
Abstract
Synaptic pathology is observed during hypoxic events in the central nervous system in the form of altered dendrite structure and conductance changes. These alterations are rapidly reversible, on the return of normoxia, but are thought to initiate subsequent neuronal cell death. To characterize the effects of hypoxia on regulators of synaptic stability, we examined the temporal expression of cell adhesion molecules (CAMs) in synaptosomes after transient middle cerebral artery occlusion (MCAO) in mice. We focused on events preceding the onset of ischemic neuronal cell death (< 48 h). Synaptosome preparations were enriched in synaptically localized proteins and were free of endoplasmic reticulum and nuclear contamination. Electron microscopy showed that the synaptosome preparation was enriched in spheres (≈650 nm in diameter) containing secretory vesicles and postsynaptic densities. Forebrain mRNA levels of synaptically located CAMs was unaffected at 3 h after MCAO. This is contrasted by the observation of consistent downregulation of synaptic CAMs at 20 h after MCAO. Examination of synaptosomal CAM protein content indicated that certain adhesion molecules were decreased as early as 3 h after MCAO. For comparison, synaptosomal Agrn protein levels were unaffected by cerebral ischemia. Furthermore, a marked increase in the levels of p-Ctnnb1 in ischemic synaptosomes was observed. p-Ctnnb1 was detected in hippocampal fiber tracts and in cornu ammonis 1 neuronal nuclei. These results indicate that ischemia induces a dysregulation of a subset of synaptic proteins that are important regulators of synaptic plasticity before the onset of ischemic neuronal cell death.
Introduction
The synapse is a specialized, asymmetric cell-cell contact formed between an axon terminal and the postsynaptic dendrite, soma, or axon. The synapse shares a number of similarities with other specialized cell-cell contacts such as the immunological synapse and the neuromuscular junction (Yamaguchi, 2002). Synapses are highly dynamic structures that are capable of adapting to a variety of physiological conditions. Synapses are gained and lost on a continuing basis in developing as well as adult brains. Generally, increased neuronal activity causes the formation of new synapses, and neuronal inactivity results in the loss of synaptic connections. Lastly, synaptic function is highly important for functional recovery after brain injury.
Synaptically localized signals, either anti- or proapoptotic, can be propagated to the cell body in both an anterograde and retrograde manner (Mattson and Duan, 1999). The ability of the synaptic compartment to initiate proapoptotic signaling has been described as synaptic apoptosis (Mattson et al, 1998). Apoptotic stimuli have been shown to induce caspase-3 activation, mitochondrial membrane depolarization, and phospholipid asymmetry in isolated synaptosomes (Mattson et al, 1998). Similarly, trophic factor withdrawal increases axonal caspase-3 activity, but not within the neuronal soma (Mattson and Duan, 1999). In hippocampal neurons, apoptotic signals initiated at the dendrites have been shown to subsequently spread toward the cell body (Mattson and Duan, 1999). Synaptic apoptosis may be a mechanism that is necessary for synaptic remodeling under nonpathological conditions as well as contributing to or initiating neuronal apoptosis during pathological conditions. Proapoptotic proteins have been found to play a role in nonpathological processes such as neurogenesis, neurite outgrowth, and synaptic plasticity (Mattson and Gleichmann, 2005). These data suggest that signals triggered at the synapse may propagate toward the cell body and instigate post-schemic (ISC) neuronal death.
Synaptic plasticity and synaptogenesis are regulated by a group of cell adhesion molecules (CAMs) that are located at synaptic junctions. Three classes of cell adhesion molecules are enriched in synaptic contacts: (1) the cadherin/catenin system, (2) the cadherin-like neuronal receptors system, and (3) the β-neurexin/neuroligin system (Brose, 1999). Both the cadherin/catenin and cadherin-like neuronal receptor systems exhibit homotypic transynaptic signaling, whereas the β-neurexin/neuroligin signaling is heterotypic in nature. The cadherin/catenin and the β-neurexin/neuroligin systems share the ability to interact with and regulate the synaptic cytoskeletal architecture. The modulatory role of α-N-catenin (Ctnna2, catenin (cadherin-associated protein), α-2 or α-catenin) on synaptic plasticity was examined by Abe et al (2004). Overexpression of α-catenins (α-N-catenin I, α-N-catenin II, α-E-catenin, or α-T-catenin), but not β-catenin (Ctnnb1, catenin (cadherin-associated protein), β-1 or β-catenin) or N-cadherin (Cdh2, cadherin 2), resulted in an increase in dendritic spine formation (Abe et al, 2004). Thus, α-N-catenin, which forms a complex with N-cadherin, β-catenin, and actin, appears to be a key regulator of catenin/cadherin-mediated control of synaptic plasticity. A number of other factors have also been implicated in regulating synaptic plasticity, including the glutamate receptor subunit GluR2 (Passafaro et al, 2003), regulators of the cytoskeleton (SPAR, Shank and Homer, Ehlers, 2002), Rho GTPases (Nakayama et al, 2000), profilin (Ackermann and Matus, 2003), drebrin (Hayashi and Shirao, 1999) as well as other signaling molecules (Syndecan-2 (Couchman, 2003), Ephrin B (Penzes et al, 2003), Oligophrenin-1 (Govek et al, 2004), and Kalirin-7 (Penzes et al, 2001)). From these studies, it becomes clear that the regulation of the cytoskeletal structure of the dendritic spine is necessary for the establishment and maintenance of a mature synaptic connection.
There is increasing interest in characterizing the role of the synapse during neuropathology conditions. In Alzheimer's disease, the functional status of synaptic connections correlates well with overall neuronal health. Synaptic pathology precedes cell death in Alzheimer's disease and is a clinical hallmark of neuropsychiatric diseases (Gasic and Nicotera, 2003). Similarly, dramatic and rapidly reversible structural changes are observed in dendritic spine morphology after excitotoxic or hypoxic stress (Hasbani et al, 2001; Park et al, 1996). It was observed that structural (Park et al, 1996) and biochemical conditions (Martone et al, 1999) at the synapse rapidly return to normal following the stressor. However, characterization of postsynaptic densities in vivo after transient ischemia revealed morphological and biochemical alterations that persisted for up to 24 h (Martone et al, 1999).
Previous studies on the effect of cerebral ischemia on synaptosomal morphology and metabolism have indicated that the synapse is highly susceptible to ISC damage (Pastuszko et al, 1982; Rafalowska et al, 1980; Sulkowski et al, 2002). Processes that occur at the synapse have a great requirement for energy, necessitating the localization of numerous mitochondria proximal to the synaptic bouton. Acute hypoxia has been shown to decrease metabolic activity in synaptosomes (Rafalowska et al, 1980) as well as the rate of neurotransmitter re-uptake (Pastuszko et al, 1982). Additionally, ischemia induces a marked depletion of synaptic vesicles as well as an increase in the number of damaged mitochondria that persists for 24 h (Sulkowski et al, 2002).
Synaptic function is positively correlated with neuronal function (Gasic and Nicotera, 2003) and viability (Deisseroth et al, 2003). Furthermore, synaptic structure and function is extraordinarily sensitive to cerebral ischemia (Hasbani et al, 2001; Park et al, 1996). Lastly, synaptic signals are important determinants of neuronal fate during pathological conditions (Mattson et al, 2001). Therefore, as synaptic cell adhesion molecules are key regulators of synaptic structure and function, they are likely to play an important role in mediating the consequences of cerebral ischemia on injured neurons. The aim of this study was to examine the expression of a subset of dendritic spine (synaptic plasticity) regulators after cerebral ischemia in an effort to correlate synaptic structure to neuronal viability.
The key findings in this paper are that synaptosomes isolated from ISC mouse brain exhibited morphological alterations, which were consistent with ischemia-reperfusion injury (depletion of synaptic vesicles and mitochondrial abnormalities). We found that there were significant differences in the expression of certain members of the cadherin/catenin system that are consistent with a dysregulation of synaptic integrity. These results are consistent with a dysregulation of synaptic adhesion after cerebral ischemia that precedes the onset of ISC neuronal death.
Materials and methods
Animal Care
A local committee for the Canadian Council on Animal Care approved all procedures using mice. The C57B mice were purchased from Charles River (Canada, St Constant, Province of Quebec). Under temporary isoflurane anesthesia, the mice (20 to 23 g) were subjected to occlusion of the left middle cerebral artery (MCA) using an intraluminal filament as described previously (MacManus et al, 1999). After 1 h of ischemia, the animals were briefly reanesthetized, the filament withdrawn and wounds sutured. Body temperatures were recorded during and after ischemia and a behavioral score (turning behavior when picked up by tail) assigned on recovery from anesthetia. After 3, 6, or 20 h of reperfusion mice were briefly anesthetized with isoflurane and the brain rapidly excised and dissected on ice.
2,3,5-Triphenyltetrazolium Chloride Staining
Middle cerebral artery occlusion-treated mouse brains were quickly removed, after a 20 h period of reperfusion, and placed on ice. Two millimeters of coronal sections were obtained by sectioning the cooled brains in a mouse brain mold. The sections were incubated in 10 mL of 2% 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich Canada, Oakville, ON, Canada) for 15 mins at room temperature. The sections were removed from the 2,3,5-triphenyltetrazolium chloride solution and photographed. Ischemic regions were identified by the absence of staining on the ipsilateral side of the coronal sections.
Synaptosome Preparation
Contralateral (CT) and ISC hemispheres from three mice were manually homogenized in 2 mL of HM buffer (0.32 mol/L sucrose, 1 mmol/L ethylenediaminetetraacetic acid, 0.25 mmol/L dithiothreitol, 1 U/mL RNasin; Promega, Madison, WI, USA) using a dounce homogenizer, 800g 13 strokes at 4°C. The homogenates were centrifuged (1,000g for 10 mins at 4°C) and the supernatant retained and re-spun as before. The supernatant was transferred to 2 mL polycarbonate tubes and centrifuged at 20,000g for 20 mins at 4°C. The resultant pellet was resuspended in 2 mL HM buffer using a dounce homogenizer. Discontinuous sucrose/percoll gradients were prepared by layering 2 mL each, in order, of 25 (percoll in HM buffer), 15, 10, and 3% into 10 mL polycarbonate centrifuge tubes. One milliliter of the sample was then layered on top of a gradient and centrifuged at 32,000g for 5 mins at 4°C. Five fractions were collected after centrifugation: F1, 3% percoll (cytoplasm); F2, interphase between 3 and 10% percoll (myelin); fraction 3, interphase between 10 and 15% percoll (small synaptosomes, myelin and mitochondria); fraction 3 (F4), interphase between 15 and 25% percoll (intact synaptosomes); F5, pellet (mitochondria). Fractions F2-F4 were made up to 3 mL with HM buffer and centrifuged at 65,000g for 10 mins at 4°C. The supernatant was removed and the pellet washed with 1 × phosphate-buffered saline (PBS), three times (each time spinning at 65,000g for 5 mins). The final pellet was resuspended in either HM buffer or 50 mmol/L Tris (pH 8.5) and 0.1% sodium dodecyl sulfate (SDS).
Transmission Electron Microscopy
Synaptosome fractions were fixed with 1.6% glutaraldehyde in 100 mmol/L sodium phosphate buffer (pH 7.4) for 1 h at room temperature. After washes with 100 mmol/L sodium cacodylate buffer (pH 7.2), the fractions were resuspended in 22% bovine serum albumin and pelleted by centrifugation at 900g for 8 mins. Pellets were then cut into 1 mm pieces, postfixed with 1% osmium tetroxide in 100 mmol/L sodium cacodylate buffer (pH 7.2) for 1 h at 4°C, stained en bloc with 2% aqueous uranyl acetate, dehydrated through graded concentrations of ethanol, and embedded in Spurr's epoxy resin. Ultrathin sections were cut and stained with lead citrate and imaged on a JEOL 1230 transmission electron microscope using ATM software.
Immunoelectron Microscopy
Synaptosome fractions were fixed with 2% paraformaldehyde and 0.5% glutaraldehyde in 100 mmol/L sodium phosphate buffer (pH 7.4) for 2 h at 4°C. After washes with 100 mmol/L sodium cacodylate buffer (pH 7.2), cell pellets were prepared as described above, dehydrated through graded concentrations of ethanol and embedded in LRWhite resin at 4°C. After polymerization in an oven at 50°C, ultrathin sections were cut, placed on nickel grids coated with Formvar, and then processed for immunocytochemistry. Sections were first blocked with 1% bovine serum albumin in PBS for 30 mins and then floated on drops of primary antibodies diluted in 1% bovine serum albumin: mouse monoclonal anti-glial fibrillary acid protein (Gfap) (1:20, clone GA-5; Neomarkers, Medicorp, Montreal, QC, Canada) or rabbit polyclonal anti-synaptophysin (Syp, 1:20; Stressgen, Assay Designs, Ann Arbor, MI, USA). After rinsing with PBS, sections were floated again on drops of secondary goat anti-mouse or goat anti-rabbit immunoglobulin 15 nm-gold-conjugated antibodies (1:100; EY Laboratories, Cedarlane Laboratories Ltd., Hornby, ON, Canada, USA). Incubation with primary antibodies was carried out at 4°C for 24 h and secondary antibodies at room temperature for 1 h, respectively. Sections were rinsed with distilled water, air-dried, and then counterstained with uranyl acetate and lead citrate. Negative controls consisted of omission of the primary antibodies. Images were captured on a JEOL 1230 transmission electron microscope using ATM software.
Catenin (Cadherin-Associated Protein) β-1 Immunoprecipitation
Synaptosomal protein samples (400 μg; F4 fraction) were resuspended at a concentration of 1 μg/μL in radioimmunoprecipitation assay buffer (50 mmol/L Tris—HCl, pH 7.5, 1 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L ethylene glycol bis(β-aminoethylether)-N,N,N′,N′,-tetraacetic acid, 1 mmol/L NaF, 1% NP40, 0.25% sodium deoxycholate, 0.1% SDS, and protease inhibitors (complete, ethylenediaminetetraacetic acid-free; Roche, Diagnostics, Laval QC, Canada). The samples were pre-cleared with 20 μL of Protein G + Agarose (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C for 1 h. The samples were centrifuged at 2,000g for 5 mins at 4°C and the supernatants were transferred to new tubes. Mouse anti-β-catenin antibody (2.5 μg; BD Bioscience, Mississauga, ON, Canada) was added to the supernatants and incubated at 4°C overnight. Subsequently, 30 μL of protein G + agarose was added and incubated for 2 h at 4°C. The samples were then centrifuged for 5 mins at 2,000g at 4°C and the supernatant transferred to new tube and retained for analysis. The pellet was then washed three times with 1 mL radioimmunoprecipitation assay buffer (with detergents) and once with radioimmunoprecipitation assay (without detergents). The pellet was then resuspended in 2 × sodium dodecyl sulfate—polyacrylamide gel electrophoresis sample buffer (0.031 mmol/L Tris, pH 6.8, 5% glycerol, 1% SDS, 1 mmol/L ethylenediaminetetraacetic acid, and 4% β-mercaptoethanol) and used in Western blotting.
Protein Detection by Western Blot
Equal amounts (10 μg) of synaptosomal protein were suspended in sample buffer (68 mmol/L Tris—HCl, pH 9.0, 2% SDS, 2% β3-mercaptoethanol, 0.01% Bromophenol blue, and 15% glycerol), heat denatured and subsequently fractionated using 10% sodium dodecyl sulfate—polyacrylamide gel electrophoresis (except for agrin (Agrn), which used 6% sodium dodecyl sulfate—polyacrylamide gel electrophoresis). The proteins were subsequently transferred to nitrocellulose (40 mA/minigel for 2 h) using a semi-dry transfer apparatus (Nova Blot, Amersham, GE Healthcare, Baie D'Urfé, QC, Canada). Immunoblots were performed using the following primary antibodies: rabbit anti-calreticulin (Calr, 1:4,000; Novus Biologicals, Littleton, CO, USA), mouse anti-Syp (1:2,000; Stressgen), mouse anti-Gfap (1:500; BD Bioscience), mouse anti-N-cadherin (Cdh2, 1:2,500; BD Bioscience), mouse anti-α-N-catenin (1:250; BD Bioscience), mouse anti-β-catenin (1:500; BD Bioscience), mouse anti-neuroligin 1 (Nlgn1, 1:1,000; Synaptic Systems, Goettingen, Germany), mouse anti-neuronal nuclear antigen (1:500, a gift from Dr RJ Mullen, University of Utah School of Medicine, Salt Lake City, UT, USA), mouse anti-Agrn-86 (1:250; Stressgen), rabbit anti-phospo-β-catenin (p-Ctnnb1, Ser33/37/Thr41; 1:1,000; Cell Signaling Technology, New England Biolabs Ltd., Pickering, ON, Canada), mouse anti-phospho-tyrosine (PY20, 1:500; Santa Cruz Technology). The blots were incubated with the primary antibody overnight at 4°C, followed by three washes of 5 mins each in PBS-T (PBS containing 0.2% and Tween-20). Blots were then incubated with a secondary antibody for 1 h at room temperature (1:5,000 donkey anti-rabbit immunoglobulin/horseradish peroxidase conjugate or 1:10,000 donkey anti-mouse immunoglobulin/horseradish peroxidase conjugate; Jackson ImmunoResearch, West Grove, PA, USA). Chemiluminescent detection was performed using the enhanced chemoluminescence plus kit (Amersham). Blots were exposed to film and scanned using a densitometer (model 300A; Molecular Probes, Invitrogen, Burlington, ON, Canada). Band densities were quantified using ImageJ software (vl.36b; http://rsb.info.nih.gov/ij/).
Semiquantitative Reverse Transcriptase-Polymerase Chain Reaction
Total RNA was isolated from CT and ISC hemispheres from one mouse were manually homogenized in Tri-Reagent LS® (Sigma-Aldrich, Oakville, ON, Canada) following the manufacturer's protocol. A260/280 readings were generally between 2.0 and 2.1 indicating high RNA purity. cDNA was synthesized from 5 μg of total RNA using Superscript II reverse transcriptase (Invitrogen, Burlington, ON, Canada) according to the manufacturer's protocol. The cDNA was then purified using Qiagen PCR purification kit and quantitated using the Oligreen method (Molecular Probes). Quantitative polymerase chain reaction was performed on an ABI Prism 7000 SDS in a 25 μL reaction containing 12.5 μL SYBR® Green PCR Master Mix, 2.5 μL primers (1.5 pmol/μL each), 10 pL cDNA (0.2 to 0.00625 and 0.15 ng/μL for standard curve and samples, respectively). Relative quantities were calculated using the standard curve method. Quantitative polymerase chain reaction optimized primers were designed using Primer Express 2.0 (Applied Biosystems, Streetsville, ON, Canada). Primers used are as follows: Cdh2 (GGCACGGTGTATGCTGTGAG, CTTTGCCTGCTCTGCAGTGA), Ctnna2 (AATGTGTGATTGCCCTGCAA, CTGTGCGATCCAGTGTGTCC), Ctnnb1 (TGCTGGTGACAGGGAAGACA, TGACGAAGAGCACAGATGGC), Nlgn1 (TTCCACACCAGTCACATCAGC, TTGCTTGGGATCATCCTGTTT), neurexin 1 (Nrxn1) (GAGTAGGTGGCCGTGAACCA, TGGACTCCCGAATCACCTCT), Agrn (CCGAGGGAAGCCACTTATGA, CTATCCCCTTCTCGCAGTGC), Actin (GGCGCTTTTGACTCAGGATT, GGGATGTTTGCTCCAACCAA).
Immunohistochemistry
Middle cerebral artery occlusion-treated animals were anesthetized with isoflurane before transcardial perfusion with fixative (4% paraformaldehyde, 0.1% glutaraldehyde, and 0.1 mol/L PBS, pH 7.4). The brains were removed, as described above, postfixed for 24 h and subsequently embedded in paraffin. Coronal brain sections were cut at a thickness of 7 μm and mounted on Superfrost®/Plus slides (Fisher Scientific Canada, Ottawa, ON, Canada). Deparafinization was conducted as follows: heat slides to 70°C for 10 mins, wash in xylene (3 × 5 mins), in 100% EtOH (5 mins), in 95% EtOH (5 mins), in dH2O (5 mins), and in PBS (5 mins). Permeabilization was performed as follows: the sections were placed in 10 mmol/L sodium citrate buffer (pH 6.0), which was subsequently brought to a boil, allowed to cool at room temperature for 1 h and washed in dH2O and PBS (5 mins each). Immunohistochemistry was conducted as follows: the sections were incubated in serum-free protein block (DakoCytomation, Mississauga, ON, Canada) for 1 h at room temperature before incubation with rabbit anti-p-Ctnnb1 (Ser33/37/Thr41; Cell Signaling Technology), diluted 1:25 in DakoCytomation antibody diluent, overnight at 4°C. The sections were then washed with PBS (5 mins × 3) before incubation with the secondary antibody (donkey anti-rabbit Cy3 (1:750 diluted in antibody diluent)) for 1 h at room temperature. The sections were then washed in PBS (5 mins × 3) and dH2O (2 mins) before mounting with Vectashield (Vector Laboratories, Burlington, ON, Canada) containing 5 μg/mL Hoechst 33258. Fluorescent images were obtained using an Axiovert 200M (Carl Zeiss Canada Ltd., Toronto, ON, Canada) inverted microscope.
Results
Characterization of Synaptosome Preparation
The efficacy and consistency of the MCAO surgeries were determined by 2,3,5-triphenyltetrazolium chloride staining (Figure 1A). The ipsilateral hemispheres exhibited substantial areas of infarct that consistently involved cortical and striatal regions, and in the majority of cases, the hippocampal regions were completely infarcted. Therefore, synaptosomes isolated from the ipsilateral hemisphere consisted mostly of infarcted tissue as well as peri-infarct and noninfarcted tissue.
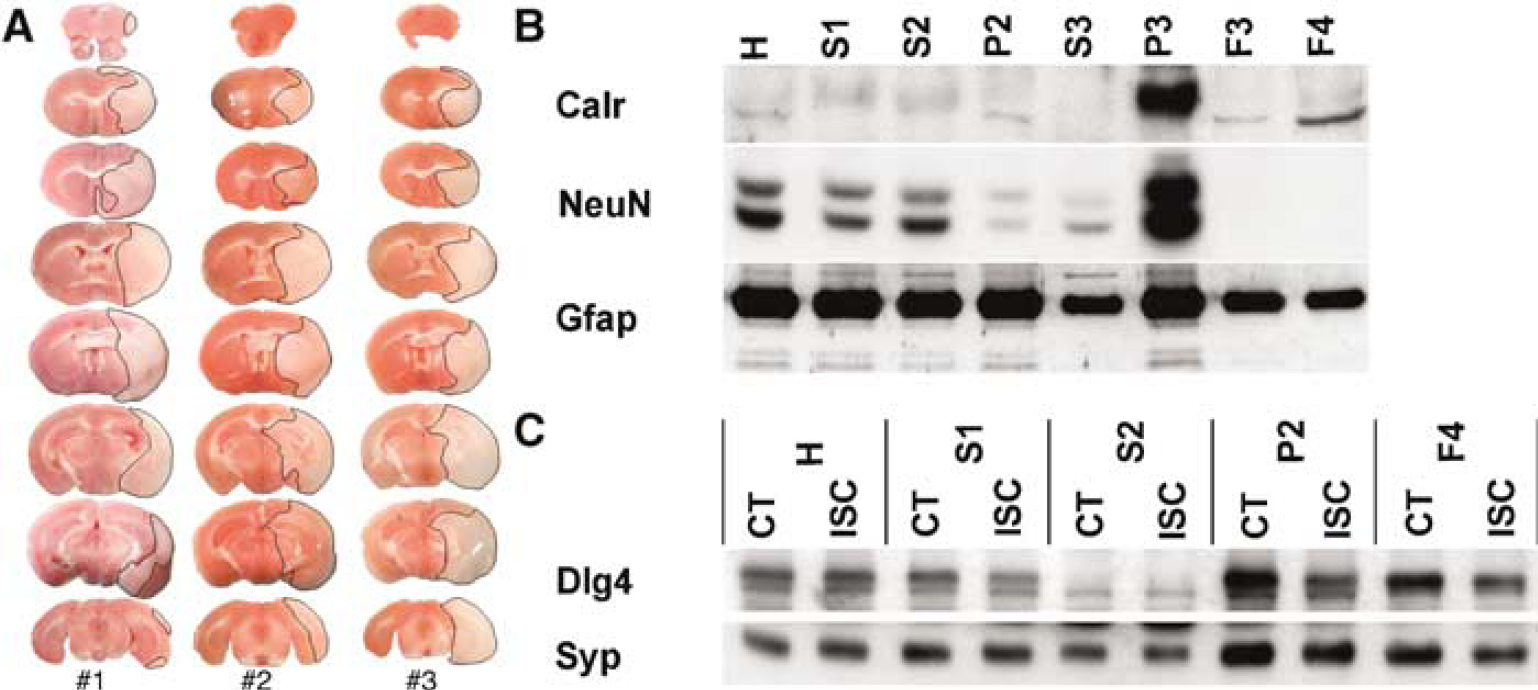
Markers of subcellular constituents were used to characterize the synaptosome preparation used in this study. Immunoblot experiments (Figure 1B) indicated that the F4 synaptosomes used throughout this study were free of nonsynaptic neuronal contaminants such as nuclei and endoplasmic reticulum. Even though there was a reduction in the amount of Gfap present in F4 synaptosomes, the persistence of glial contamination in synaptosomal preparations is consistent with observations made in previous publications (Mattson et al, 1998; Matus et al, 1980). Immunoblot experiments (Figure 1C) also showed that F4 synaptosomes were enriched in both presynaptic (Syp) and postsynaptic (discs, large homolog 4 (Psd-95)) markers. The yields of CT and ISC synaptosomal proteins (F4) were not significantly different (208.7 ± 14.24 and 194.1 ± 19.72 μg, respectively; n = 9).
Electron microscopy was used to evaluate the effect of cerebral ischemia on synaptosome morphology. Synaptosomes isolated from CT and ISC tissue exhibited features consistent with synaptic junctions (Figures 2A and 2B), namely mitochondria, synaptic vesicles, and postsynaptic densities. Contralateral synaptosomes were typically densely packed with synaptic vesicles and possessed mitochondria that were shaped as described in the literature (Figure 2C). Similar synaptosomes were also observed in ISC samples. However, the ISC samples also possessed synaptosomes that exhibited abnormal morphological characteristics. These abnormalities included sparse packing of synaptic vesicles, abnormally shaped mitochondria, and the absence of electron dense material in certain synaptosomes with sparse synaptic vesicles (Figure 2D) similar to previously described features (Sulkowski et al, 2002). In ISC samples, 55% of the synaptosomes exhibited abnormal morphology, compared with 7% in CT samples. The proportion of abnormal synaptosomes correlates well with the size of the infarcted regions (Figure 1A). Lastly, CT and ISC synaptosomes were ≈650 nm in diameter, consistent with previous observations (Sulkowski et al, 2002).
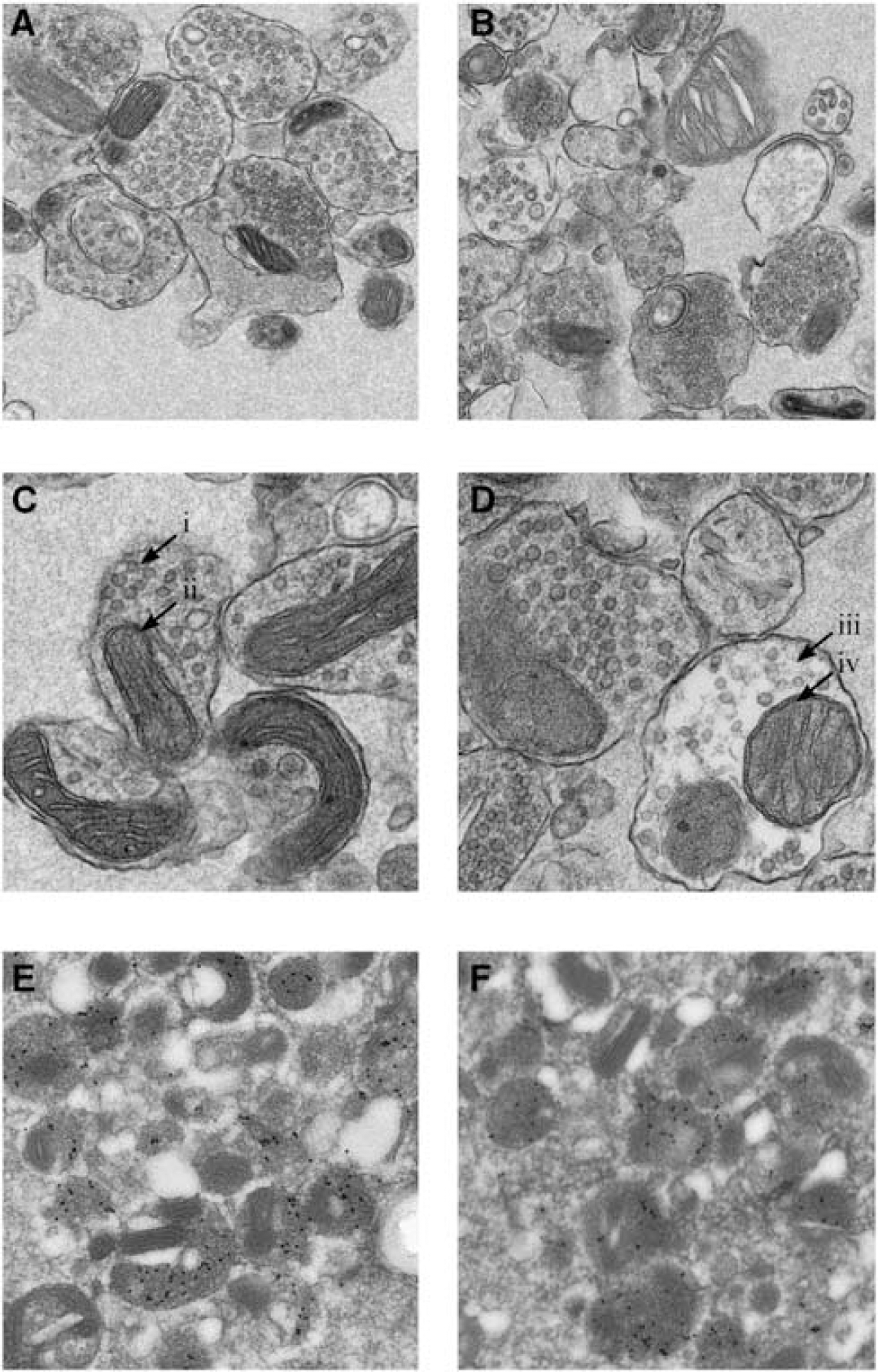
Electron microscopy reveals that cerebral ischemia induces morphological alterations in F4 synaptosomes isolated from mouse forebrain (20 h reperfusion). Synaptic vesicles and mitochondria are prominent features of F4 synaptosomes from (
Immunoelectron microscopy experiments were also conducted to confirm that the constituents of synaptosomes were of synaptic origin. The majority of synaptosomes from CT (Figure 2E) and ISC (Figure 2F) tissue exhibited extensive labeling with anti-Syp, confirming that the synaptic vesicles observed in Figures 2A to 2D were properly identified. Immunoelectron microscopy experiments using anti-Gfap resulted in very sparse deposition of gold particles, indicating that the glial cell contamination was negligible, in both CT and ISC synaptosomes (data not shown).
Ischemia Reduces Synaptic Adhesion-Related Gene Expression
The effects of cerebral ischemia on the expression of genes involved in synaptic cell adhesion, before the onset of ISC neuronal cell death, were determined using quantitative polymerase chain reaction (Figure 3). Specifically, components of the Cdh2 and the Nrxn1/Nlgn1 systems were assessed. For comparison purposes, the expression of Agrn was assessed as well. Agrn is a component of the basal lamina that is responsible for the organization of the neuromuscular junction. In the central nervous system, Agrn is membrane-bound and may function in a manner that is similar to synaptic adhesion molecules.
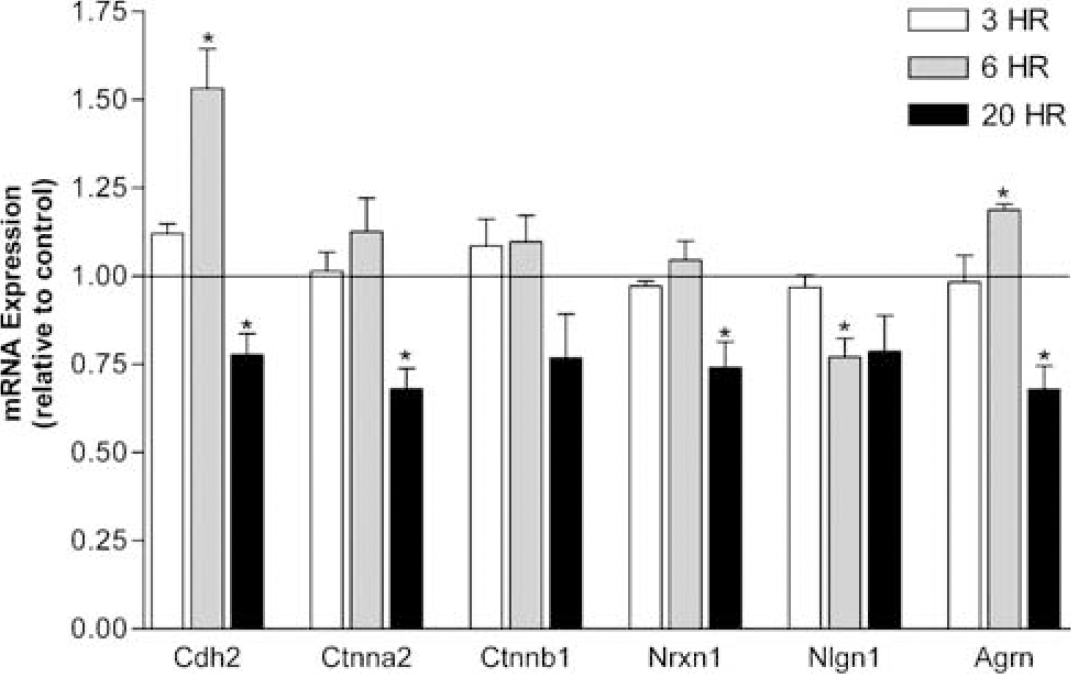
Cerebral ischemia induces alterations in the mRNA levels of synaptic adhesion molecules in mouse brain. Synaptic adhesion molecule expression was determined by quantitative polymerase chain reaction in mouse forebrain isolated from mice treated with 1 h MCAO followed by 3, 6, and 20 h of reperfusion. *P < 0.05; one sample t-test, n = 3.
At 3 h after ischemia, no significant changes in gene expression were observed. This indicates that the there is no immediate early response to ischemia for this class of genes. However, at 6 h after ischemia, Agrn expression was moderately elevated (19%; Figure 3). The expression of Cdh2 was significantly elevated (53%), whereas the expression of Ctnna2 and Ctnnb1 remained unchanged (Figure 3). While the expression of Nrxn1 was unaffected at 6 h after ischemia, the expression of Nlgn1 was significantly decreased by 23% (Figure 3). At 20 h after ischemia, all of the genes displayed a trend toward reduced expression (Figure 3). Cdh2 and Ctnna2 were both significantly reduced and Ctnnb1 was reduced to a similar extent (Figure 3). Nrxn1 and Nlgn1 were both reduced to a similar extent (Figure 3). This indicates that the expression of both synaptic adhesion systems was reduced after cerebral ischemia, indicating a reduction in the capacity for post-ISC synaptic adhesion before the onset of ISC neuronal death. Lastly, Agrn expression was significantly decreased at this time point as well.
Ischemia Induces Dysregulation of Synaptic Adhesion-Related Proteins
Plastic events at the synapse are regulated in a localized manner. Therefore, we examined the effects of cerebral ischemia on synaptically localized proteins using Western blot. Specifically, we examined a subset of those proteins that are involved in regulating synaptic structure—function relationships. Significant reductions in Cdh2 and Ctnna2, but not Ctnnb1 were observed at 3 h reperfusion (Figures 4A to 4C). Cdh2 remained decreased at 6 h and was not fully restored by 20 h (Figure 4A). Similarly, Ctnna2 was reduced at 6 h and remained significantly decreased at 20 h (Figure 4B). Conversely, Ctnnb1 exhibited a tendency toward reduction at 6 and 20 h, which did not reach significance (Figure 4C). However, levels of p-Ctnnb1 were significantly increased in ISC synaptosomes at all time points examined (Figure 4D). Expression of p-Ctnnb1 was markedly increased at the early time points with peak expression at 6 h after ischemia. Levels of p-Ctnnb1 remained elevated at 20 h after ischemia but appeared to be approaching more normal levels at this time point. These results indicate that Cdh2-mediated synaptic adhesion was adversely affected by cerebral ischemia. Moreover, this dysregulation of cadherin-mediated synaptic adhesion was evident shortly after the ISC insult.
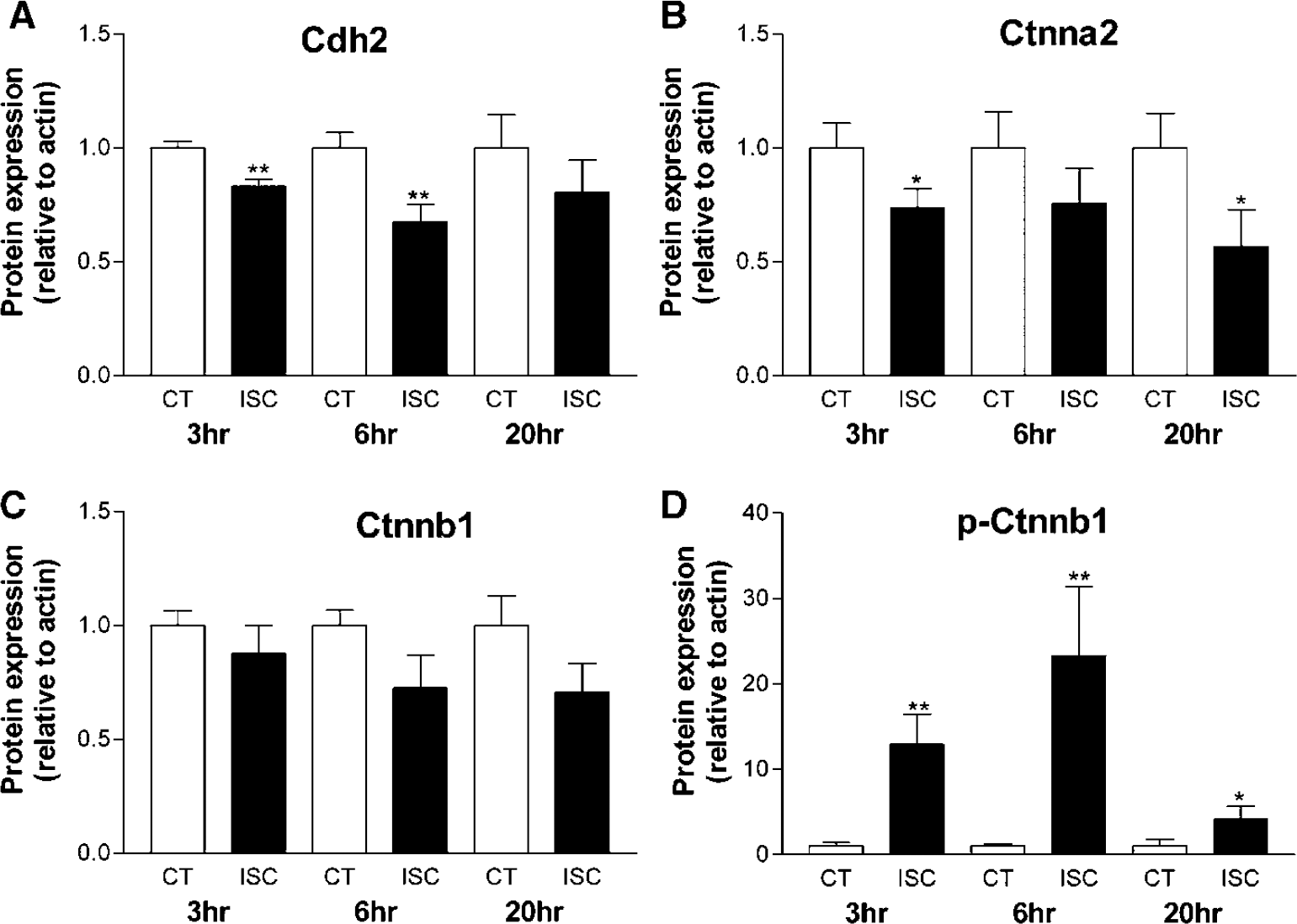
Cerebral ischemia induces dysregulation of synaptic cadherin-mediated adhesion. (
Additionally, we examined the effect of cerebral ischemia synaptic levels of the Nrxn—Nlgn adhesion molecules. We failed to detect Nrxn1 in Western blots due to limitations of the antibodies available for this molecule. Nlgn1 levels were found to decline with time in ISC synaptosomes (Figure 5A). The dramatic decrease in synaptic Nlgn1 levels at 6 and 20 h (51 and 66%, respectively) indicates that this adhesion system is highly sensitive to ISC perturbations. Post-ISC synaptosomal levels of the potential adhesion molecule Agrn were examined for comparison purposes. Figure 5B shows that cerebral ischemia had no significant effect on the expression of synaptosomal Agrn with up to 20 h of reperfusion.
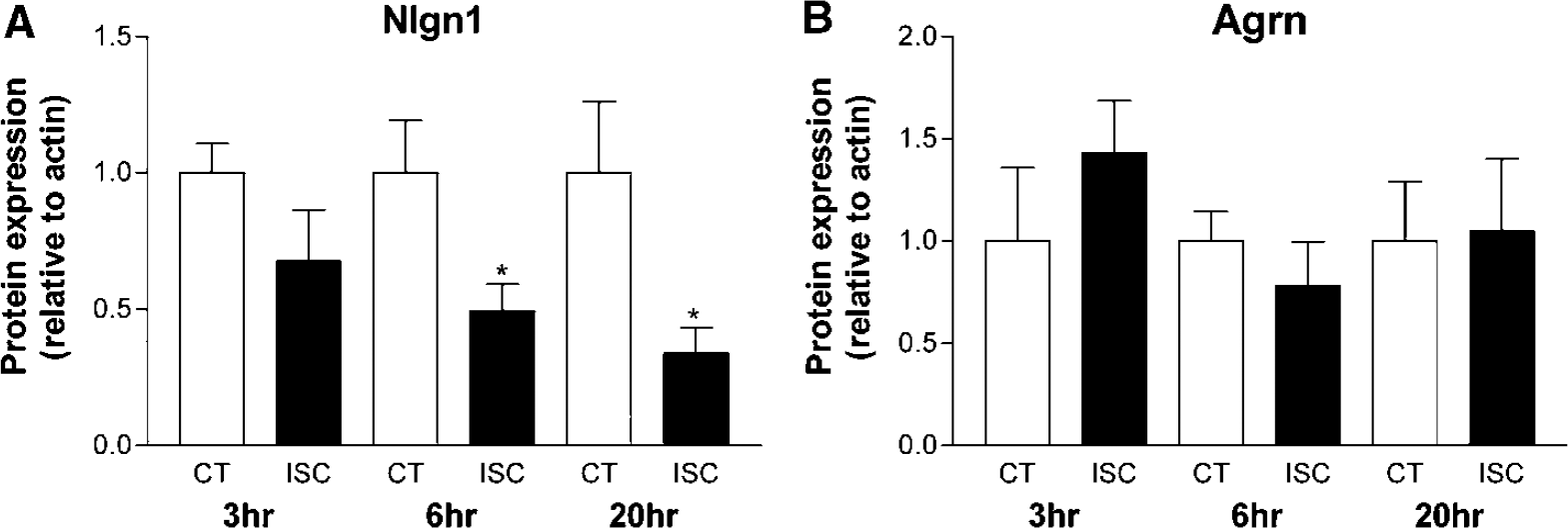
Cerebral ischemia induces dysregulation of synaptic Nlgn1, but not Agrn, mediated adhesion. (
Cerebral Ischemia-Induced Expression of Phospho-Ctnnb1 in Cortical Neurons
The effect of cerebral ischemia on p-Ctnnb1 distribution in the brain was assessed using immunohistochemistry in tissue at 3 h after ischemia (Figure 6). Figure 6A shows that p-Ctnnb1-positive cornu ammonis 1 (CA1) and thalamus neurons are few in number on the CT side of the brain. Furthermore, there is a lack of staining in the CA1 neuropil (stratum radiatum and stratum lacunosum-moleculare) as well as in the stratum moleculare and stratum oriens regions of the CT CA1. Figure 6B shows prominent p-Ctnnb1 staining in the ISC CA1 and thalamus. Phospho-Ctnnb1-positive neurons in the CA1 stratum pyramidale were plentiful, and staining in the neuropil was markedly increased. Additionally, no alterations in p-Ctnnb1 staining were observed in the stratum oriens and stratum moleculare in the ISC CA1. Neuronal death was assessed using Hoechst staining. No ischemia-induced alterations in CA1 nuclear number or morphology were observed using Hoechst staining at 3 h after ischemia (Figures 6A and 6B). Increased p-Ctnnb1 expression in the ISC caudate and cortex was also observed (data not shown). Image analysis of the ISC CA1 using z-sectioning shows that p-Ctnnb1 was present in the cell bodies and fiber tracts as well as in the nuclei (Figure 7). Phospho-Ctnnb1-positive (a) and -negative (b) neurons are indicated in Figure 7, with the cut lines clearly showing the nuclear localization of p-Ctnnb1. Lastly, the effect of cerebral ischemia on tyrosine phosphorylation of Ctnnb1 was determined (Figure 8). Probing of synaptosomal Ctnnb1 immunoprecipitates with anti-phospho-tyrosine indicated a very weak level of Ctnnb1 tyrosine phosphorylation. Furthermore, cerebral ischemia did not appear to have an effect on the level of tyrosine phosphorylation of Ctnnb1 (Figure 8).
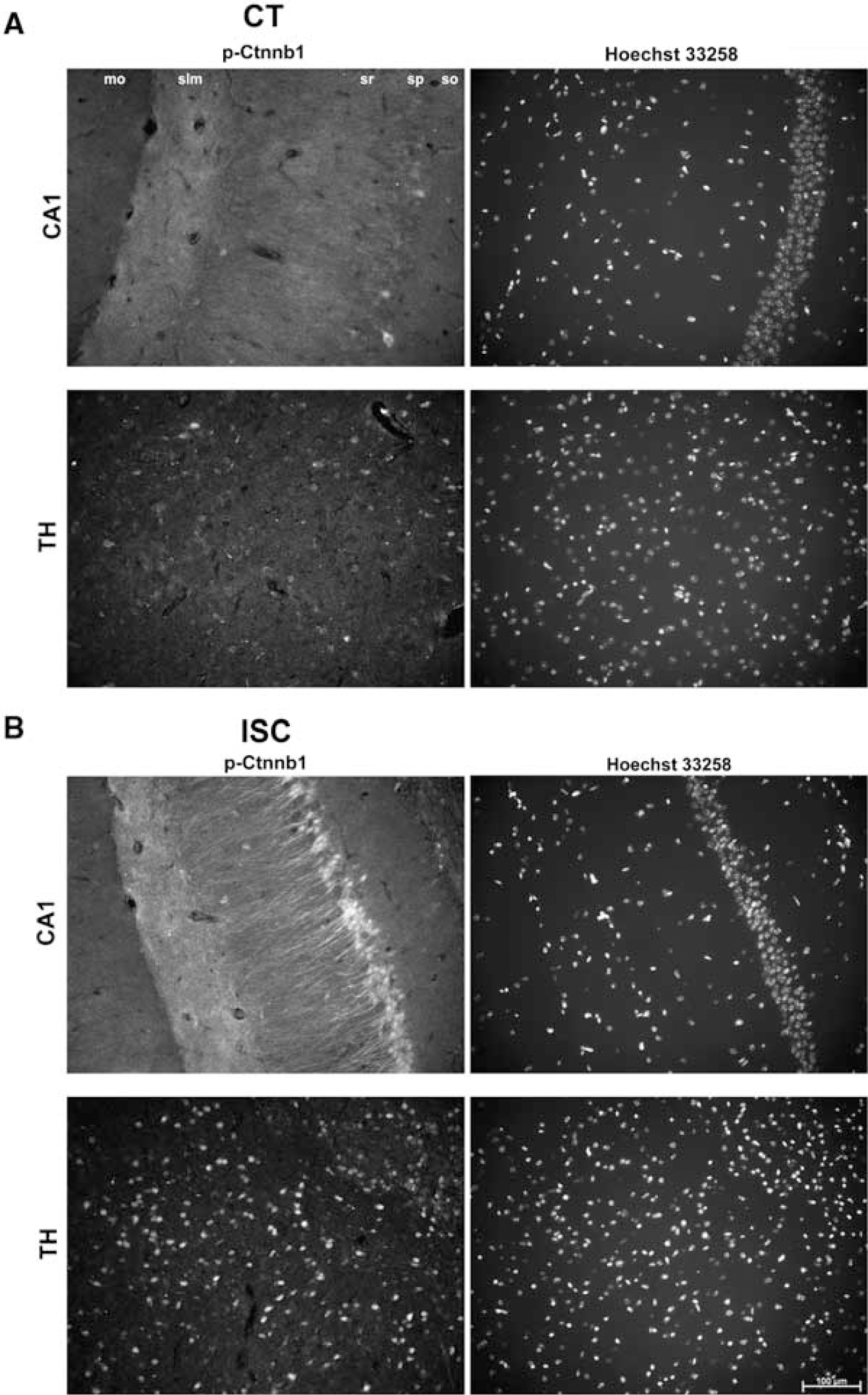
Cerebral ischemia induces p-Ctnnb1 expression in hippocampal CA1 and thalamic neurons 3 h after ischemia. (
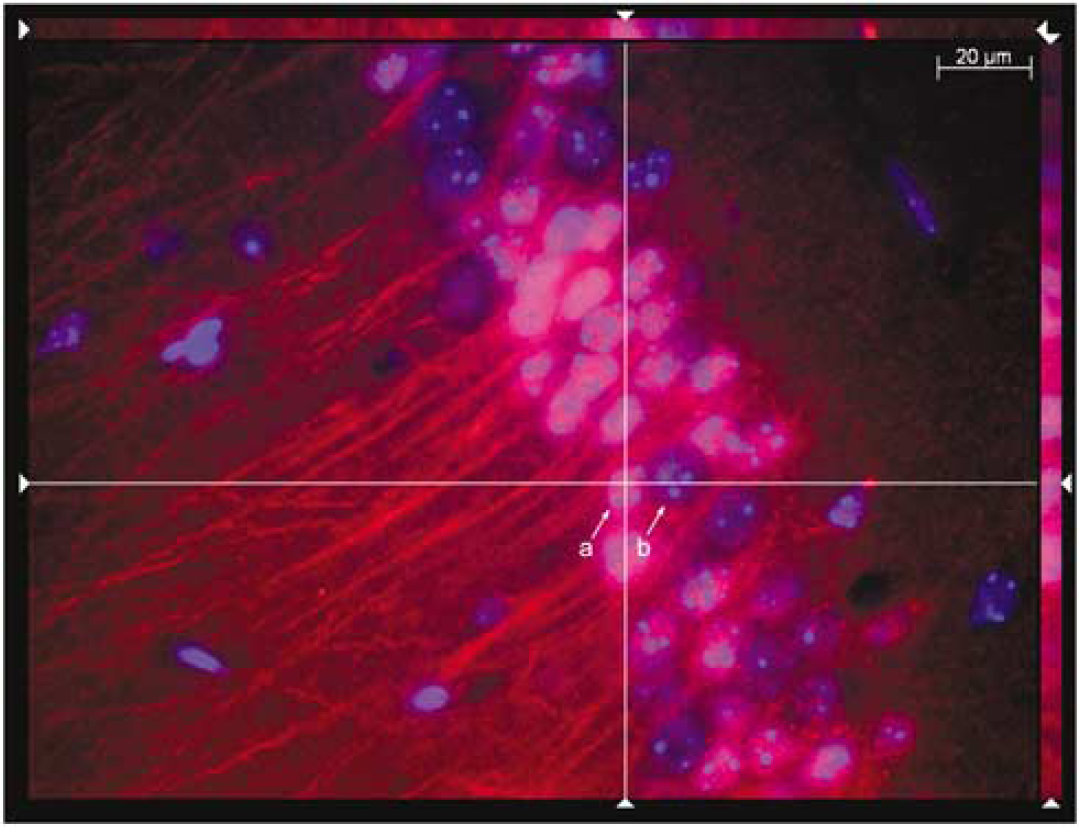
Cerebral ischemia-induced p-Ctnnb1 expression is localized in hippocampal CA1 neuronal nuclei 3 h after ischemia. Z-section imaging of the ISC hippocampus shows that p-Ctnnb1 (Cy3, red) and Hoechst 33258 (blue) staining are colocalized in CA1 neurons (intersection of cut lines). (a) p-Ctnnb1-positive nuclei and (b) p-Ctnnb1-negative nuclei.
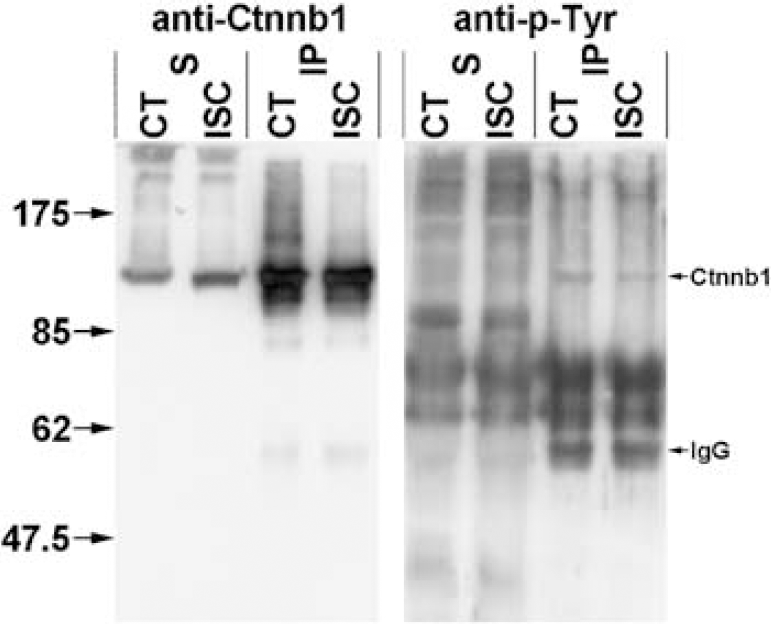
Ctnnb1 immunoprecipitation indicates that cerebral ischemia does not alter tyrosine phosphorylation of synaptosomal Ctnnb1. Western blotting with anti-Ctnnb1 shows the majority of Ctnnb1 (92 kDa) is present in the precipitate (IP) compared with the supernatant (S). Western blotting with anti-phopho-tyrosine (anti-p-Tyr) shows a very weak band corresponding to Ctnnb1 in the precipitate (IP). The presence of immunoglobulin is indicated in the figure.
Discussion
The susceptibility of neurons to hypoxia-induced damage is a well-established phenomenon that forms the basis of stroke pathology. In the MCAO model used in this study, transient cerebral ischemia is known to induce delayed neuronal cell death approximately 36 to 48 h after the insult (Kirino, 2000). The synapse appears to be particularly sensitive to hypoxia, showing structural (Hasbani et al, 2001; Park et al, 1996) and biochemical (Rafalowska et al, 1980) alterations immediately after hypoxia. Indeed, the biochemical alterations that occur at the synapse may initiate a sequence of events that propagate to the cell body and initiate ISC neuronal cell death in a process termed ‘synaptic apoptosis’ (Mattson and Duan, 1999). Because of this, we hypothesized that rapid degradation of the integrity of synaptic contacts after cerebral ischemia is possible. Therefore, we investigated the dynamics of molecules that regulate synaptic formation and maintenance. We have showed that cerebral ischemia disrupts synaptosomal adhesion molecule function before the onset of ISC neuronal death. These observations are consistent with the hypothesis that synaptic adhesion molecules participate in the phenomenon of synaptic apoptosis.
Numerous synaptically localized molecules can regulate synaptic plasticity. Our current knowledge of the mechanisms involved in the regulation of synaptic plasticity emphasizes the importance of the structure/function relationship between synaptically localized molecules and their ability to interact with the actin cytoskeleton. Many of the factors that can regulate dendritic spine formation interact directly or indirectly with the actin cytoskeleton (Tada and Sheng, 2006). Because of this, we examined the effects of cerebral ischemia on two important classes of synaptic adhesion molecules, the cadherin/catenin and Nrxn/Nlgn systems, which interact with the actin cytoskeleton. Furthermore, we focused on post-ISC events that preceded the onset of ISC neuronal cell death so that we could correlate synaptically localized alterations with crucial events, which may initiate damaging signals that could be involved in the subsequent cell death processes.
Here we examined gene expression in mouse brain and protein levels in synaptosomes, enabling comparisons of cellular expression with that of synaptic expression. For the components of the cadherin/catenin system, we found that the reduction in synaptic levels of Cdh2 and Ctnna2 at 3 h was unrelated to an alteration in gene expression (Figures 3 and 4A and 4B). This indicates that at 3 h, the decreased synaptic levels of these proteins is not due to a decrease in gene expression but probably due to either degradation of the proteins or sequestration of the proteins away from synaptic locations. Ctnna2 is a cytoplasmic protein that could easily be sequestered from the synaptic environment. Cdh2, however, is membrane-bound and would require an endocytotic process to be sequestered from the synaptic junction. At 6 h, synaptic Cdh2 levels remained significantly depressed, whereas its gene expression was significantly elevated. This would indicate that the cells were engaged in a compensatory response, which would account for the return of synaptic Cdh2 to more normal levels at the 20 h time point (Figure 4A). Conversely, mRNA levels of Ctnna2 at 6 h were not significantly altered, and the synaptic protein levels remained decreased. At 20 h, Ctnna2 mRNA and synaptic protein levels were significantly decreased, indicating a continued ischemia-induced deficit in Ctnna2's synaptic and cellular presence.
Cerebral ischemia did not have a significant effect on Ctnnb1 gene expression or synaptic protein levels (Figures 3 and 4C). However, this cannot be interpreted as a lack of cerebral ischemia effect on Ctnnb1 function. Previous studies have shown that cerebral ischemia produces an increase in the amount of p-Ctnnb1, presumably resulting from the activation of GSK3β, which results in its nuclear translocation of Ctnnb1 (Zhao et al, 2005). The relationship between Cdh2, Ctnnb1, and Ctnna2 is such that Ctnnb1 interacts with Cdh2 and Ctnna2, forming an interface between these components (Lilien and Balsamo, 2005). Tyrosine dephosphorylation of Ctnnb1 in neurons is correlated with its translocation to dendritic spines (Dunah et al, 2005; Murase et al, 2002). Conversely, phosphorylation of Ctnnb1 results in its dissociation from cadherin and a loss of cadherinbased adhesion (Lilien and Balsamo, 2005) as well as reduced localization at the synapse (Dunah et al, 2005). Furthermore, dissociation of Cdh2 and Ctnnb1 results in the degradation of Cdh2. Here, we found that post-ISC (3 to 20 h) synaptosomes are markedly enriched in serine/threonine-phosphorylated Ctnnb1 (Figure 4D). Conversely, cerebral ischemia had no effect on tyrosine phosphorylation of Ctnnb1 (Figure 8). Importantly, the greatest increases in p-Ctnnb1 were observed at 3 and 6 h, with the levels at 20 h after ischemia returning to normal. These observations indicate that cerebral ischemia induces a disruption of the cadherin/catenin-mediated synaptic adhesion system. Dysregulation of synaptic Cdh2, Ctnna2, and p-Ctnnb1 was observed shortly after the onset of reperfusion (3 h) and persisted until 20 h. While no significant changes in the levels of Ctnnb1 were observed, others have showed that cerebral ischemia induced the phosphorylation of Ctnnb1 as early as 5 h after ischemia (Zhao et al, 2005). Our findings confirm and extend this, indicating that phosphorylation of synaptically localized Ctnnb1 occurs as early as 3 h after cerebral ischemia and persists in a phosphorylated state for up to 20 h. Furthermore, immunohistochemistry experiments confirmed that p-Ctnnb1 levels were increased in the ISC hippocampus and thalamus (Figure 6). Increased p-Ctnnb1 staining was observed in the neuropil as well as in CA1 pyramidal cell bodies. Phospho-Ctnnb1-positive ISC neurons exhibited nuclear staining as well as staining in the fiber tracts (Figure 7). These results indicate that cerebral ischemia-induced disruption of cadherin-mediated synaptic adhesion precedes and persists during the onset of ISC neuronal cell death.
The Nrxn/Nlgn adhesion system also plays an important role in the establishment and maintenance of central nervous system synapses (Dean and Dresbach, 2006; Scheiffele et al, 2000). Nlgn-1 and −2 are localized primarily in excitatory and inhibitory synapses, respectively. In this study, we focused on Nlgn-1 because of the established role of N-methyl-
Agrn is an essential component of the basal lamina within the neuromuscular junction, where its organizational role is well understood (Nitkin et al, 1987). We detected Agrn in our synaptosomal preparations confirming its presence in the central nervous system synapse. The role of Agrn in the central nervous system synapse is not well understood, but may play a role in modulating synaptic adhesion by interacting with adhesion molecules and other synaptic constituents such as α-dystroglycan (Hopf and Hoch, 1996). Cerebral ischemia caused an elevation in Agrn expression at 6 h of reperfusion, followed by a reduction at 20 h of reperfusion (Figure 3). However, cerebral ischemia did not significantly alter synaptosomal Agrn protein levels between 3 and 20 h of reperfusion (Figure 5B). The lack of correlation between these observations indicates that the expression of Agrn is not likely to be limited to neurons after cerebral ischemia. A recent report has indicated that astrocytes express the soluble form of Agrn (Tournell et al, 2006), which may account for the apparent disagreement between the gene expression and synaptic protein levels. Furthermore, Solé et al (2004) observed Agrn staining in glia after cerebral ischemia. Incidentally, we did not observe the ischemia-induced 135 kDa Agrn fragment reported by Solé et al (2004) in ISC synaptosomes. This may be due to a lack of MMP3 degradation of synaptic Agrn in the 3 to 20 h time frame or loss of the nonmembrane-associated Agrn fragment during the synaptosomal isolation procedure. Regardless, the present data indicates that synaptic Agrn levels are not adversely affected by cerebral ischemia, suggesting that Agrn's role in mediating synaptic apoptosis is limited.
The results that are presented here support the contention that synaptic function is an important determinant of neuronal viability following periods of ISC injury. We have showed that systems of synaptic adhesion are adversely affected by cerebral ischemia and that the onset of effects on synaptic adhesion molecules precedes the onset of ISC neuronal cell death. These observations are consistent with the hypothesis that synaptic adhesion molecules participate in the phenomenon of synaptic apoptosis. It is possible that the observed disturbance in Cdh2 function (decreased levels of Cdh2 and Ctnna2 as well as the phosphorylation of Ctnnb1) has a negative impact on neuronal viability, as Cdh2 agonists have been shown to promote neuronal survival (Skaper et al, 2004). The neuroprotective effect of Cdh2 is dependent on fibroblast growth factor signaling, and therefore the observed ischemia-induced degradation of Cdh2 function may adversely affect fibroblast growth factor-mediated neuroprotection as well. It appears that the immediate neuronal response to cerebral ischemia is to disengage from intraneuronal signaling by weakening synaptic adhesion. At present, it is not clear whether this is done in an attempt by the cell to prevent cell death or an initiating event in the cell death process.
Footnotes
Acknowledgements
We thank Jack Daoust and Dr Craig Bihun and the animal care team for provision of mice. We also thank Dr RJ Mullen for his gift of anti-NeuN. Lastly, we thank Dr Jennifer Hill for her helpful discussions.