
Abstract
The neuroprotective effects of a systemically active, highly selective, corticotropin-releasing factor-1 (CRF1) receptor antagonist, R121920 ((7-(dipropylamino)-2,5-dimethyl-3- [2-(dimethylamino)-5-pyridyl] pyrazolo [1,5-a] pyrimidine), was assessed in two rat models of permanent focal cerebral ischemia, where the middle cerebral artery (MCA) was occluded either through the subtemporal approach or using the intraluminal suture technique. R121920 rapidly crossed the blood–brain barrier after intravenous (IV) bolus administration (10 mg/kg), with peak brain concentrations at 5 minutes (2.26 ± 0.40 μg/mL), which were approximately 2-fold greater than those in plasma (0.98 ± 0.24 μg/mL). Treatment with R121920 (10 mg/kg IV followed by 5 mg/kg subcutaneously at hourly intervals for 4 hours) significantly (P < 0.001) reduced total (by 40%) and cortical (by 37%) infarct volume at 24 hours after subtemporal MCA occlusion (MCAO). In the intraluminal suture MCAO model, IV administration of R121920 (10 mg/kg) at the time of ischemia onset (and at multiple times thereafter) reduced both hemispheric infarct volume (by 34%, P < 0.001) and brain swelling (by 50%, P < 0.001) when assessed at 24 hours. In this model of focal ischemia, significant reduction (P < 0.05) in both outcome measures was obtained when R121920 administration was delayed up to 1 hour after MCAO. These results further define the antiischemic properties of selective CRF1 antagonists in two experimental models of permanent focal cerebral ischemia.
Corticotropin-releasing factor (CRF), a 41-amino acid peptide, plays a pivotal role in integrating the human body's overall response to stress through coordinated actions on the nervous, endocrine, and immune systems (Chalmers et al., 1996; De Souza, 1995). Corticotropin-releasing factor is the major physiologic regulator of the basal and stress-induced release of adrenocorticotropin (ACTH), β-endorphin, and other proopiomelanocortin (POMC)-derived peptides from the pituitary (Vale et al., 1981, 1983). In addition to this endocrine role at the pituitary, CRF functions in the central nervous system producing a spectrum of electrophysiologic, autonomic, and behavioral effects (Chalmers et al., 1996; De Souza, 1995; Dunn and Berridge 1990; Perrin and Vale, 1999; Turnbull and Rivier, 1997).
There is increasing evidence to support the involvement of CRF in the pathogenesis of ischemic brain damage. Up-regulation of CRF mRNA has been reported in the cerebral parenchyma of rats after permanent middle cerebral artery occlusion (MCAO) (Wong et al., 1995) and traumatic brain injury (Roe et al., 1998). Moreover, CRF antagonists are beneficial in experimental models of neurodegeneration in vivo. Neuroprotective effects of the peptide CRF receptor antagonists D-phe CRF(12–41) and α-helical CRF(9–41) have been demonstrated in several animal models, including neuronal damage in rats induced by excitotoxins, seizures, head injury, forebrain ischemia, and MCAO (Loddick et al., 1998; Lyons et al., 1991; Maecker et al., 1997; Roe et al., 1998; Strijbos et al., 1994). The CRF receptor subtype(s) that mediate these effects cannot be determined from these studies as the peptide antagonists have equal affinity for both CRF1 and CRF2 receptors. Recently, however, systemic administration of a first generation, nonpeptide CRF1 antagonist NBI-27914 (Chen et al., 1996) reduced infarction and brain swelling after focal ischemia in the rat (Loddick et al., 1998; Yatsushiro et al., 1997a, 1997c). These data suggest that the CRF1 receptor mediates the neuroprotection observed in experimental models of ischemia.
The first generation of nonpeptide CRF1 -selective antagonists, although having high affinity and selectivity, suffered from low aqueous solubility and suboptimal blood–brain barrier penetration (Chen et al., 1996; Loddick et al., 1998). A second generation of CRF1 receptor antagonists has provided compounds with improved solubility and blood–brain barrier penetration (Huang et al., 2000; Mackay et al., 2000). R121920 ((7-(dipropylamino)-2,5-dimethyl-3- [2-(dimethylamino)-5-pyridyl] pyrazolo [1,5-a] pyrimidine), is a novel, potent, and highly selective second generation antagonist for the CRF1 receptor subtype (Huang et al., 2000; Mackay et al., 2000). The aim of the current study was to define the neuroprotective properties of CRF1 receptor antagonists further by investigating the ability of systemic administration of R121920 to reduce ischemic brain damage and brain swelling in two different rat models of permanent focal cerebral ischemia.
MATERIALS AND METHODS
Pharmacokinetics
Plasma and brain levels of R121920 were determined in nonoccluded male Sprague–Dawley rats (280 to 340 g). R121920, in a vehicle of water, was administered as a single intravenous (IV) dose of 10 mg/kg, and the animals were killed 2, 5, 15, 30, 60, 120, and 240 minutes later (n = 3 per time point). Brains were rapidly removed, flash frozen in liquid nitrogen, and stored at −70°C until analysis. Terminal blood samples (200 μL) were collected into tubes containing EDTA and were immediately centrifuged for 1.5 minutes at 10,000 g. The plasma was extracted, flash frozen in liquid nitrogen, and stored at −70°C until analysis. For the quantitation of R121920 concentration in plasma, 150 μL acetonitrile was added to 50 μL plasma to precipitate protein, and the sample was centrifuged. The supernatant was collected and dried in a Speed Vac at room temperature. For brain sample tissue preparation, brain tissue was first homogenized in 1 mL saline solution, and protein precipitation was achieved by adding 3 vol acetonitrile. After reconstitution, all samples were introduced into a reverse phase HPLC-UV system for analysis. The concentrations of R121920 in the samples were predicted from a linear regression of an external calibration curve of five spiked matrix standards.
Focal ischemia
Surgical preparation.
All rats were anesthetized initially with a mixture of 5% halothane, 30% oxygen, and 70% nitrous oxide in an induction chamber, and maintained thereafter with a nitrous oxide/oxygen mixture (70%: 30%) containing 1.0% to 1.5% halothane delivered through a face mask. Body temperature was monitored throughout the surgical procedure by a rectal thermometer, and the animals maintained normothermic (37 ± 1°C) through a heating blanket controlled by the thermometer.
Subtemporal middle cerebral artery occlusion model.
Permanent focal ischemia was induced in male Fischer 344 rats (240 to 280 g) through a subtemporal approach, as described by Tamura and coworkers (1981). Briefly, the craniectomy was performed at the level where the MCA crossed the lateral olfactory tract. The dura was carefully opened, the artery exposed and occluded by bipolar diathermy from its origin to the point where it crossed the inferior cerebral vein. All visible lenticulostriate branches also were occluded. The artery then was transected to confirm complete occlusion and to prevent recanalization. The incision sites were sutured closed, and the animals were allowed to recover from anesthesia.
R121920 (10 mg/kg or 20 mg/kg) was given as an initial bolus IV dose at the time of ischemia onset, followed by subsequent subcutaneous (SC) injections of R121920 (5 mg/kg or 10mg/kg, respectively) at hourly intervals thereafter for 4 hours. Control animals received injections of vehicle (water, 1 mL/kg) at the same time points.
Animals were killed 24 hours after MCAO, brains were removed, and 500-μm coronal sections were cut from the frontal pole on a vibratome. Brain sections (24 per forebrain) were incubated in a 2% solution of triphenyl tetrazolium chloride (TTC), which stains for viable tissue, at 37°C for 30 minutes. Areas of infarct in the hemisphere, cortex, and striatum on each of the 24 sections were delineated and transcribed onto scale diagrams, and quantified by automated image analysis. The volume of infarct for each of the brain regions was computed by summing the areas of damage from each section and multiplying them by the distance between the sections.
Intraluminal suture middle cerebral artery occlusion model.
The left MCA was occluded permanently in male Sprague–Dawley rats (280 to 340 g) through a cervical carotid approach using the intraluminal suture technique, as described in detail previously (Zea Longa et al., 1989). Briefly, the common carotid, external carotid, and internal carotid arteries are exposed through a midline cervical incision under an operating microscope. A 30-mm length of 3–0 monofilament nylon suture, its tip heat-blunted and coated with poly-l-lysine solution (Belayev et al., 1996), was advanced from the external carotid artery into the lumen of the internal carotid artery until mild resistance was felt (19 to 20 mm depending on body weight), thereby occluding the origin of the MCA. After closure of the skin incision, rats were allowed to recover from anesthesia.
R121920 (10 mg/kg) was administered as an IV injection at the time of MCAO, followed by additional IV injections of R121920 (10 mg/kg) at 1, 2, 3 and 4 hours later. Control animals received IV injections of vehicle (water) at the same time points. The time window for neuroprotective effect of R121920 was examined by delaying induction of the dosing regimen to 0, 0.5, and 1 hour after occlusion of the MCA.
Twenty-four hours after occlusion of the MCA, the animals were killed and the brains were carefully removed and frozen in isopentane at −40°C. Brains were cut into 20-μm serial coronal sections at 12 equidistant levels (1 mm apart, covering the entire forebrain), which were fixed for 10 minutes in 4% paraformaldehyde and stained with hematoxylin and eosin. The sections, which corresponded to 12 stereotactically predetermined coronal planes of rat brain from anterior +12.7 mm to anterior +1.7 mm relative to the interaural line (Paxinos and Watson, 1998), were examined by light microscopy. Areas of ischemic damage were delineated and transcribed onto scale diagrams of normal forebrain at each of the 12 coronal planes. Areas of infarction were quantified using a computer-based image analysis system, and the total volumes of ischemic tissue for each brain derived from integration of the areas of damage in the 12 planes and the known stereotactic coordinates of the planes. The volumes of the cerebral hemispheres ipsilateral and contralateral to the occluded MCA were determined directly from the stained histologic sections by assessment of the total surface area at the same coronal planes as used for assessing areas of ischemic damage. The difference between the two hemispheres provided a measure of brain swelling.
Statistical analysis
All data are expressed as mean ± SD. Significant differences in histologic data between two experimental groups were assessed using Student's t-test. Multiple group comparisons of ischemic damage were made using analysis of variance followed by Dunnett's post hoc test. The relation between the volume of brain swelling and volume of infarction was assessed by linear regression analysis (Pearson's). For all statistical analyses, the significance level was accepted as P < 0.05.
RESULTS
Plasma and brain levels of R121920 obtained up to 4 hours after a single IV injection (10 mg/kg) are shown in Fig. 1. These studies in the rat indicate that R121920 readily crosses the blood–brain barrier after IV bolus administration, with peak brain concentrations at 5 minutes that are approximately 2 times greater than those in plasma (Fig. 1); maximal plasma levels were observed 2 minutes after injection, the earliest time point measured (Fig. 1). On the basis of these experiments, studies examining the potential neuroprotective effects of R121920 were conducted with an initial IV bolus dose with subsequent SC or IV supplemental doses in an attempt to maintain plasma and brain drug concentrations for a period of 5 to 6 hours, the time period for which neurodegenerative mechanisms are believed to occur after MCAO.
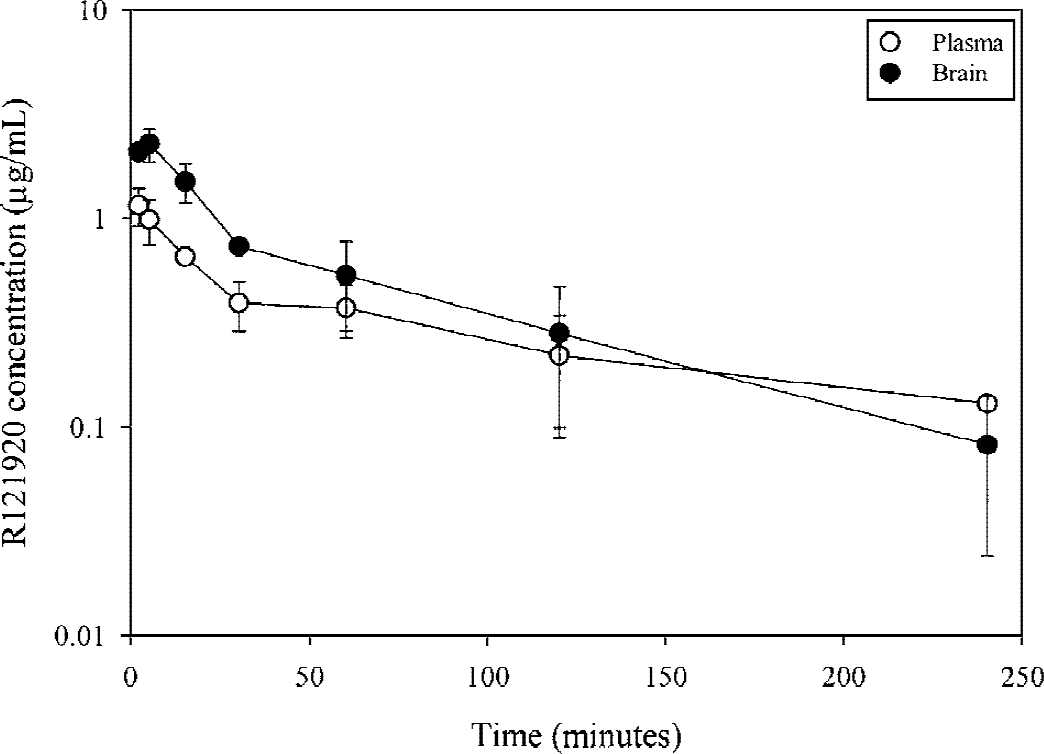
Brain and plasma concentrations of R121920 after a single intravenous (IV) bolus injection of R121920 (10 mg/kg) at time = 0. R1219290 readily crosses the blood–brain barrier after IV administration, with peak brain concentrations at 5 minutes that are approximately 2 times greater than those in plasma. Data are mean ± SD (n = 3 per group). Mean values without error bars had standard deviations too small to graph.
R121920 reduced infarct size in two models of permanent focal cerebral ischemia: the subtemporal and the intraluminal suture MCAO models. In the subtemporal model, the administration of R121920 as an IV bolus of 10 mg/kg at the time of artery occlusion (0 hour), with subsequent doses of 5 mg/kg SC at hourly intervals thereafter for 4 hours, significantly reduced the volume of total (by 40%), cortical (by 37%), and striatal (by 50%) ischemic damage compared with vehicle (Fig. 2). Increasing the IV bolus dose to 20 mg/kg at the onset of ischemia, followed by 10 mg/kg SC at 1, 2, 3, and 4 hours after occlusion significantly reduced total (by 43%), cortical (by 40%), and striatal (by 50%) infarct size to a similar degree, suggesting that a maximal neuroprotective effect was reached (Fig. 2).
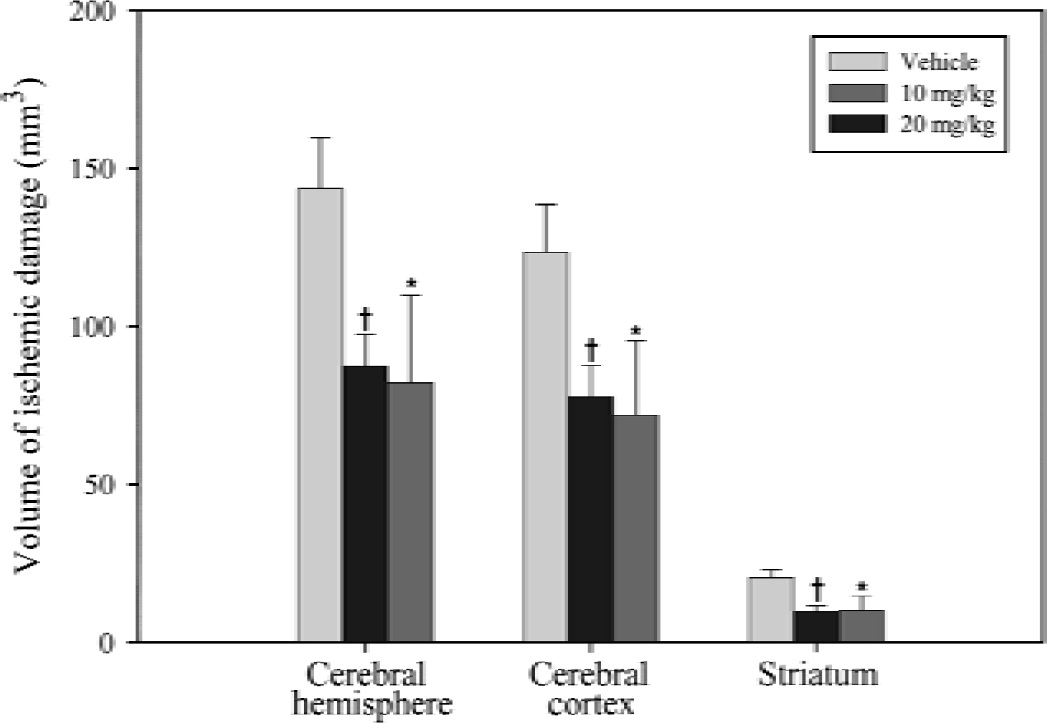
Effect of R121920 on the volume of ischemic damage assessed 24 hours after permanent transcranial middle cerebral artery occlusion in the rat. R121920 (10 mg/kg or 20 mg/kg) or vehicle (1 mL/kg) was given as an initial bolus intravenous dose at the time of ischemia onset, followed by subsequent subcutaneous doses at hourly intervals thereafter for 4 hours. Data are mean ± SD (n = 6 to 7 per group). * P < 0.01, †P < 0.001 compared with vehicle-treated control animals (analysis of variance followed by Dunnett's test).
In the intraluminal model, the IV administration of R121920 (10 mg/kg) at the time of MCAO and every hour later for 4 hours significantly reduced the volume of ischemic brain damage in the cerebral hemisphere and cerebral cortex, by 34% and 38% respectively, but not in the striatum (Fig. 3). Treatment with R121920 also significantly reduced the volume of brain swelling at 24 hours (by 50%) when compared with vehicle-treated control rats (Fig. 3). Brain swelling as a result of ischemia was directly proportional to infarction volumes in all animals, regardless of whether they received vehicle or R121920 (r = 0.915;P < 0.001).
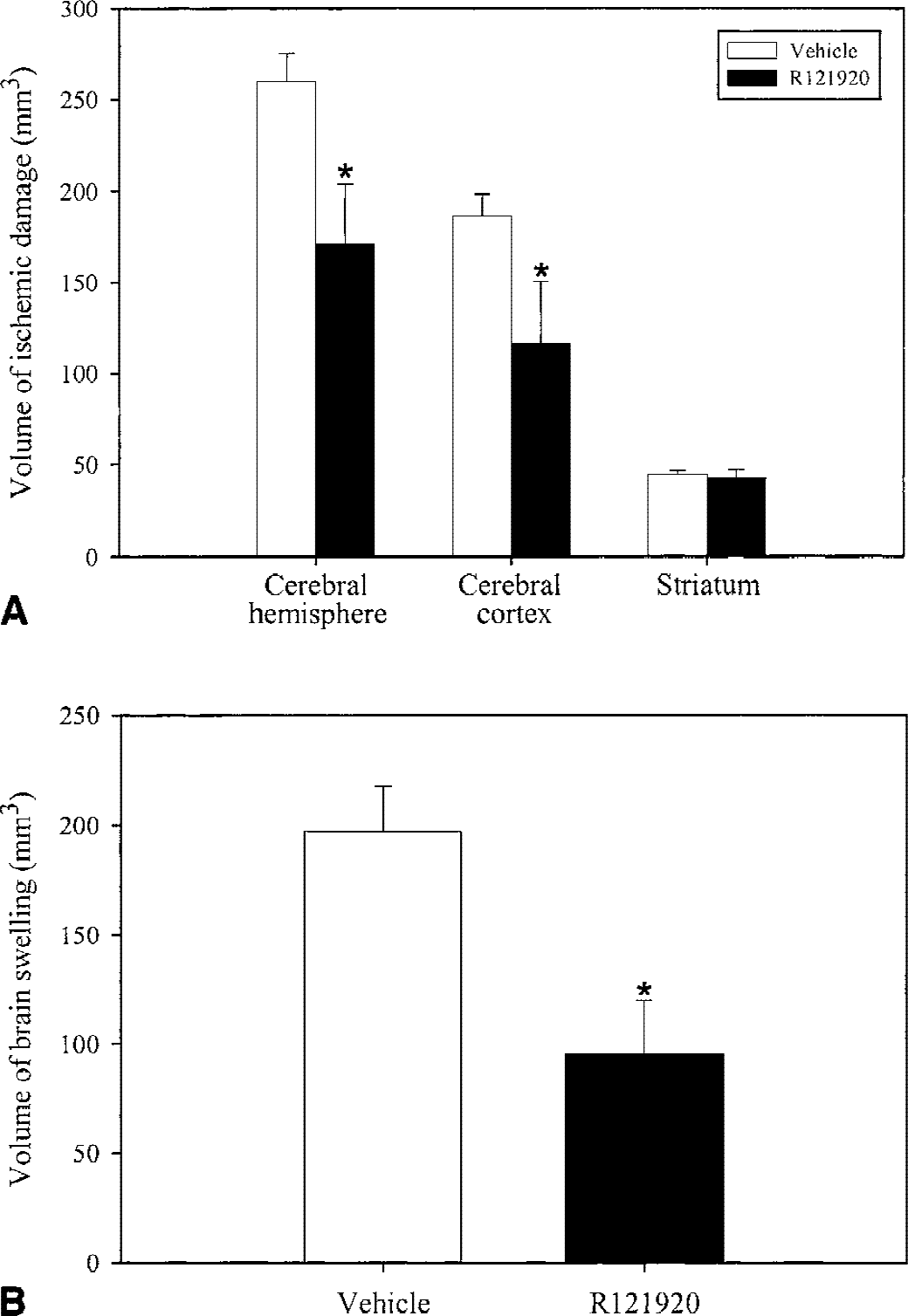
Effect of R121920 on the volume of ischemic damage
Treatment with R121920 (10 mg/kg IV and at multiple times thereafter) initiated either at the time of MCAO using the intraluminal suture technique, or delayed for 0.5 or 1 hour after occlusion, produced a significant time-dependent reduction of total and cortical infarct size that was observed in all drug-treated groups, with the most marked reduction in infarction (reduced by 39% from controls) seen when R121920 administration was started at the induction of the ischemic insult (Fig. 4). Hemispheric swelling volume showed a similar profile of reduction in the drug-treated animals versus vehicle-treated controls, the most marked reduction in swelling (by 56% from controls) again being observed when R121920 treatment was initiated at the time of MCAO (Fig. 4). There was a good linear relation between the volume of ischemic damage and the volume of brain swelling that was similar for vehicle-treated animals and all the R121920-treated groups regardless of the time delay of treatment onset (Fig. 5), with an overall correlation coefficient, r = 0.846 (P < 0.001).
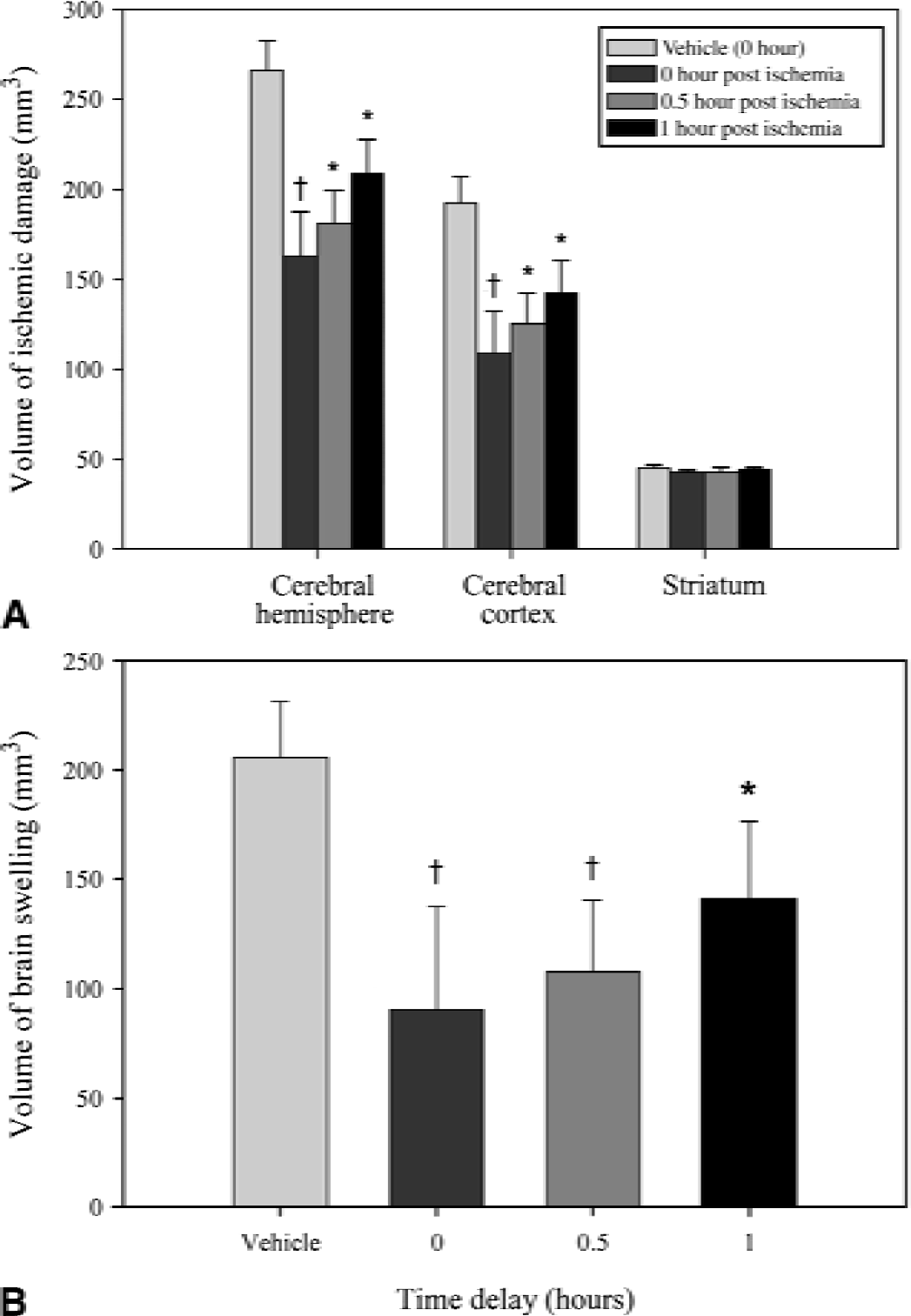
Therapeutic time window for R121920 against volume of ischemic damage
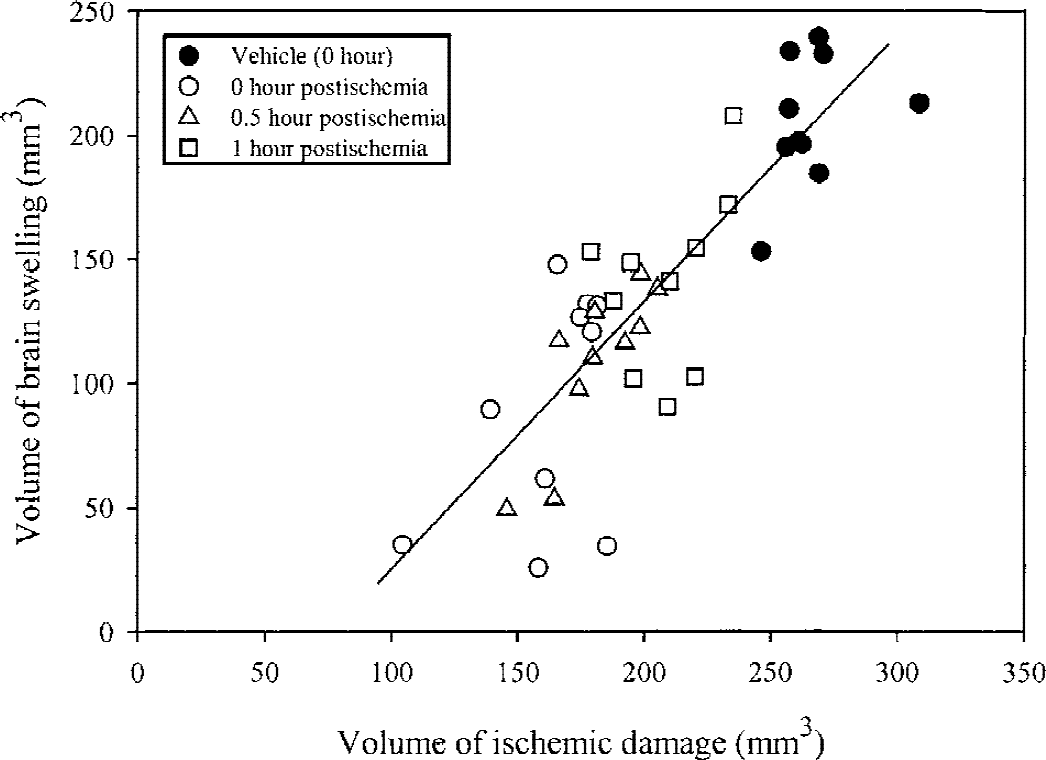
Relation between the volume of ischemic damage and the volume of brain swelling in individual animals 24 hours after the induction of ischemia. There is a good linear correlation (P < 0.001) between the two variables (r = Pearson's correlation coefficient), which is similar for all treatments.
DISCUSSION
Results of the current study demonstrate that systemic administration of the small molecule CRF1 antagonist R121920 at the time of MCAO reduces the volume of ischemic damage that develops subsequently in two rat models of permanent focal cerebral ischemia. The neuroprotection afforded by R121920 was observed when drug administration was delayed up to at least 1 hour after the induction of ischemia, although the true therapeutic time window for R121920 after MCAO may be somewhat longer. The data, therefore, substantiate and extend previous evidence that selective CRF1 antagonists ameliorate the consequences of focal ischemia in the rat (Loddick et al., 1998; Yatsushiro et al., 1997a, 1997c).
There is an increasing body of evidence describing the neuroprotective effects of CRF antagonists in experimental models of cerebral ischemia and brain injury. The peptide antagonists D-phe CRF(12–41) and α-helical CRF(9–41), which antagonize both CRF1 and CRF2 receptor subtypes, reduce infarct size in both focal and global models of ischemia when administered intracerebroventricularly (ICV) (Loddick et al., 1998; Lyons et al., 1991; Strijbos et al., 1994). D-phe CRF(12–41) has been shown to improve outcome in a model of traumatic brain injury (Roe et al., 1998). Nonselective peptide antagonists also protect against excitotoxin-induced neuronal degeneration (Strijbos et al., 1994) and ameliorate the seizure-related neuronal degeneration that is produced by kainic acid (Maecker et al., 1997). The nonpeptide CRF antagonist NBI-27914 (Chen at al., 1996), which binds with high affinity to the CRF1 receptor (Ki = 2 nmol/L), but with low affinity to the CRF2 receptor (Ki > 10 μmol/L), rendering this compound a selective CRF1 receptor antagonist, reduces ischemic brain damage after MCAO in the rat when administered systemically (Loddick et al., 1998; Yatsushiro et al., 1997a, 1997c). Data from the current study using R121920 substantiate the antiischemic effects of selective, small molecule CRF1 antagonists in MCAO models, having a degree of efficacy equal to that of the peptide CRF receptor antagonists (Loddick et al., 1998; Strijbos et al., 1994; Yatsushiro et al., 1997a, 1997c) or indeed other classes of neuroprotective agents in these models (Gill and Lodge, 1997; McCulloch, 1994). This indicates that selective blockade of CRF1 receptors is sufficient to confer the neuroprotective effects previously observed with the nonselective peptide CRF receptor antagonists.
The first generation of nonpeptide CRF1 -selective antagonists, such as NBI-27914, although having high affinity and selectivity suffered from low aqueous solubility and suboptimal blood–brain barrier penetration (Chen et al., 1996; Loddick et al., 1998). A second generation of CRF1 receptor antagonists has provided compounds with improved solubility and blood–brain barrier penetration, resulting in drugs with ideal characteristics as intravenously administered neuroprotective agents. R121920 is an example of such a compound, and has been used as a tool to define the neuroprotective properties of CRF1 receptor antagonists further in the current study. R121920 is a high affinity, CRF1 receptor–selective antagonist (Ki values (nmol/L): CRF1 = 4; CRF2 > 10,000) with good aqueous solubility (≥20 mg/mL) (Mackay et al., 2000). The pharmacokinetic profile shows that R121920 readily crosses the blood–brain barrier after IV bolus administration, with peak brain concentrations at 5 minutes that are 2 times greater than those in plasma (Fig. 1). In addition, ex vivo autoradiography studies have confirmed that systemic administration of R121920 (10 mg/kg, IV) gives substantial blockade of brain CRF1 (but not CRF2) receptors from 2 to 120 minutes after a single bolus dose (Grigoriadis et al., unpublished observations). These results, in conjunction with the neuroprotective effects of the CRF1 -selective antagonist R121920, give preliminary data to relate brain occupancy of CRF receptors to neuroprotection.
The mechanisms through which CRF receptor antagonists confer neuroprotection currently are unclear. Corticotropin-releasing factor has been shown to directly stimulate the release of the excitatory amino acid glutamate (Singewald et al., 1996), although CRF itself does not appear to be neurotoxic in rat cortical neurons in vitro or when injected into adult rat brain (Craighead et al., 2000). However, in neonatal rats, CRF provokes epileptiform activity and seizures that can result in neurodegeneration (Ribak and Baram, 1996); these effects are mediated by the CRF1 receptor subtype because they are reduced by a CRF1 -selective antagonist (Baram et al., 1997). Based on these observations in infant rats, it might be expected that CRF1 receptors mediate excitatory responses in the adult brain. Electrophysiologic studies have demonstrated excitatory effects of CRF on neurons in several brain regions, including the locus ceruleus and cerebellum (Bishop and King, 1992; Valentino et al., 1983), and also in areas highly susceptible to ischemia-induced neurodegeneration, such as the hippocampus (Aldenhoff et al., 1983). In the hippocampus, the excitatory effects of CRF appear to result from inhibition of the after-hyperpolarization, perhaps through blockade of postsynaptic potassium channels (Aldenhoff et al., 1983). Consequently, CRF release during cerebral ischemia could act directly on CRF receptors present on vulnerable neurons to potentiate excitotoxic events that are underway through overactivation of glutamate receptors. Inhibition of CRF receptor activation would remove this acceleration of ongoing deleterious events, and increase the threshold for neuronal degeneration. This would provide a plausible mechanistic hypothesis for the neuroprotective effects of CRF1 antagonists.
Corticotropin-releasing factor has been reported to elevate both heart rate and blood pressure by stimulating noradrenergic sympathetic outflow (Fisher et al., 1983) and to cause vasodilation (Lei et al., 1993). During focal cerebral ischemia in the rat brain, mRNA for CRF is increased in the occluded hemisphere (Wong et al., 1995; Yatsushiro et al., 1997c) and striking elevations are also observed in pial and parenchymal arterioles (Yatsushiro et al., 1997c). The relevance of the elevated CRF in blood vessels during ischemia is less clear. Corticotropin-releasing factor and related peptides cause vasodilation in cat cortical arterioles (Yatsushiro et al., 1997b, 1997d), and recent laser–Doppler studies in nonischemic rats indicate that this translates to a significant increase in blood flow in similar vessels (De Michele et al., 1999). During middle cerebral artery occlusion in the rat, CRF itself can increase cerebral blood flow (De Michele et al., 1999). However, the peptide antagonist D-Phe CRF (12–41) has essentially no effect on cerebral blood flow in the ischemic penumbra (De Michele et al., 1999), indicating that endogenous CRF has little or no role to play in maintaining blood flow under these conditions. Because the effects of CRF and related peptides on cerebral vessels are putatively mediated through CRF2 receptors (Yatsushiro et al., 1997b, d ), CRF1 receptor antagonists would be predicted to have no direct effect on cerebral blood flow.
A putative mechanism, which could contribute to the neuroprotective effects of CRF1 receptor antagonists, is inhibition of the hypothalamo-pituitary-adrenal axis. Marked activation of the hypothalamo-pituitary-adrenal axis in patients after acute ischemic stroke is well documented (Olsson et al., 1992). In the first few hours after stroke, patients have large elevations in ACTH and cortisol levels, the degree of which has been linked to the volume of brain injury and neurologic outcome (Fassbender et al., 1994; Olsson et al., 1992). In animals, glucocorticoids are reported to increase susceptibility to excitotoxins such as kainic acid (Stein-Behrens et al., 1994) and to global ischemia (Sapolsky and Pulsinelli, 1985). The glucocorticoid synthesis inhibitor, metyrapone, reduces neurodegeneration induced by hypoxia, focal and global cerebral ischemia, and kainic acid (Krugers et al., 1995; Smith-Swintosky et al., 1996), although one study has indicated an exacerbation of infarct size with metyrapone after MCAO in hypertensive rats (Risedal et al., 1999). The activation by CRF of the CRF1 receptor subtype on pituitary corticotrophs is responsible for the release of ACTH, and CRF1 receptor–selective antagonists inhibit the elevations of ACTH and corticosterone that are provoked by CRF administration and stress (Chalmers et al., 1996; De Souza, 1995; Dunn and Berridge 1990; Perrin and Vale, 1999; Turnbull and Rivier, 1997). Consequently, if the elevations in ACTH and glucocorticoids that occur after cerebral ischemia contribute to neuronal degeneration, treatment with a CRF1 receptor antagonist would be an effective mechanistic strategy to remove this influence and confer neuroprotection.
It is well established that there is a marked inflammatory response after MCAO in rat (Barone and Feuerstein, 1999). Corticotropin-releasing factor recently has been shown to dramatically inhibit the recruitment of leukocytes to the endothelia by blocking up-regulation of intercellular adhesion molecule-1 expression (Casadevall et al., 1999), an effect that is inhibited by astressin. Therefore, the neuroprotective effects of CRF1 receptor antagonists may be partially attributed to an alteration in the recruitment of invading leukocytes or other inflammatory mediators to the site of ischemic brain damage.
In summary, these data indicate that CRF1 receptor antagonists have robust neuroprotective effects in animal models of stroke, producing reductions in infarct size comparable with other neuroprotective agents, and are effective when drug administration is initiated after artery occlusion. Future studies with CRF1 antagonists are aimed at optimizing dosing regimes with bolus injection and IV infusion to determine the relation between IV dose, neuroprotection, plasma and brain drug levels, and ex vivo receptor occupancy, before defining more fully the therapeutic time window and possible mechanism of action in experimental models of focal ischemia. This information will be helpful in the evaluation of further generations of CRF1 receptor antagonists for their neuroprotective properties.
Footnotes
Acknowledgment:
The authors thank Mr. Keith Wilcoxen at the Department of Medicinal Chemistry at Neurocrine Biosciences Inc. for the synthesis of R121920.