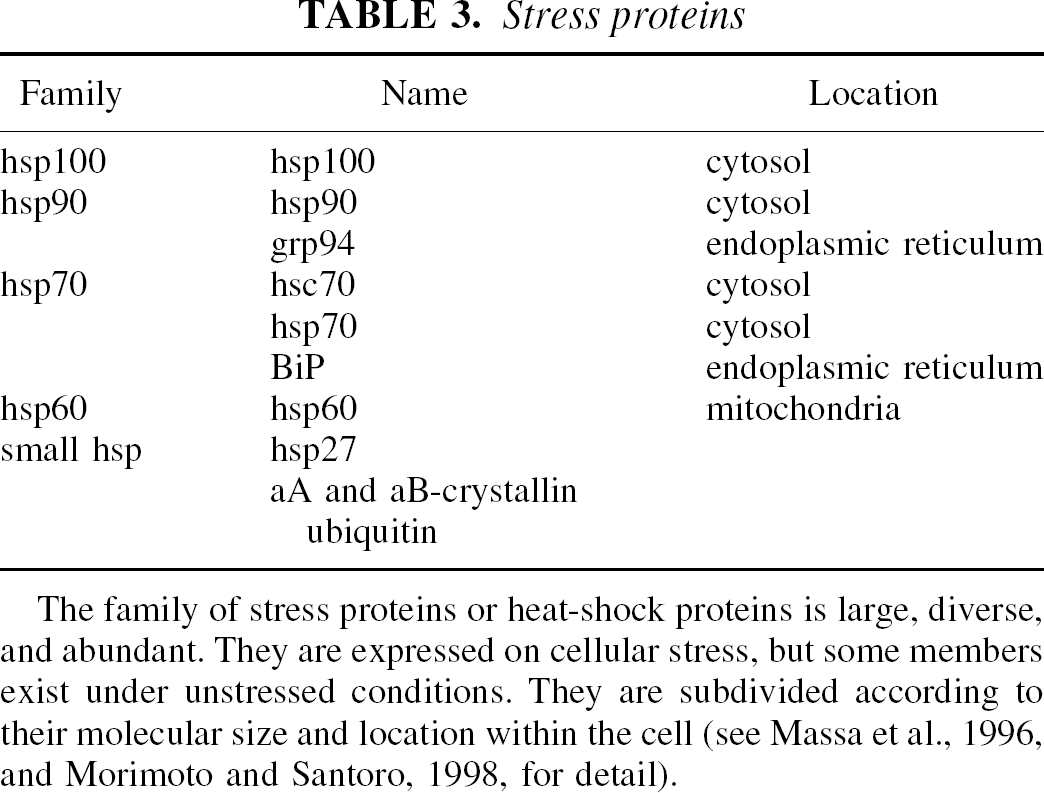
The family of stress proteins or heat-shock proteins is large, diverse, and abundant. They are expressed on cellular stress, but some members exist under unstressed conditions. They are subdivided according to their molecular size and location within the cell (see Massa et al., 1996, and Morimoto and Santoro, 1998, for detail).
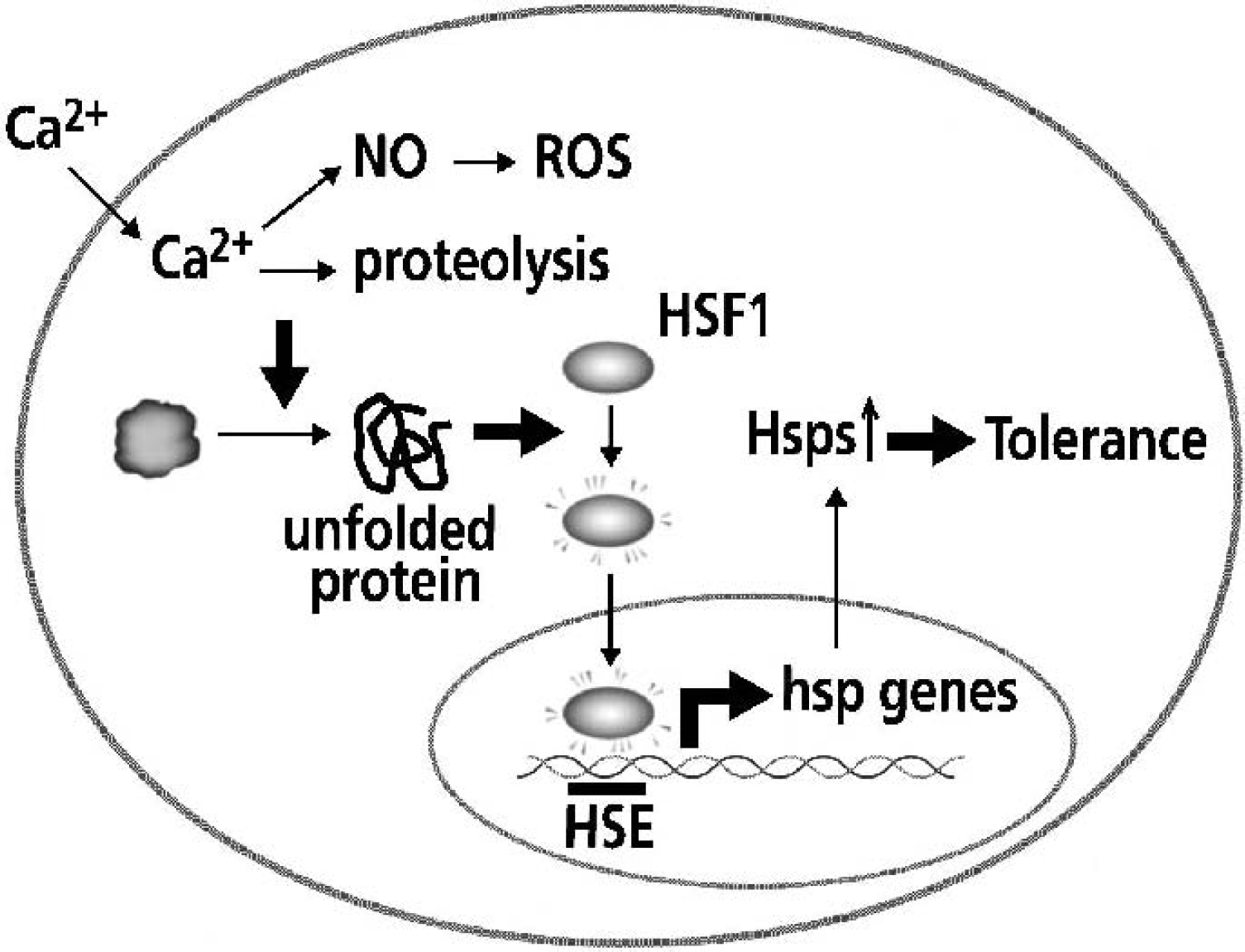
Protein molecules are one of the most sensitive sensors of stressful conditions. On ischemic stress, increased intracellular Ca2+ and nitric oxide produce damage and unfold the normal conformation of proteins within the cytoplasm and endoplasmic reticulum. These denatured protein molecules are partially renatured by chaperones. If the chaperone system cannot counter increased unfolded proteins, heat-shock factor 1 (HSF1) is activated. HSF1 then enters the nucleus and greatly enhances expression of hsp genes. When an excess amount of Hsps is produced by the stress response, it is believed to confer tolerance to subsequent stresses. Hsp, heat-shock protein; NO, nitric oxide; ROS, reactive oxygen species; HSE, heat-shock element.
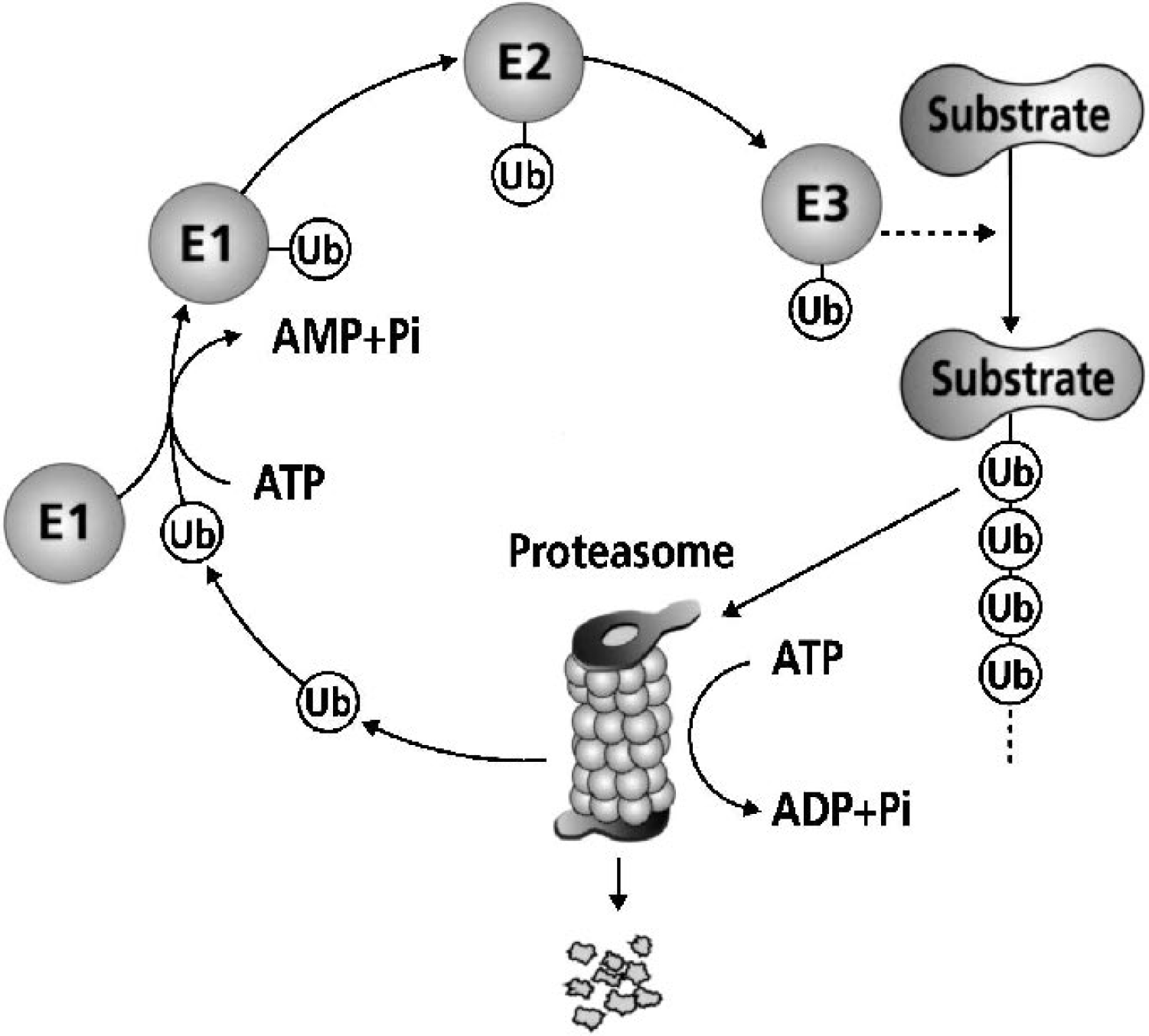
Free ubiquitin (Ub) is activated in an ATP-dependent manner by ubiquitin-activating enzyme (E1). Then, ubiquitin is transferred to ubiquitinconjugating enzymes (E2) and several ubiquitin molecules are added to the substrate proteins in a form of a multiubiquitin chain. This process usually requires the help of ubiquitin ligase (E3). Once substrate proteins are multiubiquitinated, they are degraded by the proteasome, whereas monoubiquitinated substrates have a role in the regulation of cellular functions. When cells are subjected to stressful conditions, protein molecules in the cytoplasm and in the endoplasmic reticulum are denatured and unfolded. If they are not refolded with the aid of molecular chaperones such as Hsp70, they are usually degraded by this ubiquitin–proteasome pathway. The function of this pathway is believed to be essential for cell survival.
When ischemic tolerance is induced by preceding ischemia, Hsp70 increases in the hippocampal CA1 pyramidal cells in the gerbil (Kirino et al., 1991; Nishi et al., 1993; Liu et al., 1993) and in the rat (Liu et al., 1992, 1993; Glazier et al., 1994). The amount of Hsp70 was also shown to correlate well with tolerance acquisition in a gerbil model of chronic hypoperfusion of the brain (Ohtsuki et al., 1993). Increased amounts of Hsp70 seem to be necessary for ischemic tolerance, since experimental manipulation that inhibits Hsp70 function abolishes ischemic tolerance (Nakata et al., 1993). Quercetin, a substance known to inhibit the stress response, could partially inhibit induction of ischemic tolerance when the substance was injected in the lateral ventricle in the gerbil. A similar experiment was done with an anti-Hsp70 antibody, and it showed that the antibody also partially inhibited ischemic tolerance. To interrupt increased synthesis of Hsp70, injection of MK801 (an NMDA antagonist) or anisomycin (a protein synthesis inhibitor) was examined (Kato et al., 1992). MK801 inhibited Hsp70 synthesis and abolished ischemic tolerance after 2 minutes of preconditioning ischemia. In contrast, anisomycin also inhibited Hsp70 synthesis but did not inhibit tolerance induction. This experiment could be interpreted as signifying that augmented NMDA neurotransmission is necessary for tolerance induction (Bond et al., 1999) but Hsp70 is not. Inhibition of protein synthesis, however, is in itself a stressful stimulus to cell survival, and it could function as a stressor in a different way (Lobner and Choi, 1996).
Other stress proteins were also reported to increase on tolerance induction. Concomitant increase in glucose regulated protein (grp) 75/78 mRNA and protein was observed in the rat cerebral cortex after tolerance induction. Their increase, however, did not correlate closely with that of ischemic tolerance (Chen et al., 1996). An increase of Hsp27 was demonstrated in the brain after tolerance induction (Kato et al., 1994). The direct role of Hsp27 in neuronal ischemic tolerance remains to be explained since Hsp27 expression is observed mainly in glial cells. Induction of the Hsp110/105 family was studied in rats that had acquired ischemic tolerance by transient ischemia (Yagita et al., 2001). Colocalization of Hsp110/105 and Hsp70 in the CA1 neurons suggested that expression of these stress proteins contributes to ischemic tolerance.
It is well established that protein synthesis is impaired by cerebral ischemia (Kleihues and Hossmann, 1971). When ischemic tolerance was induced in the gerbil model, the recovery phase of protein synthesis was accelerated after ischemia (Nakagomi et al., 1993; Furuta et al., 1993). Autoradiography using isotope-labeled amino acids demonstrated a good recovery, and the pattern of amino acid incorporation in the CA1 neurons returned to a normal pattern within 24 hours. Ultrastructural distribution of aggregated ribosomes was also restored to a near-normal pattern within 24 hours (Furuta et al., 1993). Stress-inducible proteins such as ubiquitin and Hsp70 do not appear after 5 minutes of ischemia in the gerbil model. After 2 minutes of ischemia that induces ischemic tolerance, however, expression of ubiquitin (Kato et al., 1993) and Hsp70 (Aoki et al., 1993) is enhanced. Ischemic tolerance also facilitated recovery of the synthesis of superoxide dismutase and, presumably, thereby protected neurons from ischemia (Kato et al., 1995; Toyoda et al., 1997). These experimental results have shown that ischemia disrupts the protein-synthesizing machinery, but ischemic tolerance can accelerate its recovery.
Contrary to the previous experiments, increased expression of stress proteins, particularly Hsp70, may not strictly be required in ischemic tolerance. Cerebral ischemia, even if it is sublethal for neurons, is now recognized to induce an increasingly complex range of gene expression (MacManus and Linnik, 1997; Wrang et al., 2001). Increase in Hsp70 could simply be a coincidental increment of gene expression on cellular stress. Evidence against the stress protein hypothesis has also been accumulated (Abe and Nowak, 1996a). Spreading depression conferred tolerance in cortical neurons to ischemia in the rat, but it failed to induce the expression of Hsp70 mRNA in regions of tolerance (Kobayashi et al., 1995). Gene expression was also studied in a gerbil ischemia model and was correlated with the timing of anoxic depolarization by DC potential in the hippocampus and acquisition of ischemic tolerance. This experiment demonstrated that the threshold depolarization that was required to induce tolerance correlated well with gene expression of transcription factors such as junB but not with Hsp70 (Abe and Nowak, 1996b; Abe and Nowak, 2000). These experiments have therefore shown that the presence of Hsp70 is not mandatory for ischemic tolerance. Immunostaining against Hsp70 should be carefully interpreted since Hsp70 protein is cryptic to immunodetection in aldehyde-fixed tissues that are obtained during early recirculation (Harrub and Nowak, 1998). Nevertheless, it is widely accepted that Hsp70 can make neurons resistant to noxious stimuli such as ischemia when it is expressed in excess. Overexpression of human inducible Hsp72 in transgenic mice also protected the heart against ischemia. Focal cerebral ischemia in the same transgenic mice revealed that overexpression of Hsp70 protects the brain from ischemic brain injury (Plumier et al., 1997; Rajdev et al., 2000).
CLINICAL IMPLICATIONS
Stressful conditions have been shown to confer tolerance to further stresses when the first stress is sufficient but sublethal for cells. The threshold for tolerance induction and that for cell injury are not usually far apart. This makes it difficult to use a tolerance induction strategy for clinical application. Drug treatment that can accelerate tolerance induction may be a potential procedure, but it should be strong enough to show a robust protection. Moderate amelioration of brain injury may not be sufficient even if it is statistically significant in experimental animals. At the same time, the substance used for tolerance induction should be safe. A number of candidate pharmacological regulators of the stress response and inducers of ischemic tolerance have been proposed (Toyoda et al., 2000; Ohtsuki et al., 1996b; Xiao et al., 1999; Morimoto and Santoro, 1998). Since overexpression of Hsp70 results in cells that are more resistant to various stresses, gene therapy for hsp70 gene transfer has been examined and it seems, at least partially, promising (Yenari et al., 1998; Amin et al., 1996). Successful clinical application must await further elucidation of the mechanisms of ischemic tolerance.