
Abstract
Brain cells produce cytokines and chemokines during the inflammatory process after stroke both in animal models and in patients. Monocyte chemoattractant protein 1 (MCP-1), one of the proinflammatory chemokines, can attract monocytes to the tissue where MCP-1 is overexpressed. However, the role of MCP-1 elevation in stroke has not been explored in detail. The authors hypothesized that elevated MCP-1 levels would lead to increased influx of monocytes and increased brain infarction size in stroke induced by middle cerebral artery occlusion with partial reperfusion. There were no differences in blood pressure, blood flow, or vascular architecture between wild-type mice and transgenic MBP-JE mice. Twenty-four to 48 hours after middle cerebral artery occlusion, brain infarction volumes after ischemia were significantly larger in MBP-JE mice than in wild-type controls and were accompanied by increased local transmigration and perivascular accumulation of macrophages and neutrophils. These results indicate that MCP-1 can contribute to inflammatory injury in stroke.
In the management of acute stroke, the major proven medical treatment is tissue plasminogen activator, which has a very short window of treatment (0–3 hours) (The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group, 1995). Proinflammatory mediators, such as cytokines and chemokines, are produced in human stroke and animal models of stroke (Asensio and Campbell, 1999). A detailed analysis of the role of these molecules may lead to an extended time window for treatment because the inflammatory processes progress over many hours after the onset of brain ischemia and contribute to evolution of tissue injury (Barone and Feuerstein, 1999). Chemokines are small, inducible, secreted, proinflammatory cytokines that act primarily as chemoattractants and activators of granulocytes, macrophages, and other inflammatory cells. Chemokines belong to a rapidly expanding family of cytokines. Their primary function is to control the positioning of cells in tissues and to recruit leukocytes to the site of inflammation. Monocyte chemoattractant protein 1 (MCP-1) is a member of the cysteine-cysteine chemokine gene family characterized by their ability to attract monocytes, memory T lymphocytes, and natural killer cells in vitro (Oppenheim et al., 1991; Stanimirovic and Satoh, 2000; Yoshimura et al., 1989a) and it induces monocyte infiltration and/or inflammation following injury in vivo. MCP-1 has also been implicated in regulating the balance of T helper type 1 (Th1) and 2 (Th2) cytokines during an inflammatory response (Siveke and Hamann, 1998). It also can regulate T cell differentiation and the balance between Th1 and Th2 cells.
Several models of transgenic expression in the mouse have validated MCP-1 as a predominantly monocytic chemoattractant in vivo (Fuentes et al., 1995; Gunn et al., 1997), but the effects of MCP-1 in ischemia have not been characterized in detail. The availability of a transgenic mouse that selectively overexpresses MCP-1 under control of the myelin basic protein promoter (i.e., in cells intrinsic to the CNS) has provided us with a unique opportunity to address the role of this chemokine in a model of experimental stroke. In this transgenic mouse, the overexpression of MCP-1 is found only in thymus and brain, but not in other organs such as heart, lung, liver, spleen, kidney, skin, and skeletal muscle (Fuentes et al., 1995). We hypothesized that elevated MCP-1 levels would lead to increased influx of monocytes and increased infarction size in the brain in stroke induced by middle cerebral artery occlusion (MCAO) with partial reperfusion. We found that increased levels of MCP-1 cause perivascular accumulation of both neutrophils and monocytes/macrophages around intraparenchymal brain vessels, or “perivascular cuffing.” This sets the stage for increased brain injury in a standard model of focal brain ischemia.
MATERIALS AND METHODS
Experimental groups
Male and female (21–30 g) MCP-1 (MBP-JE) transgenic mice, under control of the myelin basic protein promoter on a mixed C57BL/6 × DBA/2 genetic background (Fuentes et al., 1995) and wild-type C57BL/6 × DBA/2 F1 control mice were used in all experiments. The MCP-1 transgenics were developed and initially bred at Bristol-Myers Squibb (Princeton, NJ, U.S.A.) (Fuentes et al., 1995) and control mice were purchased from Jackson Laboratories (Bar Harbor, ME, U.S.A.). Additional MBP-JE transgenic mice from the line developed at Bristol-Myers Squibb were from University of Kansas Medical Center in Kansas City (KS, U.S.A.). All mice had free access to food and water prior to surgery. This protocol was reviewed and approved by Animal Care and Use Committees of the National Institute of Neurological Disorders and Stroke in the National Institutes of Health (NIH) and by the Uniformed Services University of the Health Sciences. Wild-type and MCP-1 transgenic mice were subjected to MCAO with partial reperfusion. Mice from these two groups were killed 24 or 48 hours after surgery and infarct volume measurements and immunohistochemical staining were performed on brain sections.
Permanent MCAO plus 2 hours of common carotid artery occlusion in mice (partial reperfusion model)
Mice were anesthetized with 5% isoflurane for induction and 1% to 1.5% isoflurane for maintenance in 30% oxygen and 70% nitrous oxide via facemask. Rectal temperature was measured and maintained at 37°C with a heating blanket. The surgical procedure used in the current study has been described in detail elsewhere Nawashiro et al. (1997). Briefly, the left temporoparietal region was shaved and a 5-mm incision was made between the orbit and ear. Under the operating microscope, an incision was made by dividing the temporal muscle, and the left lateral aspect of the skull was exposed by reflecting the temporal muscle and surrounding soft tissue. A small burr hole (2 mm) was made with a high-speed microdrill through the outer surface of the semitranslucent skull just over the middle cerebral artery (MCA) between the inferior cerebral vein and the lateral olfactory tract. The MCA is visible through the mouse skull. The dura was opened with 30-gauge needle to expose the MCA and MCA was occluded by bipolar electrocoagulator. The coagulated MCA segment was then transected with microscissors to ensure that the occlusion was permanent. Focal cerebral ischemia with partial reperfusion was induced as described previously (Morikawa et al., 1996), since isolated MCAO (without temporary occlusion of ipsilateral common carotid artery) did not reliably produce infarction in these mice. Briefly, the left common carotid artery (CCA) and its bifurcation were exposed. The left CCA was occluded with a metal clip (Microaneurysm clip, 10–15 g pressure, Roboz, Rockville, MD, U.S.A.) after electrocauterization and transection of MCA. The clip on the left CCA was removed to permit reperfusion 2 hours after occlusion of the left MCAO. The same procedure was used during sham operation except that no electrocauterization, transection of MCA, or clip on the left CCA was performed.
Blood pressure was measured in some of the animals (n = 3–5) during an operation in which PE 10 polyethylene tubing was inserted into the femoral artery and connected to a blood pressure analyzer (Micro-Med, Louisville, KY, U.S.A.). The vascular architecture was visualized by white latex mixed with carbon black as described previously (Maeda et al., 1998).
Measurement of infarct volume
At 24 and 48 hours after MCAO, brains were frozen on dry ice and cut into 20-μm sections. The sections were postfixed over paraformaldehyde vapors and stained with cresyl violet (Barks et al., 1995). A computerized image analysis system (NIH Image 1.62) was used to measured cross-sectional infarct areas in each coronal section (10–13 sections per brain) (Chen et al., 2001). Regional volumes were calculated by summing cross-sectional areas and multiplying these areas by the distance between sections, followed by correction for brain swelling as previously described (Leach et al., 1993; Lin et al., 1993).
Histologic, immunohistochemical, and serologic evaluation
Twenty-four or 48 hours after MCAO with 2-hour CCA occlusion, the brains were frozen on dry ice and 20-μm sections were collected. The sections were fixed for 10 minutes in acetone at 4°C and immunohistochemically stained following standard methods (Fuentes et al., 1995). Briefly, rat antimouse neutrophil (10 μg/mL, Serotec, Oxford, UK) and antimouse F4/80 (20 μg/mL, Serotec), a monocyte and microglial stain, were applied. Biotinylated donkey anti–rat immunoglobulin G was used as secondary antibody. Endogenous peroxides were blocked by 0.33% H2O2. Diaminobenzidine served as the chromogen. Two sections were taken from each brain at bregma +0.20 mm and −2.8 mm (Paxinos and Watson, 1986). For further analysis of perivascular cuffing, two adjacent sections were stained with anti-F4/80 and antineutrophil antibodies, respectively. The samples were analyzed with a light microscope (40× objective) and the number of immunohistochemically positive cells within the ischemic hemisphere of each of these sections was counted in a blinded manner. In negative control experiments (e.g., without primary antibody and substitution of rat immunoglobulin G), no positive staining was detected. The neutrophil-stained sections were then counterstained with hematoxylin.
At the time of death, sera were also collected from individual animals and circulating MCP-1 levels were measured by ELISA (Opt-EIA Kit, Pharmingen, San Diego, CA, U.S.A.) precisely as recommended by the manufacturer.
LPS-induced lethality
Mice were injected intraperitoneally with 12-mg/kg proteinfree Escherichia coli K235 LPS prepared by the method of McIntire et al. (1967). Lethality was scored for 72 hours after injection.
Statistics
Means and standard deviations were calculated and differences were tested by means of two-factor analysis of variance for infarction volume and analysis of variance followed by a Fisher PLSD at a significance level of P<0.05 for number of F4/80-positive cells. Unpaired t-test was used for the level of MCP-1 in the serum of mice.
RESULTS
Overexpression of MCP-1 results in increased sensitivity to systemic and localized inflammatory insults
A previous study reported that transgenic mice that express MCP-1 under the control of the myelin basic protein promoter exhibit increased infiltration of monocytic cells into the brain, which could be exacerbated by systemic administration of a potent inflammatory stimulus, such as gram-negative lipopolysaccharide (LPS) (Fuentes et al., 1995). Since macrophages have been implicated as the principal producers of the inflammatory cytokine response to LPS (Vogel and Hogan, 1990), we hypothesized that the MCP-1 transgenic mice described in the original publication by Fuentes et al. (1995) would also exhibit increased sensitivity to LPS-induced lethality. To test this hypothesis, wild-type and MCP-1 transgenic mice were challenged with high doses of LPS and monitored for survival over a 3-day period. The MCP-1 transgenic mice were significantly more sensitive to the lethal effects of LPS injection: all 10 MCP-1 trangenics died within 24 hours after i.p. injection of 12-mg/kg LPS, while only 2 of 10 wild-type mice succumbed to this same dose of LPS over 3 days. Even at one tenth of this dose of LPS (1.2 mg/kg), two of five MCP-1 transgenic mice died. These data support the hypothesis that overexpression of MCP-1 results in a greatly exaggerated immune response to a subsequent inflammatory insult.
Since MCP-1 has been shown to attract primarily monocytic cell types into areas of inflammation (Gu et al., 1999) and levels of MCP-1 messenger RNA have been reported to be upregulated in response to stroke in the brain (Ivacko et al., 1997; Kim et al., 1995; Wang et al., 1995), we next sought to test the hypothesis that overexpression of MCP-1 in the CNS results in greater ischemic damage after MCAO and partial reperfusion. The data in Fig. 1 illustrate the infarct volumes measured 24 and 48 hours after surgery in wild-type vs. MCP-1 transgenic mice subjected to MCAO and reperfusion. In the MCP-1 transgenic mice, infarct volumes (mean ± SD) were 17.02 ± 13.06 mm3 at 24 hours and 17.32 ± 15.37 mm3 at 48 hours after MCAO plus 2 hours reperfusion, which was 67% and 102% larger than the corresponding values measured in wild-type mice (10.22 ± 6.25 mm3 at 24 hours and 8.55 ± 5.47 mm3 at 48 hours). Two-factor analysis of variance revealed that the infarct volume is significantly greater in the MCP-1 transgenics than in wild-type mice (P<0.05), but no significant difference was observed between the 24- and 48-hour time points.
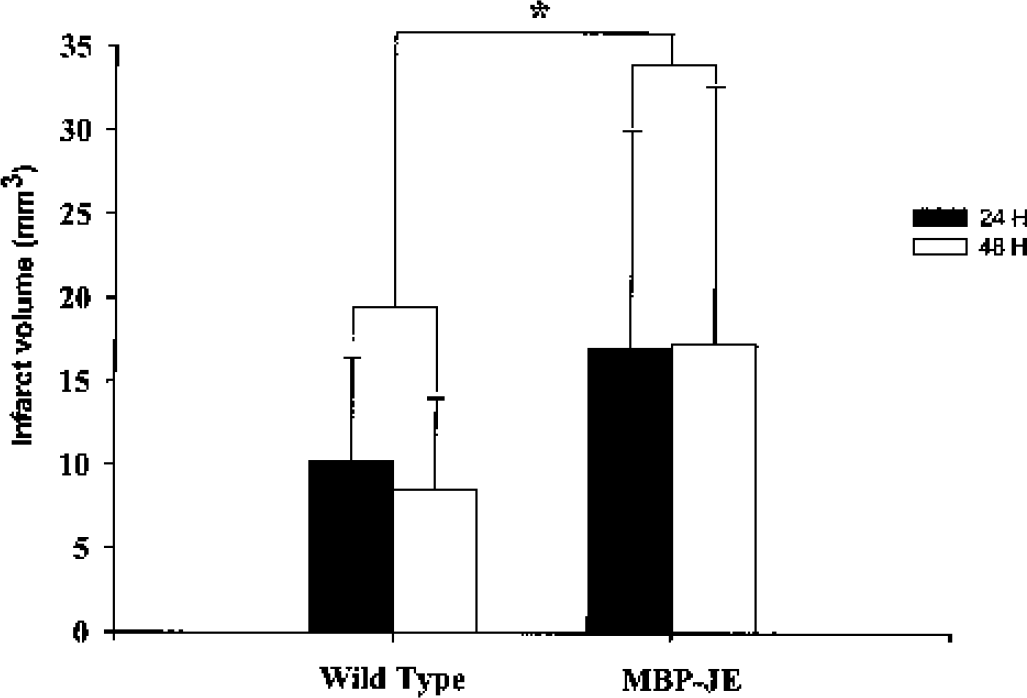
Infarct volumes (corrected for edema) of ischemic hemispheres in wild-type and MBP-JE transgenic mice 24 and 48 hours after middle cerebral artery occlusion with 2 hours of temporary common carotid artery occlusion. A significant difference was found between wild-type and MBP-JE transgenic groups (n = 6–11 mice per treatment per time point) (*P < 0.05, two-factor analysis of variance), with no significant effect of time. Data are presented as mean ± SD.
After the mice were killed, sera from each mouse were collected and MCP-1 levels were determined using enzyme-linked immunosorbent assay. MCP-1 transgenic mice exhibited circulating MCP-1 levels that were approximately 40-times greater than that exhibited by wild-type control mice (2,400.3 ± 1,496.6 vs. 61.6 ± 19.0 pg/ml, P<0.01, unpaired t-test), consistent with overexpression of this transgene in the MCP-1 transgenic mice.
Basal blood pressure, cerebral blood flow, and intracranial vascular anatomy
Mean arterial blood pressure (mean ± SD) under anesthesia was 96 ± 11 mm Hg in wild-type mice and 92 ± 2 mm Hg in MBP-JE mice. These values were not significantly different. Brain blood flow was 63.0 ± 5 mL/min per 100 g tissue in wild-type mice and 63.3 ± 12.5 mL/min per 100 g tissue in MBP-JE mice before MCAO, and was 23.3% and 22.8% of the basic level in wild-type and MBP-JE mice at 20 minutes after MCAO, respectively. These flow rates were not significantly different. Visual inspection of the anatomy of intracranial arteries (circle of Willis, MCA) did not reveal obvious differences among the strains. No difference in the distance from midline to line of anastomoses between the MCA and anterior cerebral artery was seen (data not shown) (Maeda et al., 1998).
Effect of MCP-1 overexpression in the CNS on inflammatory cell infiltration following MCAO and partial reperfusion
Previous studies have demonstrated that the MCP-1 transgenic strains used in this study selectively attract cells of the mononuclear phagocyte lineage into the brain, and that this influx of mononuclear cells was augmented after LPS administration (Fuentes et al., 1995). Immunohistochemical staining for cells of monocytic lineage using a monoclonal antibody directed against the F4/80 antigen revealed that positively stained cells were widely distributed in the ischemic hemisphere with an increased concentration in the periinfarct region. Fewer cells were found in the contralateral hemisphere or in sham-operated transgenic mice corresponding to the slight increase of monocyte/macrophage penetration into brain noted by Fuentes et al. (1995). At 24 hours after stroke, there was no significant difference seen in the average number of F4/80-positive cells in brain sections from wild-type compared with MCP-1 transgenic mice. However, by 48 hours, the overexpression of MCP-1 had a statistically significant impact on the accumulation of F4/80-positive cells (P<0.05). Neutrophils were widely expressed in the ischemic hemisphere, but the average number of neutrophils showed only a trend for an increase in MCP-1 transgenic mice at the two time points (Fig. 2B). However, the perivascular distribution of inflammatory cells was distinctly different between wild-type and transgenic mice. F4/80 and antimouse neutrophil immunoreactive cells clustered around intraparenchymal blood vessels in both hemispheres of sham-operated and MCAO transgenic mice (Figs 3A, 3C–3F, and 4C–4F). Wild-type mice did not display this perivascular distribution of invading inflammatory cells after sham operation or MCAO (Figs. 3B and 4B). Perivascular cuffing was much more prominent with stronger staining in the periinfarct region of MCP-1 transgenics subjected to MCAO plus partial reperfusion (Figs. 3C–3F and 4C–4F). The perivascular cuffing stained by anti-F4/8 or antineutrophil antibodies in two adjacent sections showed colocalization of monocytes/macrophages and neutrophils around scattered vessels in the brain (Fig. 5).
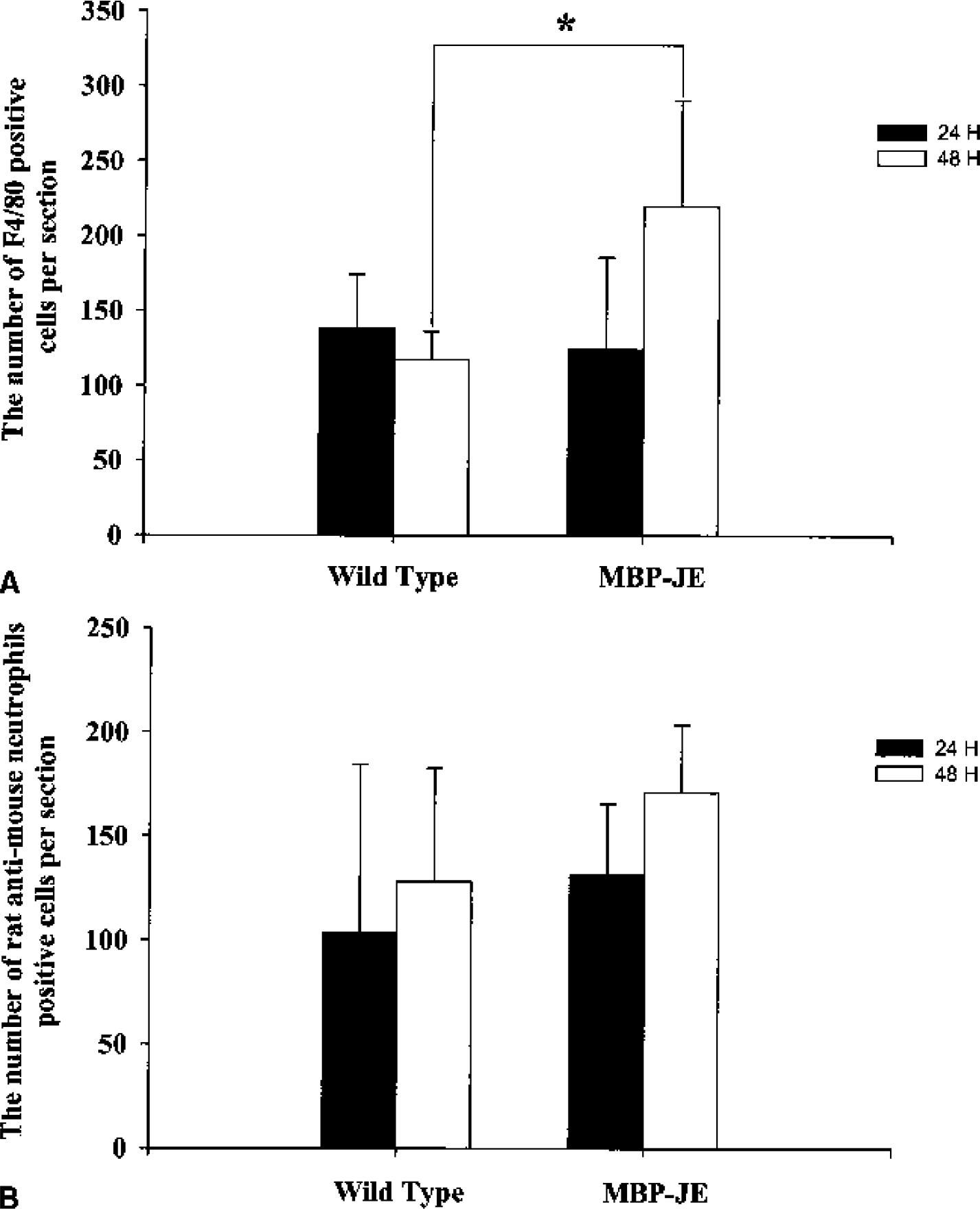
(A) The number of F4/80-positive monocyte/macrophages per section in the ischemic hemisphere in wild-type and MBP-JE transgenic groups (n = 6 mice per treatment time point). The overexpression of MCP-1 had a statistically significant impact on the accumulation of F4/80-positive cells (*P < 0.05) 48 hours, but not 24 hours, after stroke.
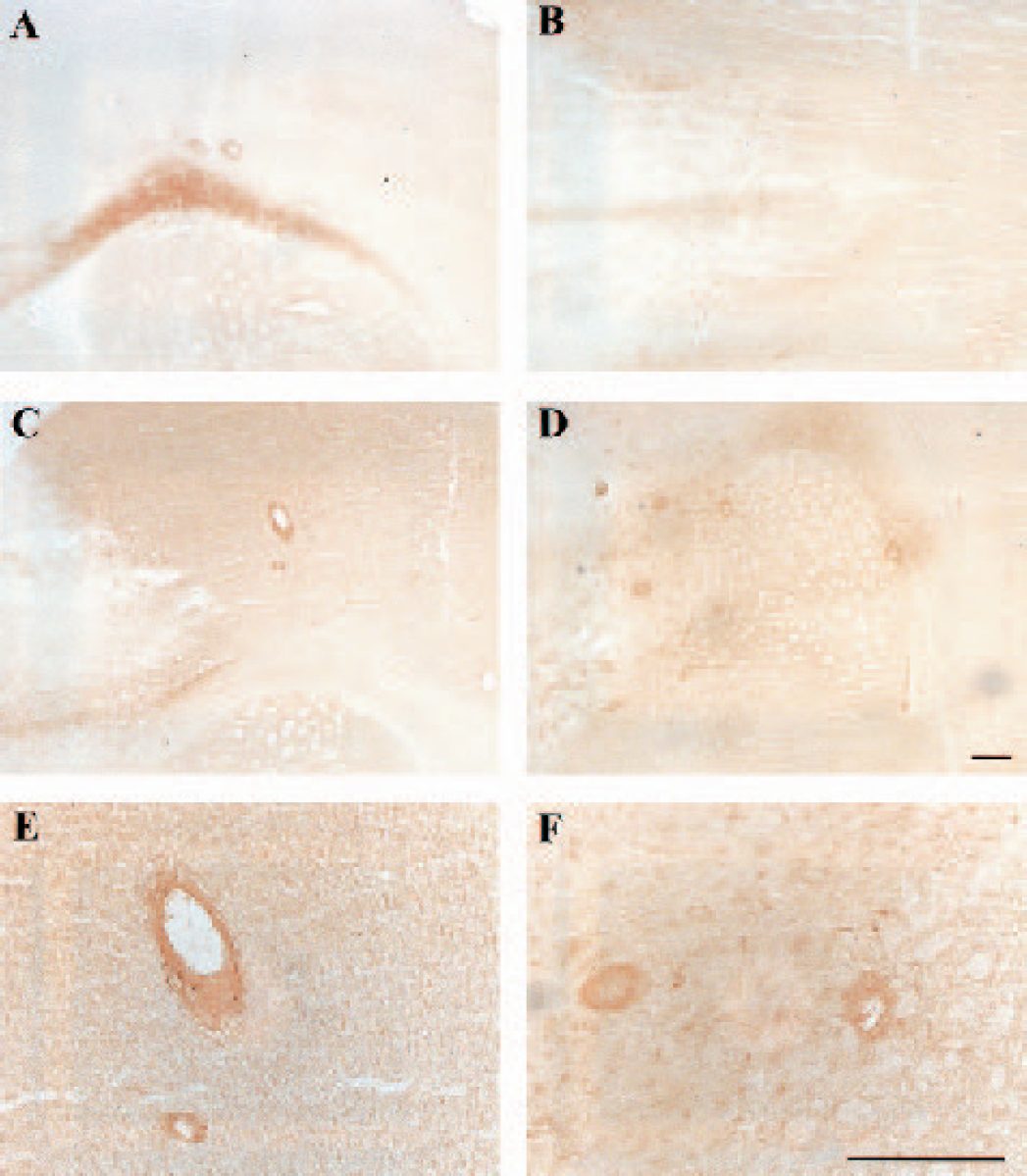
Perivascular mononuclear cell cuffing in the periinfarct region of mouse brain after middle cerebral artery occlusion plus 2 hours partial reperfusion and in sham-operated MBP-JE mice. The slides were stained with F4/80. (
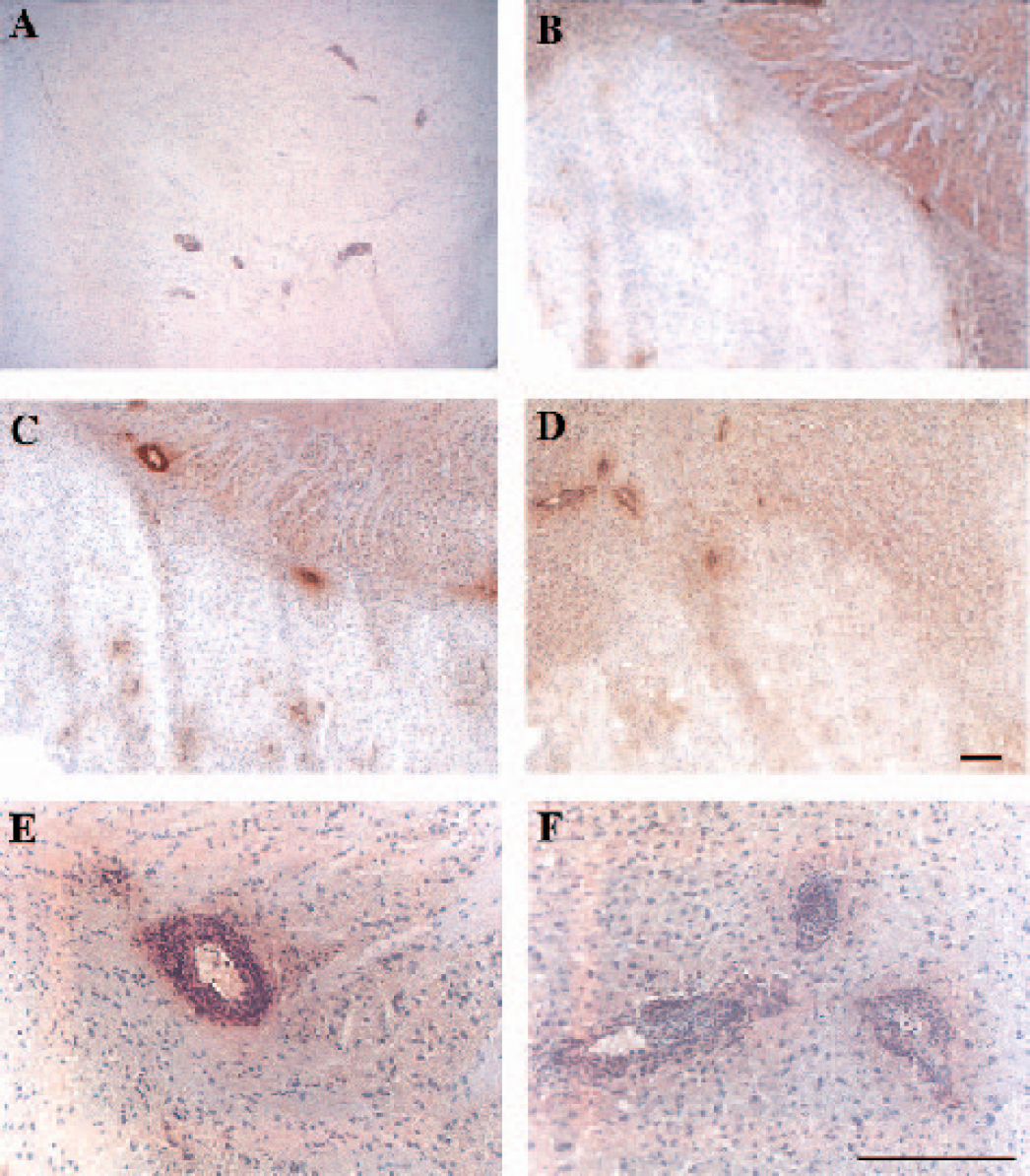
Perivascular neutrophil cuffing in the periinfarct region of mouse brain after middle cerebral artery occlusion plus 2 hours partial reperfusion and sham-operated MBP-JE mice. The slides were stained with antimouse neutrophil antibody and counterstained with hematoxylin. (
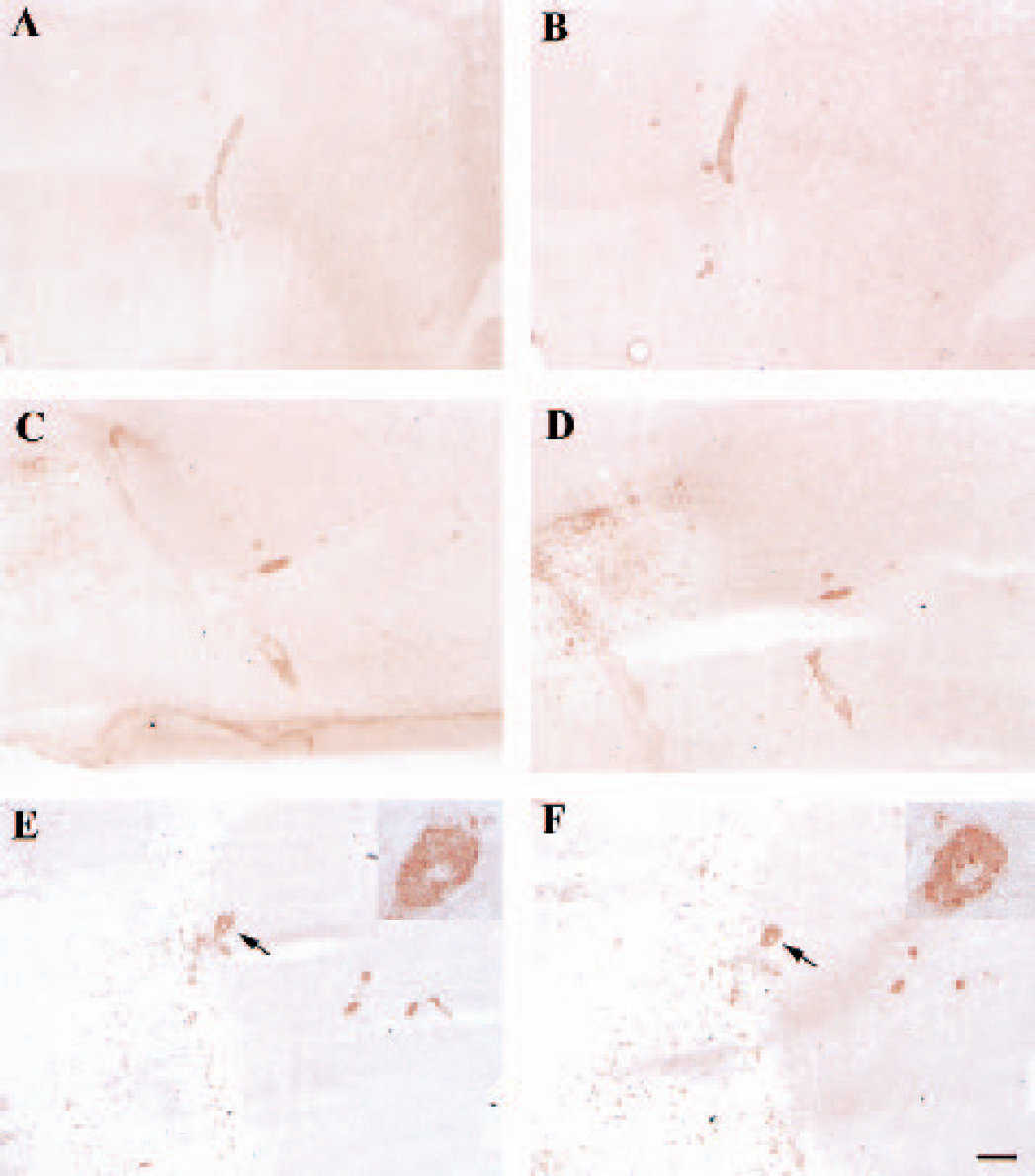
Perivascular cuffing in the periinfarct region of mouse brain after middle cerebral artery occlusion plus 2 hours partial reperfusion and in sham-operated MBP-JE mice. The two adjacent sections were stained with F4/80 (
DISCUSSION
Monocyte chemoattractant protein 1 was originally identified as an immediate early gene JE induced by platelet-derived growth factor in mouse fibroblast cells (Cochran et al., 1983). It has cytokinelike properties, and attracts monocytes to the wound area with little effect on granulocytes or lymphocytes (Cochran et al., 1983; Yoshimura et al., 1989b). MCP-1 regulates monocyte accumulation in several transgenic experimental models (Fuentes et al., 1995; Gunn et al., 1997). It can also stimulate monocytes, inducing an increase in cytosolicfree calcium, rapid induction of arachidonic acid release, and the respiratory burst characteristic of the action of chemotactic agents on phagocytes (Baggiolini et al., 1989; Locati et al., 1994; Rollins et al., 1991; Sozzani et al., 1994), but it does not stimulate neutrophils (Rollins et al., 1991). MCP-1 messenger RNA peaked 24 hours after injury and was no longer detected at 48 hours in a neonatal hypoxia–ischemia model (Ivacko et al., 1997). In an adult stroke model, MCP-1 messenger RNA expression was detected at 6 hours, and the peak was observed at 24 to 48 hours in endothelial and macrophagelike cells (Kim et al., 1995; Wang et al., 1995). MCP-1 protein has been noted to increase in ischemia (Yamagami et al., 1999), particularly in hypertensive rats (Wang et al., 1995), and is also inducible by LPS (Thibeault et al., 2001), acute excitotoxic injury (Galasso et al., 2000b), and mechanical (aspiration) cortical injury (Hausmann et al., 1998; Muessel et al., 2000).
In vitro, MCP-1 can induce chemotaxis of monocytes and interleukin 2–activated natural killer cells at subnanomolar concentrations. In vivo studies have confirmed MCP-1's pathophysiologic role in monocyte infiltration in inflammatory diseases (Gu et al., 1999).
It has been shown that a decrease in levels of MCP-1, induced in mice by overexpression of transforming growth factor β1, results in neuroprotection during stroke (Pang et al., 2001). Direct antileukocyte therapy mitigates CNS ischemic injury in preclinical stroke models (Hallenbeck and Kochanek, 1998). These agents appear to be more effective in CNS reperfusion models than in models of permanent ischemia and exert beneficial effects even when administered up to 3 hours after the onset of CNS ischemia (Clark and Zivin, 1997). The purpose of the current study was to examine whether MCP-1 plays a role in the progression of brain damage during ischemia and to identify mechanisms that contribute to its effects. We have found that infarction volumes after standardized brain ischemia are significantly larger in the MBP-JE mouse than in wild-type controls. These data provide direct evidence for a pathobiologic role of MCP-1 in infarct development after focal brain ischemia. During the preparation of this manuscript, Hughes et al. (2002) reported the converse relationship of MCP-1 to infarct volume (i.e., a decrease in infarct volume in mice with a targeted null mutation in MCP-1). Taken together, these two studies provide complementary evidence for a role of MCP-1 in the regulation of ischemic damage in a model of stroke.
The monocyte has been shown to provide appropriate membrane sites for efficient assembly coagulation proteases, which activate blood coagulation to induce more damage (Hallenbeck and Kochanek, 1998). F4/80 antigen is specific for monocytes and all known macrophage populations including microglia (Hume et al., 1984). In MCP-1 knockout mice, F4/80-positive cells in contact hypersensitivity lesions were decreased threefold compared to wild-type mice (Lu et al., 1998) and had a delayed transmigration into the thalamus of mice following cortical injury (Muessel et al., 2002). MCP-1 actually plays a significant role in the migration of macrophages into the lesion in cerebral ischemia (Yamagami et al., 1999). We have found that MBP-JE transgenic mice produce increased levels of circulating MCP-1 and have increased numbers of F4/80-positive cells in the ischemic hemisphere, which are associated with larger infarcts as compared to wild-type mice.
Blood pressure, blood flow, and vascular architecture are very important parameters in stroke, and such parameters could differ among different strains (Maeda et al., 1998) and contribute to the observed differences in infarct volume after MCAO. We monitored the blood pressure and blood flow before and after operation, and compared the anatomy of vessels in the brains of the two groups of mice. No differences between wild-type and genetically modified mice were found.
MCP-1 could play an important role in regulating the balance between Th1 and Th2 cytokine expression, which could also influence the ischemic injury (Muessel et al., 2001). Consistent with this notion is the recent observation that MCP-1 has also been shown to increase the production of interleukins 1 and 6 in monocytes (Rollins, 2000). The perivascular cuffing observed in MCP-1 transgenic mice that had not undergone MCAO indicates that MCP-1 overexpression leads to monocyte and macrophage accumulation around vessels even in the absence of brain ischemia. Since MCP-1 does not directly activate and attract neutrophils, the perivascular accumulation of these cells can be attributed to local activation of endothelium by proinflammatory cytokines that have been elaborated by surrounding mononuclear cells. In support of this, perivascular cuffing stained by anti-F4/80 and antineutrophil antibodies showed the colocalization of monocytes/macrophages and neutrophils in sham-operated and MCAO transgenic mice (fig.5). It is on this substratum that focal ischemia induces florid inflammation with increased accumulation of inflammatory cells and increased brain damage in the MCP-1 transgenic mice. It has also been reported that recombinant MCP-1 injected together with N-methyl-D-aspartate exacerbated brain injury, while coadministration of N-methyl-D-aspartate with MCP-1 neutralizing antibody attenuated injury (Galasso et al., 2000a). Taken together, these data demonstrate that MCP-1 can contribute to the progression of tissue damage in acute ischemic injury. This cytotoxicity is intensified in MCP-1 transgenic animals.
In summary, we showed that MBP-JE transgenic mice, which overexpress MCP-1, have larger infarct volumes after MCAO with partial reperfusion than wild-type control mice. The differences are associated with chemoattraction of monocytes and macrophages, and secondarily of neutrophils, into the ischemic region, and with increased sensitivity to LPS systemically. No differences in blood pressure, blood flow, or vascular architecture between wild-type and MBP-JE mice could explain these results. Antagonism of MCP-1 could serve as one aspect of a novel approach to improve outcome of stroke.
Footnotes
Acknowledgments:
The authors thank Mrs. Mary Crawford and Sandra Taubenkibel for their excellent secretarial assistance.