
Abstract
Ischemic preconditioning (IPC) via protein kinase C epsilon (PKCε) activation induces neuroprotection against lethal ischemia. Brain-derived neurotrophic factor (BDNF) is a pro-survival signaling molecule that modulates synaptic plasticity and neurogenesis. Interestingly, BDNF mRNA expression increases after IPC. In this study, we investigated whether IPC or pharmacological preconditioning (PKCε activation) promoted BDNF-induced neuroprotection, if neuroprotection by IPC or PKCε activation altered neuronal excitability, and whether these changes were BDNF-mediated. We used both in vitro (hippocampal organotypic cultures and cortical neuronal-glial cocultures) and in vivo (acute hippocampal slices 48 hours after preconditioning) models of IPC or PKCε activation. BDNF protein expression increased 24 to 48 hours after preconditioning, where inhibition of the BDNF Trk receptors abolished neuroprotection against oxygen and glucose deprivation (OGD) in vitro. In addition, there was a significant decrease in neuronal firing frequency and increase in threshold potential 48 hours after preconditioning in vivo, where this threshold modulation was dependent on BDNF activation of Trk receptors in excitatory cortical neurons. In addition, 48 hours after PKCε activation in vivo, the onset of anoxic depolarization during OGD was significantly delayed in hippocampal slices. Overall, these results suggest that after IPC or PKCε activation, there are BDNF-dependent electrophysiologic modifications that lead to neuroprotection.
Introduction
Cerebral ischemia (i.e., from cardiopulmonary arrest or stroke) is a leading cause of death and disability in the United States, 1 where survivors have a diminished quality of life, as a consequence of the ischemia-induced neuronal damage. 2 Although only tissue plasminogen activator and hypothermia have been successfully used clinically to treat cerebral ischemia, more viable therapies with fewer logistical limitations are needed. Possible therapies may emerge from the field of ischemic preconditioning (IPC). Ischemic preconditioning occurs when a sublethal ischemic insult followed by a recovery period induces neuroprotection against future lethal ischemic events. Neuroprotective mechanisms induced by IPC include the modulation of synaptic activity,3, 4 improved synaptosomal mitochondrial respiration, 5 activation of extracellular signal-regulated kinase pathways,4, 6 and the modulation of transcriptional modification factors involved in neuroprotection (i.e., mRNA and gene expression).7, 8 Ischemic preconditioning-induced neuroprotection can also be emulated pharmacologically via activation of protein kinase C epsilon (PKCε), 9 which activates analogous IPC-induced neuroprotective pathways.3, 6, 10, 11 Interestingly, inhibition of PKCε after IPC abolished neuroprotection, suggesting PKCε activation is required for IPC-induced neuroprotection. 9 In addition, we previously found that IPC- and PKCε activation-induced neuroprotection depended on a functional modification of γ-aminobutyric acid (GABA) synapses, 3 suggesting that preconditioning induces electrophysiologic modifications for neuroprotection. In addition to neuroprotection, PKCε has been shown to modulate voltage-gated Na+ channels (Nav),12, 13 which are responsible for the production of action potentials and neuronal integrative properties such as threshold, firing frequency, and neurotransmitter release. 12
Ischemic preconditioning activates numerous protective pathways including members of the neurotrophic family of proteins, which consists of different nerve growth factors involved in neurite outgrowth and cell survival during periods of stress.8, 14 These factors include nerve growth factor, brain-derived neurotrophic factor (BDNF), neurotrophin-3, and neurotrophin-4/5, which activate either the Trk family of receptor tyrosine kinases or the p75 neurotrophin receptor. 15 Brain-derived neurotrophic factor is ubiquitously expressed in the brain with the highest levels found in the hippocampus and cerebral cortex and its actions are primarily mediated through neurotrophic TrkB receptors. 15 Since BDNF expression is enhanced in neurons by various stressors (e.g., epilepsy, hypoglycemia, ischemia, and trauma 16 ), chronic exposure to BDNF has been used as an agent for neuroprotection in vitro17, 18 and in vivo.19, 20 In addition to the pro-survival mechanism(s), BDNF is a signaling molecule that modulates synaptic plasticity21, 22 and neurogenesis. 15 Interestingly, BDNF mRNA levels are increased after IPC, 8 suggesting that the protective mechanisms induced by BDNF and neuroplasticity modulations may be present after preconditioning stimuli. Therefore, we hypothesized that activation of IPC via PKCε activation results in modulation of neuronal excitability that requires BDNF for neuroprotection.
Materials and Methods
All animal procedures were approved by the Institutional Animal Care and Use Committee (University of Miami) and performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
Reagents
Minimum essential medium, Hank's balanced salt solution, and fetal bovine serum were purchased from Gibco/Life Technologies (Grand Island, NY, USA). Protein kinase C epsilon activator (psi-epsilon RACK; ΨεRACK) and Tat carrier peptide were a generous gift from Dr Daria Mochly-Rosen (Stanford University, CA, USA). All other chemicals were obtained from Sigma-Aldrich (St Louis, MO, USA) unless otherwise noted.
Animals and Acute Slice Preparation
Male or female Sprague Dawley rats (21 to 32 days old) from Charles River Laboratories (Wilmington, MA, USA) were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine intraperitoneally. The animals were then decapitated, the brain was rapidly removed, and the hippocampus was dissected from the brainstem and overlying cortex. Slices of 300 μm thickness were sectioned using a Leica VT1000S microtome in sucrose artificial cerebrospinal fluid (aCSF; in mmol/l: 206 sucrose; 2.8 KCl; 1.0 CaCl2; 1.0 MgCl2; 2.0 MgSO4; 26 NaHCO3; 1.25 Na2HPO4; 0.4 ascorbic acid; 10 glucose; and oxygenated with 95% O2 and 5% CO2) at 4°C. Slices were then transferred into aCSF (in mmol/l: 126 NaCl; 3.5 KCl; 2.0 CaCl2; 2.0 MgSO4; 26 NaHCO3; 1.25 Na2HPO4; 0.4 Na-ascorbate; 10 glucose; and oxygenated with 95% O2 and 5% CO2) at 34°C for 30 minutes before being transferred/stored at room temperate in aCSF for an additional 30 minutes. Individual slices were then transferred to a submerged recording chamber, where they were perfused with oxygenated (95% O2 and 5% CO2) aCSF (33°C±1°C) using a Warner Instruments (Hamden, CT) automated temperature controller (TC-344) at a rate of 2 ml/min.
Cortical Neuronal-Glial Cocultures
Cortical neuronal-glial cocultures were prepared as previously described. 11 In brief, a glial monolayer was prepared from 1- to 2-day-old neonatal rats. The cerebral cortices were isolated, enzymatically dissociated and the resulting cellular suspension plated on glass coverslips or 6-well plates at a density of 3 hemispheres per 24-well plate in minimum essential medium containing 10% fetal bovine serum, 10% horse serum, 20 mmol/l glucose, and 2 mmol/l glutamine. After 10 days in culture, neuronal cells where isolated from the cortices of E18- to 19-day rats and plated on the confluent monolayer of glial cells at a density of 2.5 hemispheres per 24-well plate in minimum essential medium containing 5% fetal bovine serum, 20 mmol/l glucose, and 2 mmol/l glutamine. Cultures were maintained for 14 days before experiments were performed.
Organotypic Slice Cultures
Organotypic hippocampal slice cultures were prepared as previously described. 9 In brief, rats were anesthetized by intraperitoneal injection of ketamine (1.0 mg, ∼80 to 100 mg/kg) and the brains were rapidly removed. Transverse slices (400 μm) were dissected from the hippocampi and placed in Grey's balanced salt solution that was supplemented with 6.5 mg/ml glucose at 4°C. After 1 hour, slices were placed onto one membrane insert that was 30 mm diameter (Millicell-CM, EMD Millipore, Billerica, MA, USA). Individual inserts were placed into a six-well culture dish containing 1 ml per well of culture medium (50% minimum essential medium, 25% Hank's balanced salt solution, 25% heat-inactivated horse serum supplemented with 6.5 mg/ml glucose, and 1 mmol/l glutamine). The slice cultures were maintained at 36°C, in 5% CO2 for 14 to 15 days before experiments were performed.
Induction of Ischemic Preconditioning, PKCε Preconditioning, and Lethal Ischemia
For in vivo pharmacological preconditioning (specific PKCε activation), ΨεRACK (0.2 mg/kg) 11 or Tat (control peptide) were administered intraperitoneally 48 hours before acute slice preparation. Cultures were exposed to IPC or oxygen and glucose deprivation (OGD) as previously described.3, 11 Organotypic slices or cocultures were washed three times with aglycemic Hank's balanced salt solution (in mmol/l: 1.26 CaCl2·2H2O; 5.37 KCl; 0.44 KH2PO4; 0.49 MgCl2; 0.41 MgSO4·7H2O; 136.9 NaCl; 4.17 NaHCO3; 0.34 mmol/l Na2HPO4·7H2O; and 15 sucrose; pH 7.4) and exposed to an oxygen-free environment (90% nitrogen, 5% hydrogen, and 5% CO2, 37°C) using a COY anaerobic chamber (COY Laboratory Products, Grass Lake, MI, USA). To induce IPC, slices and cocultures were exposed to OGD for 15 or 55 minutes, respectively. Pharmacological preconditioning was performed by exposing cultures to the specific PKCε agonist (ΨεRACK; 200 nmol/l) for 1 hour, after which the slices (organotypic or naive acute slices) or cocultures were returned to normal culture media (or aCSF for acute slices). To induce lethal ischemia, slices and cocultures were exposed to OGD for 45 minutes or 4 hours, respectively. After IPC or lethal ischemia, cells and slices were transferred to normal culture media and placed back in the incubator. In some experiments cultures were treated with the Trk receptor antagonist K252a, 17 (200 nmol/l; Chemicon, Temecula, CA, USA) or dimethyl sulfoxide (0.02%) for 48 hours after IPC exposure and before lethal ischemic exposure.
Assessment of Cell Death
Organotypic hippocampal cell death was determined by propidium iodide staining as previously described.
9
In brief, slices were incubated in culture medium supplemented with 2 μg/ml propidium iodide for 1 hour before imaging. Micrographs of the organotypic slice propidium iodide staining were captured: (1) before preconditioning or sham treatment (baseline); (2) 24 hours after lethal ischemia to assess ischemic damage; and (3) 24 hours after N-methyl-
Immunohistochemistry Staining of Organotypic Slice Culture
Slices were collected 48 hours after preconditioning or sham treatment. Slices were fixed in 4% paraformaldehyde for 4 hours and permeabilized in phosphate-buffered saline (PBS) containing 0.8% Triton X-100 (PBST). After blocking with 10% goat serum, slices were incubated with anti-BDNF antibody (Chemicon, Temecula, CA, USA) in PBST overnight at 4°C. After overnight washing with PBST, the sections were incubated with fluorescein isothiocyanate-conjugated antirabbit secondary antibody (Santa Cruz Biotechnology, Dallas, TX, USA), for 24 hours at 4°C. The sections were washed in PBST and mounted using ProLong Antifade solution (Molecular Probes, Eugene, OR, USA). Slice images were captured on a Carl Zeiss Laser Scanning Microscope 510 (Zeiss, Jena, Germany) and analyzed using LSM 5 image browser (Zeiss).
Western Blot Analysis
Western blot analysis was performed as previously described. 11 Slices and cocultures were lysed in 1% Nonidet P-40, 20 mmol/l Tris (pH 8.0), 137 mmol/l NaCl, 0.5 mmol/l ethylenediaminetetraacetic acid, 10% glycerol, 10 mmol/l sodium pyrophosphate, 10 mmol/l sodium fluoride, 1 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mmol/l vanadate, and 1 mmol/l phenylmethylsulfonyl fluoride. Equal amounts of protein were separated on a 12% SDS—PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) gel and electrophoretically transferred to nitrocellulose membranes blocked in 5% milk and incubated overnight with antibodies against BDNF (Santa Cruz Biotechnology) and β-actin. Membranes were washed with Tris-buffered saline with Tween followed by incubation with secondary antibodies (Pierce Thermo Scientific; Rockford, IL, USA) for 1 hour at room temperature. Proteins were detected using enhanced chemiluminescence system (Pierce Thermo Scientific). Western blot densitometry was analyzed using ImageJ software (National Institute of Health, Bethesda, MD, USA). Quantification was performed by using a normalization to a fixed point or control protocol. 23
Whole-Cell Recordings
Whole-cell recordings were made from hippocampal cornu ammonis 1 (CA1) and excitatory cortical neurons using an Axopatch 700B amplifier (Molecular Devices, Sunnyvale, CA, USA) and Digidata1322A (Molecular Devices). One neuronal recording was made from individual slices or coverslips. Recordings were made using pipettes pulled from borosilicate glass with a resistance of 2 to 5 MΩ. The intracellular solution contained (in mmol/l): 140 K+-gluconate; 5 KCl; 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; 10 ethylene glycol tetraacetic acid; 2 MgCl2; 2 Adenosine 5′-triphosphate magnesium; and 0.5 guanosine 5′-triphosphate sodium, adjusted to a pH of 7.4. Recordings were not corrected for a 14.8 mV junction potential. To minimize variability between cells, current-clamp protocols were initiated using similar sequences and timing procedures. Cortical neuronal action potentials were recorded in the presence of 10 μmol/l 6-cyano-7-nitroquinoxaline-2,3-dione and 20 μmol/l
After obtaining GΩ seal, whole-cell configuration was obtained and maintained with a series resistance <20 MOhm. Recordings were excluded if the series resistance changed >20%. Resting membrane potential was not modified. For CA1 neurons, 50 pA increments were made from −250 to 450 pA, whereas for cortical neuronal cultures 20 pA increments from −100 pA to 300 pA in current clamp mode after bridge balance for 800 ms. No significant differences were observed in resting membrane potential when recorded immediately after whole-cell configuration or membrane properties after action potential recordings between experimental groups. Data were acquired using pClamp 9.0 (Molecular Devices), where action potentials were analyzed offline using custom written software in IGOR Pro (Wavemetrics, Lake Oswego, OR, USA). Threshold was determined as the point where the membrane potential dV/dt exceeded 10 V/s. Action potential amplitude was determined as the difference in mV between the maximum action potential amplitude and threshold. The after-hyperpolarization minimum was determined as the minimum membrane potential after an action potential.
Statistical Analysis
All data are expressed as mean±s.e.m. Statistical analysis between two groups was primarily performed using an unpaired Student's t-test. A Student's paired t-test was used to determine when the resting membrane potential was significantly different from the onset of OGD. Statistical analysis between more than two groups was performed using a one-way analysis of variance with Bonferroni multiple comparison post hoc test. All statistical tests were performed using SPSS statistical software (Chicago, IL, USA). P0.05 was considered statistically significant.
Results
Ischemic Preconditioning or PKCε Activation Increases Brain-Derived Neurotrophic Factor Protein Expression
Since BDNF mRNA expression was enhanced after IPC in the brain, 8 we hypothesized that IPC (via PKCε activation) increases BDNF protein levels. Ischemic preconditioning was induced in cortical neuronal-glial cocultures and BDNF protein expression was investigated using western blot analysis after IPC. Brain-derived neurotrophic factor protein levels were significantly increased 24 hours after IPC (1.8±0.3-fold from sham) and continued to increase to 3.2±0.3-fold as compared with sham-treated neuronal-glial cultures 48 hours after IPC (Figures 1A and 1B; P<0.05). To define if BDNF also increased in hippocampus, we induced IPC in hippocampal organotypic slices. Ischemic preconditioning induction significantly increased BDNF levels in organotypic hippocampal slices 1.5±0.2-fold from sham after 24 hours and increased further to 2.0±0.2-fold as compared with sham 48 hours after IPC (Figures 1C and 1D; P<0.05). Brain-derived neurotrophic factor protein levels in hippocampal organotypic cultures were also measured after ΨεRACK (200 nmol/l) exposure. Twenty-four hours after PKCε activation with ΨεRACK, BDNF levels were significantly increased to 1.7±0.2-fold as compared with sham-treated controls, and continued to increase (2.1±0.3-fold from sham) 48 hours after ΨεRACK treatment (Figures 1E and 1F; P<0.05). Finally, to confirm that BDNF levels increased in CA1 pyramidal neurons, we performed immunohistochemistry on organotypic hippocampal slices 48 hours after either IPC or ΨεRACK treatment. Brain-derived neurotrophic factor protein expression was increased in the CA1 region of the hippocampus after IPC or ΨεRACK treatment as compared with sham slices (Figure 2).
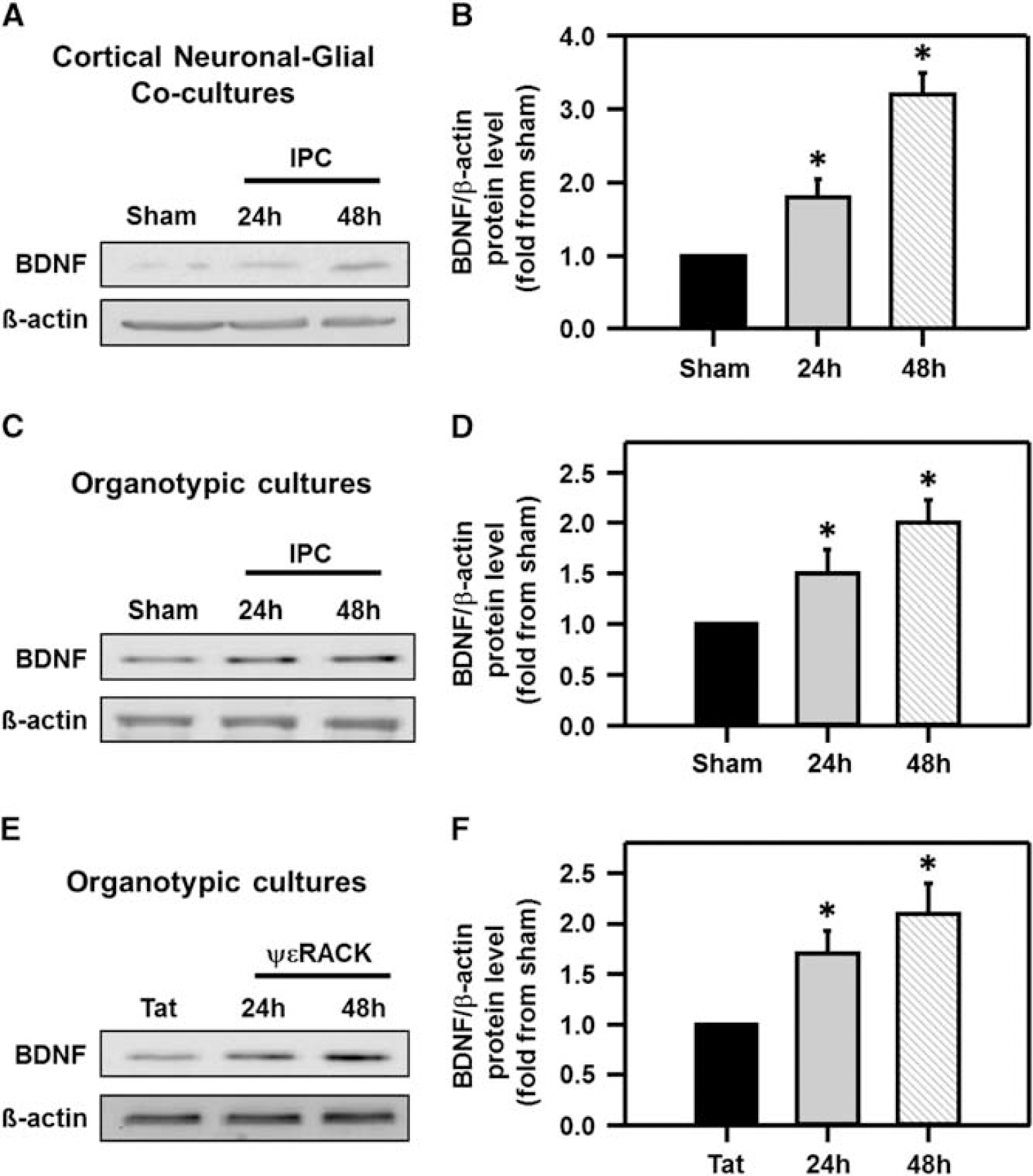
Increased brain-derived neurotrophic factor (BDNF) protein expression after preconditioning. Western blot images depicting increased BDNF protein expression from (
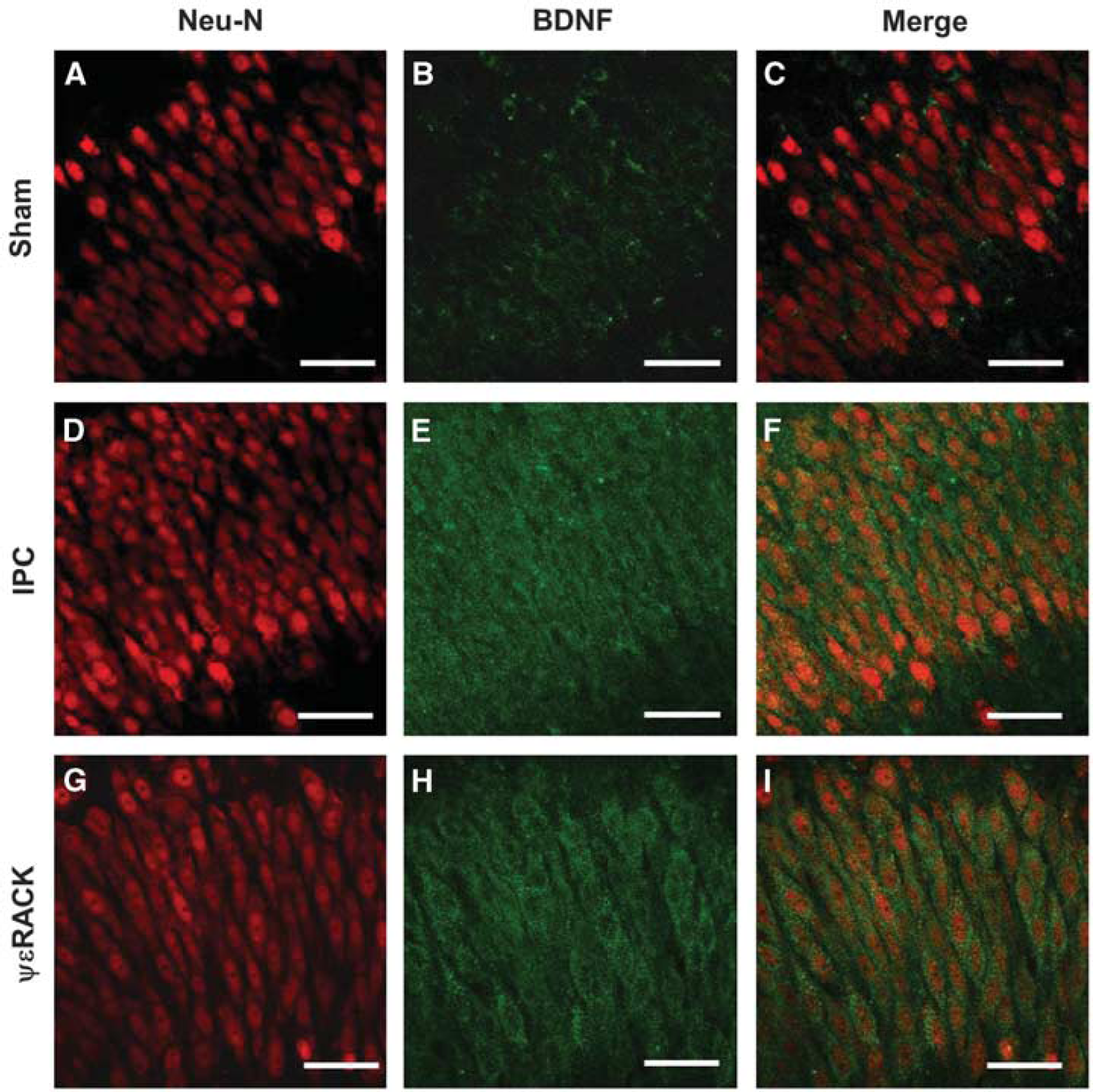
Preconditioning increases brain-derived neurotrophic factor (BDNF) protein expression in the cornu ammonis 1 (CA1) region of hippocampus. Typical micrographs from the CA1 region of organotypic hippocampal slices with (
Brain-Derived Neurotrophic Factor Signaling is Required for Preconditioning-Induced Neuroprotection
Our next hypothesis was that BDNF signaling is required for IPC- or ΨεRACK-induced neuroprotection. Since BDNF exerts its main effects via the TrkB receptor, the Trk receptor antagonist, K252a (200 nmol/l), was administered after IPC and ΨεRACK treatment. Hippocampal organotypic cultures exposed to lethal OGD alone resulted in 52%±3% cell death in the CA1 region of the hippocampus 24 hours after OGD as compared with NMDA-induced cell death (maximal neuronal cell death). The induction of IPC 48 hours before OGD resulted in significant neuroprotection in the CA1 region of the hippocampus, reducing cellular death to 18%±1%. Inhibition of BDNF signaling (via K252a) abolished IPC-induced neuroprotection against OGD (50%±3% cell death; Figure 3A; P<0.05). Protein kinase C epsilon activation in hippocampal organotypic cultures with ΨεRACK 48 hours before OGD resulted in significant neuroprotection, as cell death was limited to 25%±4%. This protection was abolished when K252a was administered (58%±2%; Figure 3B; P<0.05). Inhibition of Trk receptors after IPC also abolished neuroprotection in cortical neuronal-glial cocultures. Exposing cocultures to lethal OGD resulted in 76%±7% cell death in sham-treated cells, as compared with 38%±3% cell death in cells that were exposed to IPC 48 hours before lethal OGD. Addition of K252a after IPC resulted in an increase of cell death (71%±11%; Figure 3C; P<0.05).
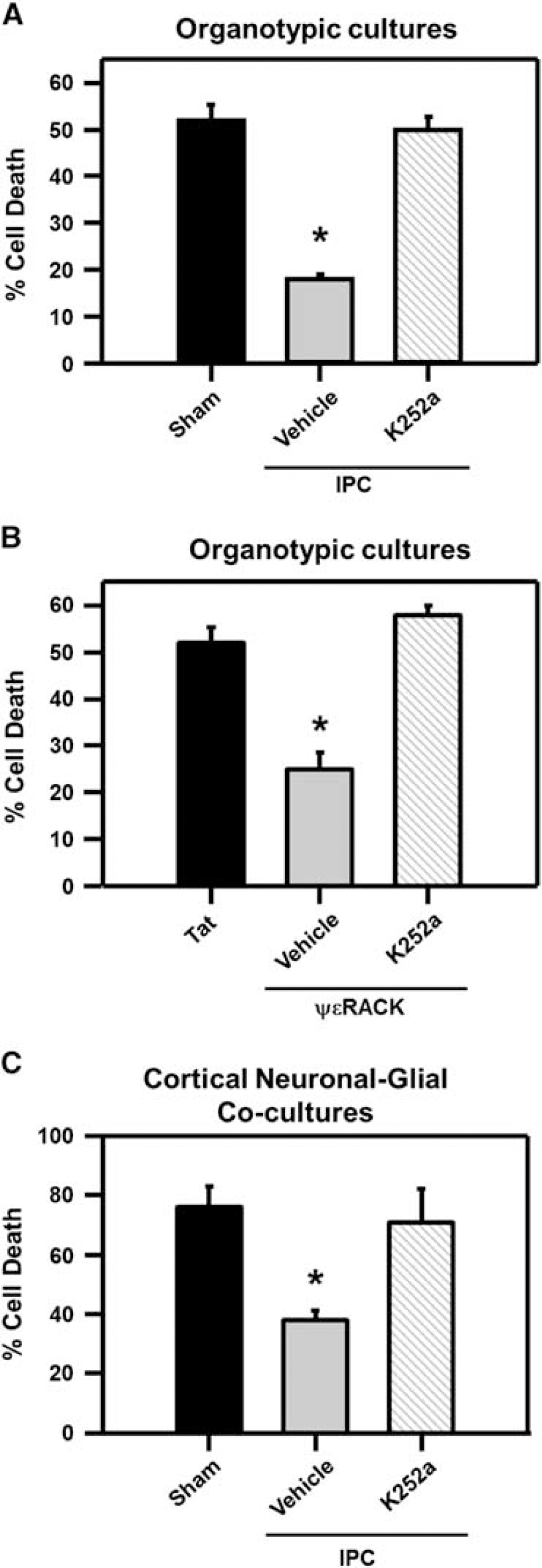
Brain-derived neurotrophic factor (BDNF) receptor activation is required for ischemic preconditioning (IPC)- and protein kinase C epsilon (PKCε) activation-induced ischemic tolerance. (
Cornu Ammonis 1 Neuronal Firing Frequency and Threshold Potential is Modulated by PKCε Activation
Since, we previously found that IPC-induced neuroprotection required PKCε activation 9 and that both IPC- and PKCε activation-induced electrophysiologic modifications for neuroprotection (i.e., increased GABAergic transmission 3 ), and two other studies showed evidence that PKCε can modulate Nav,12, 13 we hypothesized that activation of PKCε alters neuronal integrative properties. Therefore, acute hippocampal slices were isolated 48 hours after intraperitoneal injection of Tat or ΨεRACK peptide (0.2 mg/kg, PKCε activation). Cornu ammonis 1 neuronal excitability was investigated using whole-cell patch-clamp using a series of current injections from −250 to 450 pA in 50 pA increments (Figure 4A). Action potential firing frequency (1 Hz) was plotted against current injections. There was a significant reduction in the number of action potentials at or above current injections of 250 pA in the ΨεRACK-treated neurons, with a 35% reduction at 250 pA, 25% at 300 pA, 19% at 350 pA, 16% at 400 pA, and 15% at 450 pA (Figure 4B; P<0.05). There were no significant differences between the resting membrane potential on cellular access (−66.50±0.64 mV in the Tat-treated groups versus −67.29±0.69 mV after ΨεRACK treatment) or cellular membrane properties.
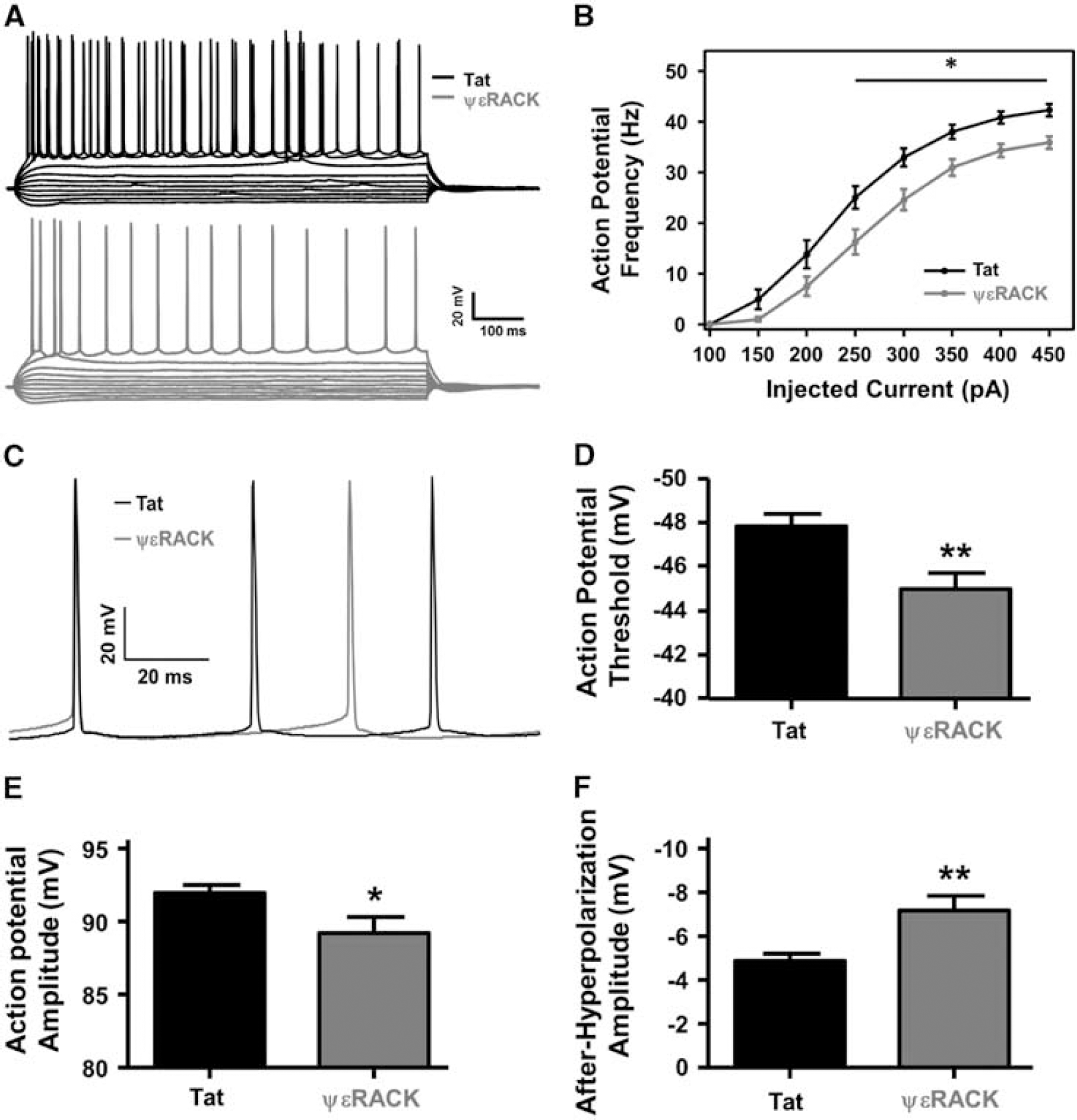
Protein kinase C epsilon (PKCε) activation decreases cornu ammonis 1 (CA1) neuronal firing frequency and excitability. (
Because of the reduction in action potential firing frequency, action potential characteristics from the first three action potentials were averaged for each CA1 neuron (Figure 4C). There was a significant increase in threshold potential from −47.83±0.58 mV in the Tat-treated groups as compared with −45.00±0.69 mV in the ΨεRACK-treated groups (Figure 4D; P<0.01). Action potential amplitude significantly decreased from 92.00±0.52 mV in the Tat-treated groups compared with 89.24±1.08 mV in the ΨεRACK-treated animals (Figure 4E; P<0.05). In addition, the after-hyperpolarization amplitude significantly increased from −4.85±0.34 mV in the Tat-treated versus −7.17±0.65 mV in ΨεRACK-treated neurons (Figure 4F; P<0.01).
Brain-Derived Neurotrophic Factor Signaling is Required for Neuronal Threshold Modulation after Ischemic Preconditioning or PKCε Activation
Based on the aforementioned findings and since BDNF modulates synaptic plasticity,21, 22 we next tested the hypothesis that the electrophysiologic modifications observed in Figure 4 were mediated by IPC- or ΨεRACK-induced increases of BDNF. To inhibit BDNF signaling, K252a was applied after sham or IPC treatment for 48 hours before recording cortical neuronal action potentials from cortical neuronal-glial cocultures (Figure 5A). Cortical neurons exposed to IPC (vehicle-treated cells) resulted in a significant increase in the threshold potential (−29.48±0.63 mV) as compared with sham+dimethyl sulfoxide-treated controls (−32.66±0.55 mV). This change in threshold potential was lost in IPC+K252a-treated neurons (−34.00±1.07 mV; Figure 5B; P<0.05). In addition, neuronal-glial cocultures were exposed to Tat or ΨεRACK treatment with/without K252a 48 hours before action potential recordings (Figure 5C). Cortical neuronal threshold potential was significantly increased after ΨεRACK+dimethyl sulfoxide treatment (−25.37±1.09 mV) as compared with Tat+dimethyl sulfoxide-treated neurons (−29.31±0.90 mV). Inhibition of Trk receptors (via K252a) abolished ΨεRACK's threshold modulation, as the neuronal threshold potential returned to −30.27±1.00 mV (Figure 5D; P<0.05).
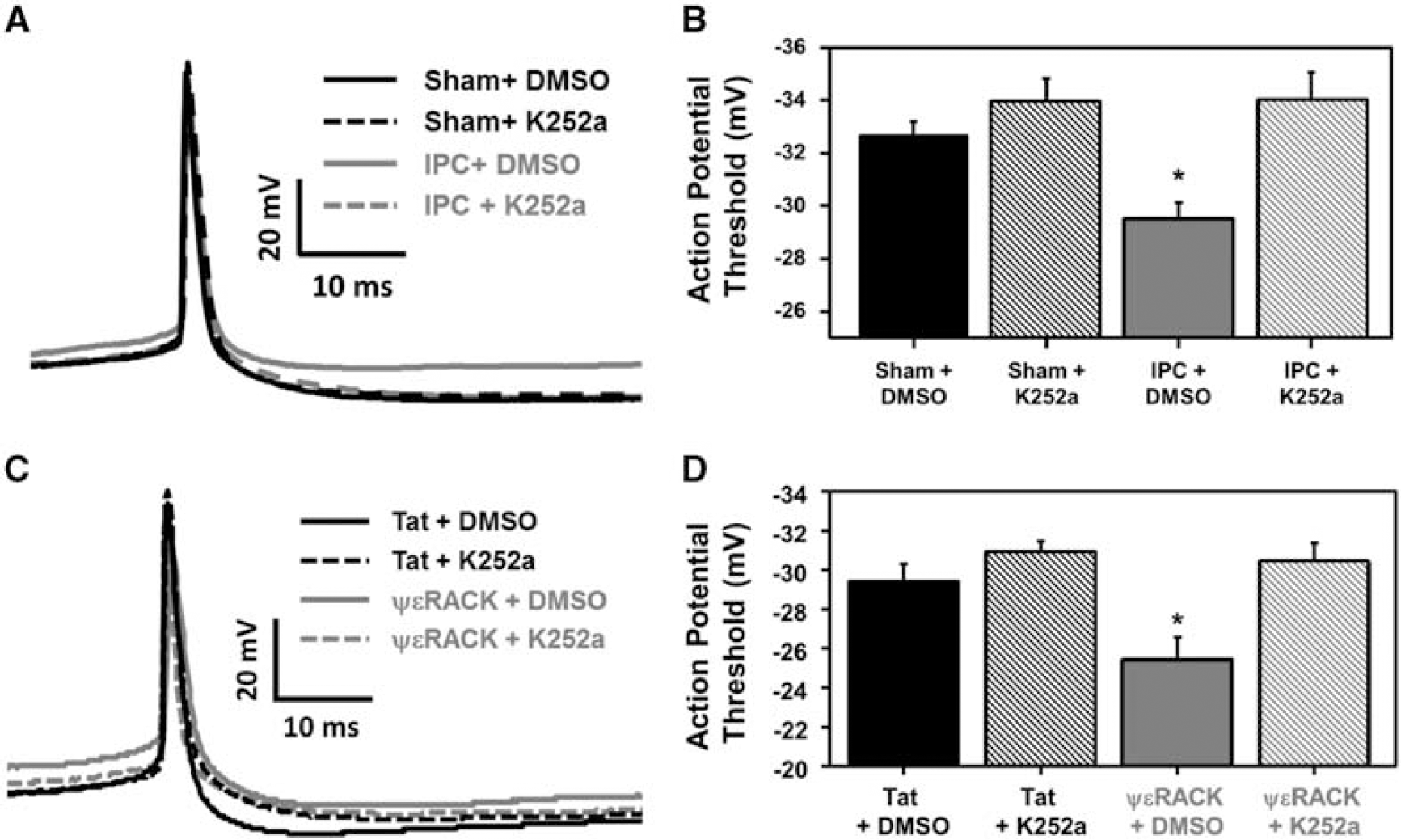
Preconditioning modulation of neuronal threshold potential is mediated by brain-derived neurotrophic factor (BDNF). (
Oxygen and Glucose Deprivation-Induced Anoxic Depolarization was Delayed 48 hours after PKCε Activation
Based on our findings that IPC or PKCε activation decreased neuronal excitability and induced neuroprotection, we next ask whether this treatment affected anoxic depolarization ex vivo. Acute hippocampal slices harvested 48 hours after in vivo injection of Tat or ΨεRACK (0.2 mg/kg) were subjected to continuous OGD after whole-cell patch configuration. The neuronal membrane potential was measured every 30 seconds during OGD, where the average membrane potential was depolarized at 5 minutes and 30 seconds in Tat-treated neurons (Figure 6A; P<0.05). This depolarization continued until each neuron reached ∼0 mV. In ΨεRACK-treated neurons, the average membrane potential was depolarized at 10 minutes and 30 seconds (Figure 6A; P<0.05), which also continued to depolarize under continuous OGD. The amount of time the neurons maintained (or were hyperpolarized) their resting membrane potential during OGD for Tat-treated neurons was 4.75±0.76 minutes compared with 8.50±0.83 minutes for ΨεRACK-treated neurons (Figure 6B; P<0.001). Next, the membrane potential during OGD for each neuron was normalized to the resting membrane potential at the start of OGD (Figure 6C). The hyperpolarization phase after ΨεRACK treatment was enhanced as compared with Tat-treated neurons. In addition, the maximum hyperpolarization measured during OGD was significantly larger in the ΨεRACK group (−2.55±0.18 mV) as compared with the Tat-treated neurons (−1.02±0.42 mV; Figure 6D; P<0.01).
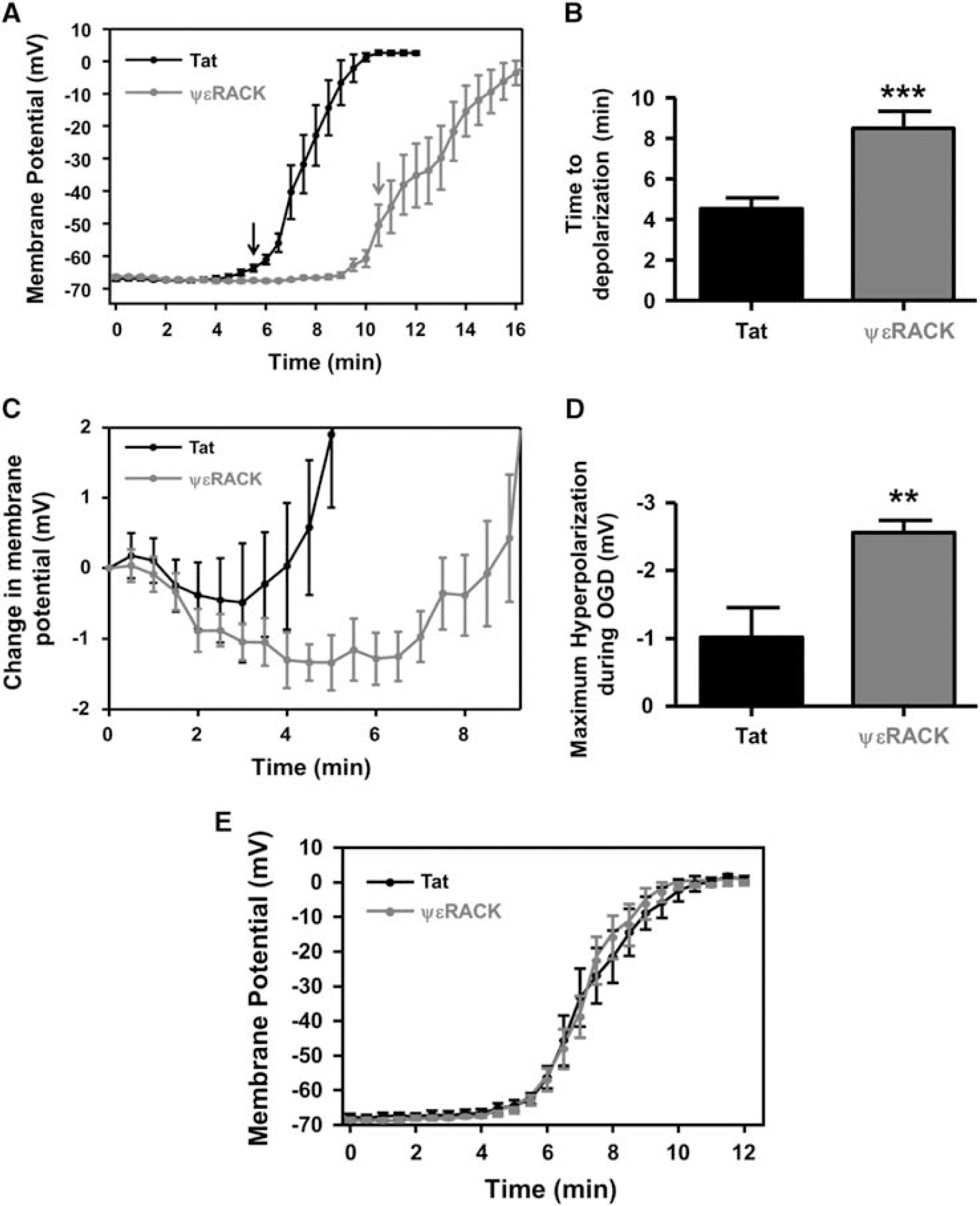
Protein kinase C epsilon (PKCε) activation delays the anoxic depolarization of hippocampal cornu ammonis 1 (CA1) neurons. (
Acute activation of PKCε can induce neuroprotection when administered immediately before middle cerebral artery occlusion; 24 therefore, to determine if acute PKCε activation induces a delay in anoxic depolarization, whole-cell recordings were obtained from naive hippocampal slices that were pretreated with Tat or ΨεRACK for 1 hour. After whole-cell patch configuration, continuous OGD was performed and the membrane potential was measured every 30 seconds (Figure 6E). There was no significant difference in the time to anoxic depolarization between ΨεRACK- or Tat-treated slices, suggesting that this is not an acute effect of the ΨεRACK peptide.
Discussion
The main goal for this study was to further define the inherent preconditioning response after IPC or pharmacological preconditioning (PKCε activation via ΨεRACK) leading to ischemic tolerance. One novel finding from the current study was that IPC or direct activation of the novel PKC isozyme, PKCε, upregulated BDNF protein levels 24 to 48 hours after a single treatment. To our knowledge, this is the first study in which a transient activation of PKCε (IPC or ΨεRACK) promoted an upregulation of BDNF 2 days later, which led to ischemic neuroprotection. This neuroprotective pathway induced ischemic tolerance via BDNF-dependent activation of Trk receptors. Furthermore, our results point to a novel preconditioning mechanism in which IPC and PKCε activation promote a decrease in neuronal excitability and delay the time to anoxic depolarization, which may lead to neuroprotection against OGD. Overall, these data suggest that BDNF is a common signaling pathway activated after IPC or PKCε activation, which is responsible for neuroprotection in the hippocampus and cortex.
Brain-derived neurotrophic factor signaling occurs through activation of the TrkB receptor, via receptor dimerization and autophosphorylation of tyrosine residues within the TrkB cytoplasmic domain. 25 These phosphorylated tyrosine residues serve as docking sites for adapter protein binding allowing for the initiation of numerous pro-survival signaling pathways including mitogen-activated protein kinase/extracellular signal-regulated protein kinase, phospholipase Cγ, phosphoinositide 3-kinase and small G proteins. 25 Interestingly, IPC-induced neuroprotection is dependent on the activation of extracellular signal-regulated kinase 1/24, 6 and phospholipase C. 6 Direct application of BDNF is neuroprotective against various models of cerebral ischemia in vivo, such as middle cerebral artery occlusion and global cerebral ischemia.19, 20 However, in those studies continuous intraventricular administration of BDNF was required for neuroprotection, whereas in the present study we only administered ΨεRACK one time. Two days after ΨεRACK treatment, BDNF protein expression increased and ischemic neuroprotection was achieved using an in vitro model that emulates cerebral ischemia (OGD; Figures 3 and 4).
Protein kinase C epsilon activation has previously been implicated in the upregulation of BDNF, 26 however, these studies administered bryostatin-1, (PKCε and PKCα activator) two to three times per week (up to 5 weeks) to promote synaptogenesis and improve learning during aging or after global cerebral ischemia. Bryostatin-1 is a partial agonist of the PKC family of isozymes that is relatively selective for PKCε and PKCα activation. The binding of bryostatin-1 to PKC results in PKC activation, autophosphorylation, and translocation to the cell membrane, followed by a bryostatin-1-bound PKC downregulation by ubiquitination and degradation by proteasomes. 27 In the current study, 2 days after a single ΨεRACK 1-hour treatment, BDNF protein expression was increased and neuroprotection was achieved (Figures 1). In addition, inhibition of Trk signaling after IPC or ΨεRACK treatment abolished neuroprotection during lethal OGD (Figure 3). In another model of cellular stress (sublethal exposure to NMDA), enhanced BDNF expression was observed in cerebellar granule cells. 14 Therefore, BDNF appears to be a common signaling pathway necessary for neuroprotection as IPC, PKCε activation, and NMDA receptor activity all increase the expression of BDNF. In addition, IPC- and NMDA-mediated preconditioning are dependent on PKCε activity, 9 further establishing that PKCε is a ubiquitous signaling pathway that is activated during cellular stress necessary for BDNF-mediated neuroprotection.
Protein kinase C epsilon modulation of Nav has been well-established for various Nav isoforms, such as Nav1.6, 12 Nav1.7, 13 and Nav1.8; 28 however, PKCε-mediated-Nav regulation may differ according to location. For example, acute activation of PKCε enhances the current of peripheral channels (Nav1.8), 28 but a reduction in Na+ current was found in hippocampal Nav1.6 channels. 12 Interestingly, the authors state that the decreased Nav1.6 current could affect the integrations of depolarizing inputs in dendrites, threshold, and action potential frequency in the axonal initial segment and cell bodies. 12 In fact, decreased expression of Nav channels in the axon initiation segment induces an increase in threshold potential. 29 In addition, activation of PKCε decreases the mRNA half-life and cell surface Nav expression in adrenal chromaffin cells, 13 where the downregulation of Nav expression has been suggested as a potential neuroprotective pathway. 30 Therefore, based on these studies, our data suggest that the decrease in CA1 neuronal firing frequency and modulation of the action potentials by PKCε (Figures 4 and 5) or IPC (Figure 5) may be because of, in part, via a decrease Nav expression. In addition, this decreased neuronal excitability may reduce neuronal activity during periods of stress leading to neuroprotection.
Brain-derived neurotrophic factor has also been implicated in the regulation of Na+ channel expression 21 and a reduction in Na+ currents in Nav1.2 channels. 31 The attenuation of BDNF signaling through Trk receptor antagonism (via K252a) resulted in the loss of IPC- and ΨεRACK-induced threshold modifications (Figure 5). It should be noted that K252a can also inhibit PKCε; however, the IC50 for K252a-induced-PKCε-inhibition is 4.5 μmol/l, far greater than the 200 nmol/l of K252a administered in our study. 32 Thus, these data suggest that BDNF signaling is essential for preconditioning-induced threshold modification and neuronal firing alterations.
In addition to the potential modulation of Nav channels by PKCε and BDNF, neuronal firing has been suggested to be modulated through the spontaneous activity of GABAA receptors. 33 In this study, background GABAA receptor signaling was shown to alter the excitability and spike interval timing of cortical pyramidal neurons. 33 Previously, we showed that IPC- or PKCε activation-induced neuroprotection depended on a functional modification of GABA synapses 48 hours after preconditioning, where IPC induction increased the frequency and amplitude and PKCε activation increased the amplitude of GABAA receptor-mediated miniature postsynaptic currents. 3 Therefore, in addition to a decreased Nav expression, an increase in GABA signaling could further modulate neuronal firing for neuroprotection.
Anoxic depolarization is a large depolarization that occurs during ischemia, 34 resulting in cellular death in the long-term absence of oxygen and glucose. Since anoxic depolarization does not occur from the accumulation of glutamate or its receptor activation, 35 investigators have focused on compounds outside of traditional glutamate receptor modulators such as sigma 36 or α7 nicotinic acetylcholine 37 receptors, or nonspecific treatment modalities such as the use of anesthetics. 38 However, these studies have been limited to treatments administered immediately before ischemia. In contrast, in the present study, a single ΨεRACK treatment (pharmacological preconditioning) significantly increased the time to anoxic depolarization 48 hours later, suggesting that activation of PKCε has long-lasting synaptic modifications that delays anoxic depolarization. This treatment also significantly increased the time and maximum amplitude of the hyperpolarization phase before the ischemia-induced anoxic depolarization (Figure 6). Although the exact mechanism of delayed anoxic depolarization has remained elusive, preconditioning has been suggested to alter numerous pro-survival pathways for neuroprotection that could account/contribute to the ‘delay’ in the ischemia-induced anoxic depolarization.9, 39 A possible explanation for the ischemia-induced anoxic depolarization-mediated neuroprotection could be a shift from excitatory to inhibitory transmission, as IPC induction increases basal synaptic GABA transmission 3 and increases GABA release after ischemia. 40 In addition, this delay could arise from increased mitochondrial respiration, as increased synaptic PKCε levels 48 hours after IPC has been associated with increased mitochondrial respiration upon PKCε activation. 5 In addition, increased activity of the Na+/K+ ATPase occurs after IPC in organotypic hippocampal cultures 41 or in vivo; 42 therefore, an increase in Na+/K+ ATPase activity during ischemia could contribute to maintaining the resting membrane potential. All of these potential mechanisms (increased GABA signaling, ATP synthesis, or Na+/K+ ATPase activity) could produce an increase in the hyperpolarization phase, reduce neuronal signaling, and prolong the time to anoxic depolarization for neuroprotection.
A primary goal of preconditioning research is to establish pharmacological-based targets or therapies that can be used for neuroprotection against cerebral ischemia in a prophylactic manner. One limitation to this study is the usage of culture and young animal models, where the primary targets for clinical therapies against cerebral ischemia are adults or aged humans. Future studies to confirm the activation of these pathways after IPC or PKCε activation in aged animal models are necessary. In addition, further understanding of the molecular pathways and neuronal modifications by which preconditioning induces neuroprotection against cerebral ischemia is essential. The therapeutic potential of BDNF as treatment against ischemia in the clinic has been greatly limited because of the short half-life in plasma (∼1 hour) and poor penetration of the blood—brain barrier. 43 However, the results from the present study point to an alternative approach to increase BDNF brain levels pharmacologically (i.e., ΨεRACK), which could lead to ischemic neuroprotection.
Footnotes
The authors declare no conflict of interest.