
Abstract
Interleukin-6 (IL-6) is a neurotrophic cytokine expressed in both neurons and glia. The present study shows that cerebral ischemia produced by permanent occlusion of the middle cerebral artery (MCAO) produces a dramatic increase in IL-6 bioactivity in the ischemic hemisphere within 2 hours of MCAO (167 ± 55 IU versus sham: 50 ± 35 IU), with further increases at 8 hours (3,456 ± 1,162 IU) and 24 hours (6,088 ± 1,772 IU). In a separate series of experiments, intracerebroventricular injection of recombinant IL-6 (3,100 or 31,000 IU) significantly reduced ischemic brain damage after MCAO (to 52% and 65% of controls, respectively). The large increase in endogenous IL-6 bioactivity in response to ischemia, together with the marked neuroprotection produced by exogenous IL-6 suggest that this cytokine is an important endogenous inhibitor of neuronal death during cerebral ischemia.
Study of the mechanisms underlying neuronal death due to cerebral ischemia has recently indicated the importance of immune/inflammatory proteins (cytokines) in this complex pathology. Despite numerous reports of increased expression of various cytokines after experimentally induced cerebral ischemia, relatively few (e.g., interleukin-1 [IL-1]; tumor necrosis factor; nerve growth factor; transforming growth factor-B) have been directly implicated in ischemic brain damage (see Rothwell et al., 1995; Feuerstein et al., 1995).
IL-6, first identified as B-cell stimulating factor (Kishimoto, 1989), is also synthesized within neurons and glia, and cerebral expression is increased in a wide variety of CNS disorders, including ischemia (Woodroofe et al., 1991; Yan et al., 1992; Taupin et al., 1993; Wang et al., 1995). IL-6 is important to the survival of CNS neurons in culture (Hama et al., 1991), and plays an important role in peripheral nerve regeneration (Hirota et al., 1996). Furthermore, IL-6 reduces excitotoxic-induced neuronal death in hippocampal in vitro (Yamada and Hatanaka, 1994) and striatal cholinergic in vivo (Toulmond et al., 1992) and prevents learning disability and delayed neuronal loss in gerbils after forebrain ischemia (Matsuda et al., 1996). However, transgenic mice overexpressing IL-6 in the brain develop severe neurologic disease, suggesting a complex role for IL-6 in the pathology of CNS disorders (Campbell et al., 1993).
Recently, Wang et al (1995) reported that cerebral ischemia, produced by permanent occlusion of the middle cerebral artery (MCAO) causes increased cortical expression of IL-6 mRNA. The present study was undertaken to clarify the role of IL-6 in permanent cerebral ischemia. We measured brain IL-6 bioactivity after MCAO in the rat, and determined the effect of recombinant human IL-6 (rhIL-6) administration on neuronal death 24 hours after MCAO. Because IL-6 is a potent pyrogen (LeMay et al., 1990b) and body temperature may influence outcome after MCAO (Ginsberg et al., 1993; Meden et al., 1994), the effect of rhIL-6 administration on the body temperature of ischemic rats was also determined.
MATERIALS AND METHODS
All experiments were performed on male, Sprague-Dawley rats (Charles River, United Kingdom), weighing 230 g to 270 g. Animals were housed at an ambient temperature of 21°C with a 12-hour light/dark cycle (lights on 7
Cerebral ischemia was induced under halothane anesthesia by permanent unilateral occlusion of the left middle cerebral artery using an adaptation (Bederson et al., 1986) of the method originally described by Tamura et al (1981) (see Loddick and Rothwell, 1996, for detailed description). Sham-operated animals were subjected to the same procedure, except the artery was not occluded.
To determine the effect of MCAO on brain IL-6 bioactivity, animals were subjected to MCAO or sham surgery, killed by decapitation 2 hours (MCAO: n = 4; sham: n = 4), 8 hours (MCAO: n = 4; sham: n = 3), or 24 hours (MCAO: n = 4; sham: n = 4) later and the brains immediately removed and frozen. Each hemisphere was homogenized in 1 mL of phosphate-buffered saline, then spun at 14,000g on a bench-top microfuge (Eppendorf 5402; VWR, Brisbane, CA, U.S.A.). The supernatant was retained and assessed for IL-6 bioactivity using the 7TD1 cell line, as previously described (Turnbull and Rivier, 1996). Specificity of this assay for IL-6 was shown by absence of proliferation of cells when an antibody to the α-chain of the IL-6 receptor (Genzyme, USA) was co-incubated with samples. Results are expressed as International Units/hemisphere (International standard 89/548, National Institute of Biological Standards and Controls, United Kingdom). The detection limits for individual samples varied between 2.77 and 8.15 IU/hemisphere.
In a second series of experiments, the effects of injection of rhIL-6 (Sandoz, USA, biological activity = 6.2 × 107 IU/mg) on lesion size and body temperature (Tb) after MCAO were determined. Animals were equipped with intracerebroventricular guide cannulae to permit intracerebral injections and intraperitoneal radio transmitters (Data Sciences, USA) to allow the recording of Tb by remote radio telemetry (as described in Loddick and Rothwell, 1996). The recording of Tb was commenced 6 days later at 8
All data are expressed as mean ± SD. Statistical differences in IL-6 bioactivity were analyzed using the paired Students t-test by comparing log10 transformed values (LeMay et al., 1990a) in the two hemispheres. Differences between lesion volume were analyzed using one-factor analysis of variance (ANOVA), followed by Tukey's post-hoc test. Temperature data were analyzed by multivariate ANOVA with repeated measures.
RESULTS
MCAO produced a rapid, dramatic and sustained increase in IL-6 bioactivity in the ischemic hemisphere (Table 1). This increase was apparent by 2 hours (167 ± 55 IU/hemisphere; versus 10 ± 8 IU in contralateral hemisphere), more marked by 8 hours (3456 ± 1162 IU/hemisphere), and further increased at 24 hours (6096 ± 1772 IU/hemisphere). Much smaller (23-fold to 25-fold less) increases in IL-6 activity were also apparent in both the contralateral hemisphere of rats subjected to MCAO and in the ipsilateral hemisphere of sham-operated animals.
Effect of MCAO or sham surgery on brain IL-6 biactivity
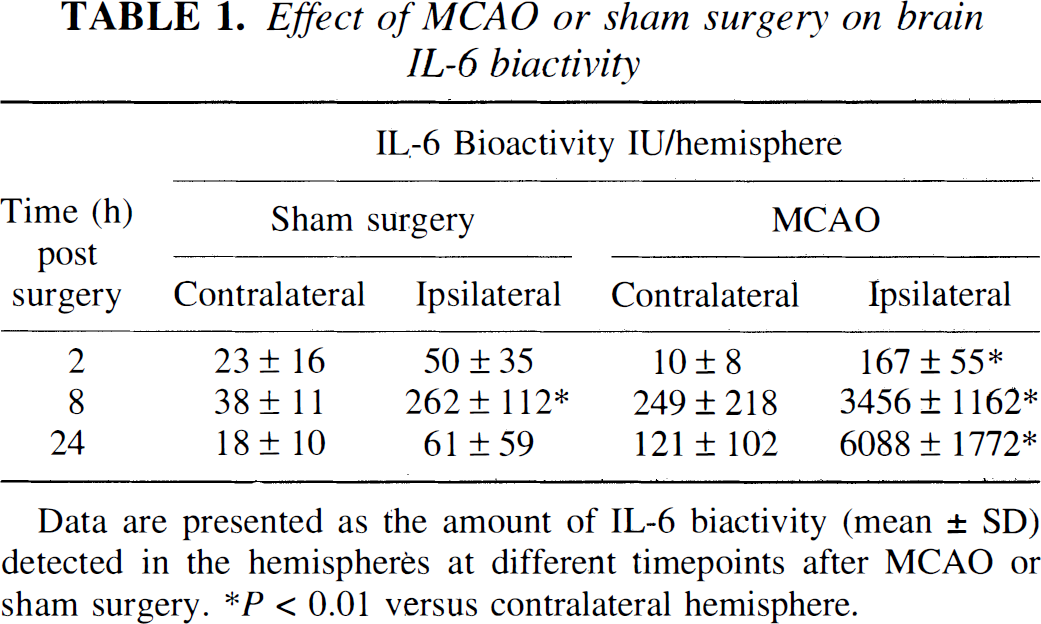
Data are presented as the amount of IL-6 biactivity (mean ± SD) detected in the hemispheres at different timepoints after MCAO or sham surgery. *P < 0.01 versus contralateral hemisphere.
Intracerebroventricular injection of rhIL-6 dramatically reduced ischemic brain damage measured 24 hours after MCAO (Fig. 1). This was evident as a significant reduction (versus vehicle treated) in total, (50 ng: 48 ± 4%; 500 ng: 35 ± 6%), cortical (50 ng: 45 ± 5%; 500 ng: 34 ± 6%), and striatal lesion volume (50 ng: 62 ± 6%; 500 ng: 37 ± 5%) compared to animals injected with saline.
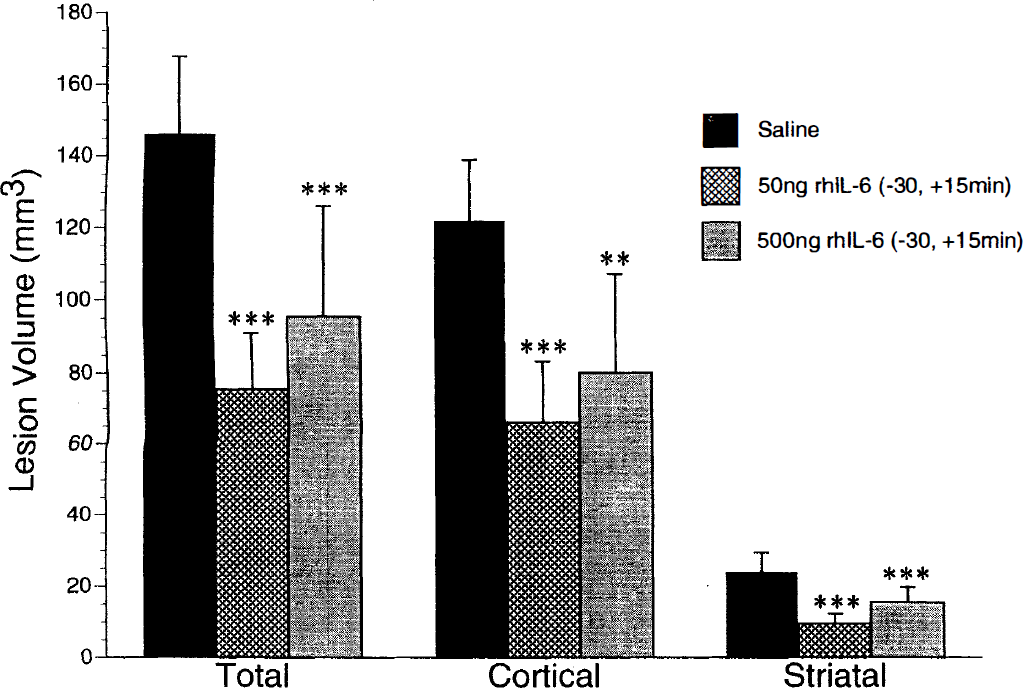
Lesion volume measured 24th after middle cerebral artery occlusion (MCAO) in rats injected intracerebroventricularly with either saline (4 μL, n = 9), 50 ng recombinant human interleukin-6 (rhIL-6) (n = 8) or 500 ng rhIL-6 (n = 8) 30 minutes before and 15 minutes after MCAO. Values denote the mean, and error bars indicate SD. ***P < .001 versus saline-treated group; **P < 0.01 versus saline-treated group.
The effect of rhIL-6 on Tb of rats subjected to MCAO is presented in Table 2 as the change in Tb after ischemia relative to the values obtained during the previous days recordings, thus avoiding the influence of circadian variation. Before surgery, Tb was similar in all groups (saline: 37.4 ± 0.1 °C; 50 ng rhIL-6: 37.4 ± 0.1 °C; 500 ng rhIL-6: 37.4 ± 0.1°C). A transient hypothermia (<1°C) was observed in all groups as animals recovered from anesthesia. However, within 4 hours, the Tb of saline-injected rats returned to values similar to those recorded on the previous day, and remained constant thereafter. rhIL-6 produced a dose-dependent increase in Tb that was statistically significant only at the 500-ng dose and was apparent by 4 hours, peaked at 5 hours, and returned to values similar to saline-injected rats by 10 hours.
Effect of rhIL-6 on body (peritoneal) temperature (Tb)
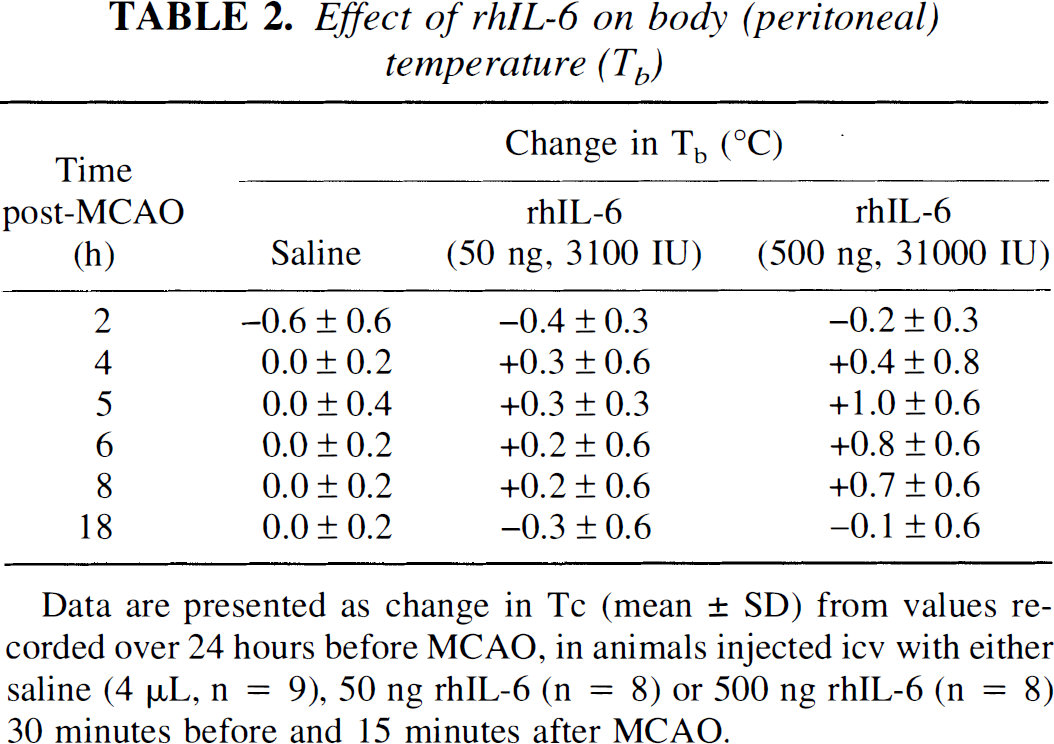
Data are presented as change in Tc (mean ± SD) from values recorded over 24 hours before MCAO, in animals injected icv with either saline (4 μL, n = 9), 50 ng rhIL-6 (n = 8) or 500 ng rhIL-6 (n = 8) 30 minutes before and 15 minutes after MCAO.
DISCUSSION
This study shows that permanent focal cerebral ischemia produces a dramatic increase in brain IL-6 activity, and exogenous IL-6 reduces ischemic damage after MCAO in the rat. The present study does not identify the cellular or even tissue source of IL-6. The rapid induction of IL-6 by ischemia suggests production by constitutive brain cells. Infiltrating inflammatory cells are not detected in the brain until 12 hours after MCAO (Clarke et al., 1993). Thus, although these cells could contribute to IL-6 bioactivity detected at 24 hours, they cannot account for the profound increase in bioactivity 8 hours after MCAO. Although neurons and neurovasculature are capable of producing IL-6 (Schöbitz et al., 1992; Rott et al., 1993), astrocytes and/or microglia are the likely source(s) after brain injury/ischemia (Woodroofe et al., 1991; Maeda et al., 1994). We also observed a modest increase in IL-6 bioactivity after sham surgery, and in the contralateral hemisphere after MCAO, consistent with a previous observation of increased IL-6 expression in both hemispheres after mechanical brain injury (Taupin et al., 1993). Because it is unlikely that the IL-6 detected in the contralateral hemisphere is derived from the ischemic hemisphere due to the different time-course of IL-6 expression, this observation suggests that the contralateral hemisphere synthesizes IL-6. The reason for this synthesis is unknown, but it may be due to physical deformation of tissue resulting from swelling of the ischemic hemisphere.
To determine if IL-6 present in ischemic brain could influence neuronal survival, we investigated the effect of administration of rhIL-6 on ischemic damage after MCAO. Based on the amount of endogenous IL-6 bioactivity found in ischemic hemispheres (3,260 IU to 5,868 IU), two doses of rhIL-6 were tested, equivalent to 3,100 IU and 31,000 IU, respectively. Both doses caused a dramatic reduction in ischemic damage throughout both the cortex and striatum. The higher dose was less effective at reducing ischemic damage, which may relate to the dramatic increase in Tb, which may have exacerbated neuronal death (Ginsberg et al., 1993; Meden et al., 1994). Regardless of effects on T b , exogenous rhIL-6 was clearly neuroprotective at doses (3,100 IU to 31,000 IU) similar to the amount of endogenous IL-6 bioactivity found in ischemic hemispheres (3,260 IU to 5,868 IU). Although the effects of neutralization of endogenous IL-6 were not tested here because of lack of suitable reagents, that administration of physiologically relevant doses of IL-6 provides protection suggests that cerebral IL-6 generated during ischemia may be an important endogenous neuroprotective factor.
The present study did not investigate the mechanism by which IL-6 influences neuronal survival after ischemia. A modification of T b is unlikely to account for the observed effects because the fever produced by IL-6 would be expected to exacerbate rather than inhibit neuronal death (Ginsberg et al., 1993; Meden et al., 1994). An effect of rhIL-6 on neuronal viability secondary to changes in physiologic variables such as blood pressure and blood flow is possible. However, IL-6 protects hippocampal neurons in vivo without affecting hippocampal blood flow (Matsuda et al., 1996) and protects against excitotoxicity in vitro (Yamada and Hatanaka, 1994) suggesting a direct action of IL-6 on neuronal viability. In the rat, mRNA encoding IL-6 and both its receptor subunits (IL-6 receptor α-chain and gp130) share similar distributions in the brain (Schöbitz et al., 1992; Schöbitz et al., 1993; Watanabe et al., 1996), and are all present in the cortex and striatum, areas where rhIL-6 reduced ischemic damage after MCAO. IL-6 may produce neuroprotection by an action directly on or secondary to release of intermediates by glial cells because IL-6 receptors are present in both cell types (Schöbitz et al., 1992; Watanabe et al., 1996). Furthermore, that rhIL-6 protects against excitoxicity both in vivo and in vitro suggests that its neuroprotective effects during ischemic brain damage may be downstream of excitatory amino acid receptor activation.
The effects of IL-6 on neuronal death after MCAO are in marked contrast to those of a related cytokine, IL-1. We and others have provided convincing evidence that IL-1 exacerbates ischemic brain damage and is an endogenous neurotoxic factor in cerebral ischemia (Relton and Rothwell, 1992; Yamasaki et al., 1995; Loddick and Rothwell, 1996) and excitatory amino acid-induced excitotoxicity (Relton and Rothwell, 1992). Although IL-6 and IL-1 share many common actions (e.g., fever, acute phase protein synthesis, activation of the pituitary-adrenal axis), IL-6 can inhibit IL-1 synthesis (Schindler et al., 1990) and stimulates production of the endogenous receptor antagonist (Tilg et al., 1994), actions that may also contribute to its neuroprotective effects.
In conclusion, we have shown for the first time that cerebral administration of rhIL-6 produces a profound reduction in neuronal death due to permanent cerebral ischemia in the rat. Because our work also shows that the concentration of endogenous IL-6 in ischemic brain tissue is markedly increased, we propose that IL-6 is an important endogenous neuroprotective factor.