
Abstract
Insulin-like growth factor (IGF-1) markedly increases myelination and glial numbers in white matter after ischemia in near-term fetal sheep; however, it is unclear whether this is due to reduced cell loss or increased secondary proliferation. Brain injury was induced in near-term fetal sheep by 30 minutes of bilateral carotid artery occlusion. Ninety minutes after the occlusion, fetuses were given, intracerebroventricularly, either a single dose of IGF-1 (either 3 or 30 μg), or 3 μg followed by 3 μg over 24 hours (3 + 3 μg). White matter was assessed 4 days after reperfusion. Three micrograms, but not 30 μg of IGF-1 prevented loss of oligodendrocytes and myelin basic protein density (P < 0.001) compared to the vehicle-treated ischemia controls. No additional effect was observed in the 3 + 3 μg group. IGF-1 treatment was associated with reduced caspase-3 activation and increased glial proliferation in a similar dose-dependent manner. Caspase-3 was only expressed in oligodendrocytes that showed apoptotic morphology. Proliferating cell nuclear antigen co-localized with both oligodendrocytes and astrocytes and microglia. Thus, increased oligodendrocyte numbers after IGF-1 treatment is partly due to suppression of apoptosis, and partly to increased proliferation. In contrast, the increase in reactive glia was related only to proliferation. Speculatively, reactive glia may partly mediate IGF-1 white matter protection.
The causes and treatment of white matter injury after hypoxia–ischemia at term has been relatively neglected, but it is now known to be a significant contributor to developmental neuropathology and sequelae (e.g., epilepsy, cerebral palsy, and neurobehavioral deficits) (Marin-Padilla, 1997; Hack, 2000; Petersson et al., 2002). Contrary to early reports, recent experimental evidence has now shown that cerebral white matter, in both adult and immature brains, is highly vulnerable to ischemia (Pantoni et al., 1996; Ikeda et al., 1998; Petty and Wettstein, 1999; Guan et al., 2001). We have recently reported that insulin-like growth factor I (IGF-1) can markedly reduce postischemic demyelination and loss of bioactive oligodendrocytes (Guan et al., 2001). The mechanisms mediating this protection remain unclear.
The prevailing hypothesis is that cell rescue with exogenous IGF-1 is primarily mediated by its potent receptor mediated antiapoptotic effects (Yin et al., 1994; Galli et al., 1995; Parrizas et al., 1997; Mason et al., 2000). Of particular relevance for white matter injury, IGF-1 is a potent mitogen and survival factor for oligodendrocytes (Barres et al., 1992). The window of opportunity for neuroprotection with IGF-1 after hypoxic–ischemic injury appears to be short (Guan et al., 2000), suggesting that its actions are limited to early events after ischemic injury (Mason et al., 2000), although this has not been tested in developing white matter. Consistent with this hypothesis, apoptosis is a significant factor in white matter injury in the developing brain, both in the human infant and immature animal models (Edwards et al., 1997; Pulera et al., 1998; Guan et al., 2001; Yue et al., 1997). Although multiple pathways are likely to be involved in such postischemic apoptosis, caspase-3, one of the family of cysteine proteases, is reported to play a crucial role in immature brains (Hu et al., 2000; Wang et al., 2001).
However, IGF-1 is also a potent mitogen for a variety of cell types, particularly the oligodendrocyte lineage (Goddard et al., 1999; McMorris and Mckinnon, 1996), but also astrocytes (Chernausek, 1993) and, as recently shown, microglia (O'Donnell et al., 2002). Interestingly, we found a marked increase in reactive glia, including reactive microglia in association with IGF-1 white matter protection in the fetal sheep (Guan et al., 2001). This raised the speculation that part of the protective effect of IGF-1 may have been indirectly mediated through these reactive glia, and this is particularly striking because microglia are most often suggested to have a negative effect on survival of other cell types (O'Donnell et al., 2002). However, it was unclear whether this increase in white matter glial phenotypes was due to promotion of glial survival and/or glial proliferation, or indeed a nonspecific cellular response to IGF-1 treatment.
Therefore, in the present study we examined the relationship between dose and timing of IGF-1 delivery and white matter protection after transient cerebral ischemia in the near-term fetal sheep. The effects of IGF-1 on postischemic white matter apoptosis and proliferation after 4 days' recovery from ischemia were assessed.
METHODS
Animals and surgery
All procedures were approved by the Animal Ethics Committee of the University of Auckland, New Zealand. Romney-Suffolk fetal sheep, while under general anesthesia (2% halothane in O2), were instrumented at 117 to 124 days of gestation (term = 147 days) using sterile techniques. Ewes were given 5 mL benzylpenicillin/streptomycin (Streptopen, Pitman-Moore, Wellington, New Zealand) intramuscularly for prophylaxis. Polyvinyl catheters were placed in both brachial arteries. The vertebral–occipital anastomoses were ligated bilaterally to restrict vertebral blood supply to the carotid arteries. A doubleballooned inflatable occluder cuff was placed around each carotid artery. Two pairs of electroencephalographic electrodes (AS633–5SSF, Cooner Wire, Chatsworth, CA, U.S.A.) were placed on the dura over the parasagittal parietal cortex (5 mm and 15 mm anterior and 10 mm lateral to bregma), with a reference electrode sewn over the occiput. A 17-mm-long cannula was inserted into the left lateral cerebral ventricle at 6 mm anterior and 4 mm lateral to bregma. The fetus was then returned to the uterus, and the antibiotic Gentamicin (80 mg, Roussel, Auckland, New Zealand) was administered into the amniotic sac before closure of the uterus. All catheters and electrodes were exteriorized through the maternal flank. A maternal femoral vein was catheterized.
Postsurgery sheep were housed together in separate metabolic cages with access to water and food ad libitum. They were kept in a temperature-controlled room (16°C, 50% humidity), in a 12-hour day/night cycle and they recovered for 3 to 4 days before experiments began. Antibiotics were administered daily to the ewe (600 mg benzylpenicillin [Crystapen], Biochemie, Vienna, Austria) intravenously for 4 days, and 80 mg gentamicin intravenously daily for the first 3 days. Vascular catheters were maintained patent by continuous infusion of heparinized saline (40 U/mL at 0.2 mL/h). The lateral ventricle cannula was maintained patent by daily flushing with 200 μL artificial cerebrospinal fluid (aCSF). Fetal arterial blood was taken daily before, during, and after ischemia for pH and blood gas analysis to determine fetal well-being.
Experimental procedures
Fetuses were randomly assigned to either sham ischemia with no infusion (sham control group), or to ischemia groups that received intracerebroventricular infusions of 1 mL of either vehicle (aCSF) or recombinant human (rh) IGF-1 (Chiron, Emeryville, CA, U.S.A.) dissolved in 1 mL of vehicle. Reversible cerebral ischemia was induced by inflation of both carotid cuffs with sterile saline for 30 minutes. Successful occlusion was confirmed by the onset of an isoelectric electroencephalographic signal within 30 seconds of inflation. Before administration, the pH of the infusate was buffered to 7.33 to 7.39 with 1 mol/L NaHCO3, as previously described (Guan et al., 2001). The dead space in the lateral ventricle catheter (0.7 mL) was primed with either rhIGF-1 or vehicle by infusion over 45 minutes. Ninety minutes after reperfusion, either rhIGF-1 (3 or 30 μg in 1 mL) or vehicle was infused in a volume of 1 mL during 1 hour. In an additional group, an infusion of 3 μg (in 1 mL aCSF) was given during a further 24 hours after an initial bolus of 3 μg of rhIGF-1 (3 + 3-μg group). At the end of the experiment (4 days after ischemia), the ewe and fetus were killed by an intravenous overdose of sodium phenobarbital. The fetuses were rapidly removed through an abdominal incision, and the brain was perfusion fixed in situ with normal saline, followed by 10% phosphate-buffered formalin. Each brain was removed from the skull and fixed in the same fixative for an additional 7 days before processing and embedding using a standard paraffin tissue preparation.
Immunohistochemistry
Immunohistochemical staining was performed on coronal sections (6 μm) at the level of the parietal cortex. The sections were cut and mounted onto chrome alum-coated slides and dried at 37°C overnight. The sections were dewaxed in xylene, dehydrated in a series of ethanol, and washed in phosphate-buffered saline (PBS, 0.1 mmol/L) for three times for 5 minutes. For antigen unmasking [for caspase-3 and antiproliferating cell nuclear antigen (PCNA) staining], sections were heated in 10 mmol/L sodium citrate buffer (pH 6.0) by microwave oven for 1 minute at high power. Sections were pretreated with 1% H2O2 in 50% methanol for 30 minutes to quench endogenous peroxidase activity. To block nonspecific background staining, sections were incubated with 1% to 2% normal serum of the species that the secondary antibody was raised in for 1 hour at room temperature.
The following primary antibodies were applied to sections at 4°C for 12 to 48 hours: monoclonal mouse anti-glial fibrillary acidic protein (GFAP) antibody (Sigma, St. Louis, MO, U.S.A., diluted 1:500); polyclonal rabbit anti-caspase-3 p17 antibody [Cleaved Caspase-3 Antibody, detects only the large fragment of activated caspase-3 (17–20 kDa), Cell Signaling Technology, Beverly, MA, U.S.A., diluted 1:1,000]; mouse PCNA antibody (Dako, Cambridge, UK, diluted 1:100); mouse antimyelin basic protein (MBP) antibody (Roche, Mannheim, Germany, diluted 1:100). Sections were then incubated with biotinylated horse anti-mouse or goat anti-rabbit secondary antibody (Vector Laboratories, Burlingame, CA, U.S.A., diluted 1:200) at 4°C overnight. For peroxidase staining, sections were incubated with the avidin–biotin complex (ABC, Vector Laboratories, Peterborough, England, diluted 1:100) for 3 hours at room temperature and reacted with reconstituted Sigma FAST diaminobenzidine tablets to produce a brown reaction product. The sections with caspase-3 staining were lightly counterstained with thionine. Sections were dehydrated in a series of alcohol to xylene and coverslipped with mounting medium. For fluorescence staining of PCNA and caspase-3, sections were incubated with streptavidin-Alexa Fluor-568 (Molecular Probes, Eugene, OR, U.S.A.; 5 μg/mL) instead. Fluorescence staining for GFAP was carried out by using Alexa Fluor-488 conjugated rabbit anti-mouse (Molecular Probes, 10 μg/mL) as the secondary antibody. The sections were mounted with glycerol/PBS and viewed using confocal microscopy. Control sections were processed in the same way except the primary antibody was replaced with PBS. After each incubation in the above procedure, sections were washed in PBS three times for 5 minutes.
Lectin histochemistry
Microglia were identified with labeled lectin from Griffonia simplicifolia seeds (Sigma). For peroxidase staining, deparaffinized and rehydrated sections were pretreated with 1% H2O2 in 50% methanol for 30 minutes and incubated with peroxidase conjugated isolectin B4 (Sigma, 10 μg/mL in Tris-buffered saline) overnight at 4°C. Diaminobenzidine was used as the chromogen as above. For fluorescence staining, deparaffinized and rehydrated sections were directly incubated with fluorescein conjugated isolectin B4 (Sigma, 250 μg/mL in Tris-buffered saline) overnight at 4°C.
Nonradioactive in situ hybridization
To label oligodendrocytes, nonradioactive in situ hybridization was performed using digoxigenin (DIG)-labeled anti-sense RNA probes specific for sheep proteolipid-protein (PLP) gene as described previously (Guan et al., 2001). DIG signals were visualized using either alkaline phosphatase conjugated sheep anti-DIG-Fab fragments (Roche, 1:300 diluted) and its substrate 4-nitro blue tetrazolium chloride/5-bromo-4-choro-3-indoyl-phosphate (Promega, Madison, WI, U.S.A.) or sheep anti-DIG (Roche, 1:200 diluted) and Alexa Fluor-488 conjugated anti-sheep (Molecular Probes, 5 μg/mL).
Tissue assessment and analysis
GFAP, IB4, and PCNA immunoreactive cells and PLP mRNA-positive cells were counted in three areas in the intragyral white matter of the parasagittal cortex by light microscopy (×20) (Guan et al., 2001). Within the intragyral region, counts were made using full cross sections of the tract for a fixed length of 0.54 mm. The density of MBP from the same areas and their background was measured using image analysis (Sigmascan, SPSS Inc., Chicago, IL, U.S.A.). The difference between the MBP density and the background reading from adjacent gray matter was calculated and used for data analysis. Different staining from each brain was assessed in the same area of adjacent sections. The number of cells positive for GFAP, IB4, PLP, and PCNA and the MBP density were averaged from the three selected intragyral areas. Because of the relatively low density of caspase-3p17 positively stained cells, the measurement represented the total number of caspase-3p17 positive cells in the white matter of the first parasagittal gyrus. Data are presented as mean ± SD. Treatment effects were analyzed using one-way analysis of variance followed by multiple comparisons (Dunnett multiple comparison test, Prism 3.0). The significance level was set at P < 0.05. Fluorescentstained sections were photographed by a Leica TCS 4D system (Leica, Wetzlar, Germany) equipped with a confocal microscope.
RESULTS
Proteolipid-protein mRNA expression
In the sham control group, the PLP mRNA signal was diffusely distributed from the corona radiata toward the intragyral regions of the parasagittal white matter tracts. Morphologically, PLP staining was peripherally distributed within the cytoplasm of oligodendrocytes in a bipolar distribution, as described previously (Guan et al., 2001).
There was a significant loss in the number of PLP mRNA-containing cells in the vehicle-treated group (47.3 ± 47.3, n = 8) compared to the sham control group (132.1 ± 22.3, n = 5, P < 0.001, Fig. 1A). Treatment with 3 μg IGF-1 (108.7 ± 54.9, n = 9) significantly increased the number of PLP mRNA-containing cells compared to the vehicle-treated group (P < 0.001 Fig. 1A). The 3 + 3 μg IGF-1 group (81.6 ± 28.9, n = 7, P > 0.05) showed a trend towards an increase in PLP mRNA-positive cells, but there was no effect after treatment with 30 μg IGF-1 (43.6 ± 26.6, n = 7) compared to the vehicle-treated group.
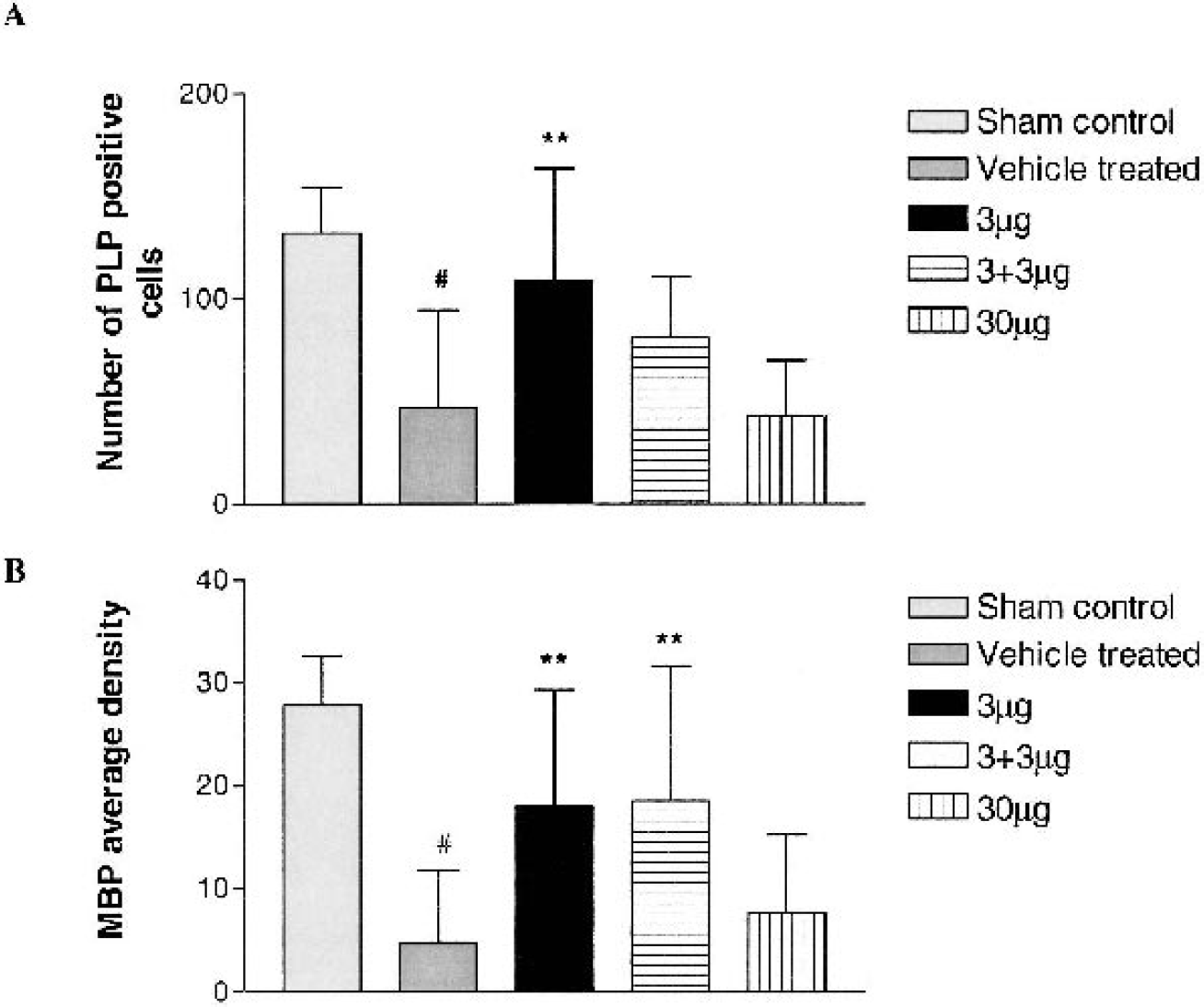
The number of proteolipid protein (PLP) mRNA-positive cells and the density of myelin basic protein (MBP) immunostaining measured in the intragyral cerebral white matter tracts.
Myelin basic protein
Myelin basic protein staining was located in the myelin sheath without obvious staining in the cell bodies. Consistent with the loss of PLP mRNA-containing cells, ischemia resulted in a severe loss of MBP staining in the intragyral white matter tracts, with morphologic changes similar to those described previously (Guan et al., 2001). The average density of MBP immunostaining in the vehicle group (4.7 ± 7.1, n = 8) was significantly reduced compared to the sham control group (27.8 ± 4.8, n = 5, P < 0.001, Fig. 1B). Treatment with both 3 μg (18.0 ± 11.2, n = 9) and 3 + 3 μg (18.5 ± 13.0, n = 7) rhIGF-1 significantly increased the average MBP density compared to the vehicle-treated group (P < 0.001, Fig. 1B). There was no difference in myelin density between the 30 μg IGF-1-treated group (7.6 ± 5.6, n = 7) and the vehicle-treated group.
Glial reaction
Both GFAP and isolectin B4 immunostaining were located in cell cytoplasm and processes. Morphologically, we observed hypertrophic astrocytes with an increased GFAP staining of cell bodies and processes in the white matter tracts. Hyperplasia of GFAP-positive cells was also found in the white matter. Figure 2A shows the numbers of the constitutive GFAP-positive cells in the white matter of the sham control group. In the vehicle-treated group, no increase in GFAP-positive cells was observed (35.08 ± 15.39, n = 8) compared to the sham controls (24.87 ± 13.39, n = 5, Fig. 2A). Treatment with either 3 μg (70.59 ± 15.99, n = 9) or 3 + 3 μg (53.71 ± 11.56, n = 7) IGF-1 significantly increased the numbers of GFAP-positive cells compared to the vehicle-treated group (P < 0.001) but no effect was seen for the 30 μg IGF-1 treatment group (35.57 ± 14.89, Fig. 2A). In contrast to the GFAP-containing cells, there were no isolectin B4-positive cells in the sham control group. The number of isolectin B4-positive cells was significantly increased in the vehicle-treated group (18.04 ± 8.47, n = 8) compared to the sham control group (nil, n = 5, Fig. 2B, P < 0.001). Treatment with either 3 μg (42.22 ± 28.11, n = 9) or 3 + 3 μg (46.33 ± 10.74, n = 7) IGF-1 significantly increased the numbers of isolectin B4-positive cells when compared to vehicle-treated group (P < 0.05). There was no effect after 30 μg IGF treatment compared to the vehicle-treated group (Fig. 2B).
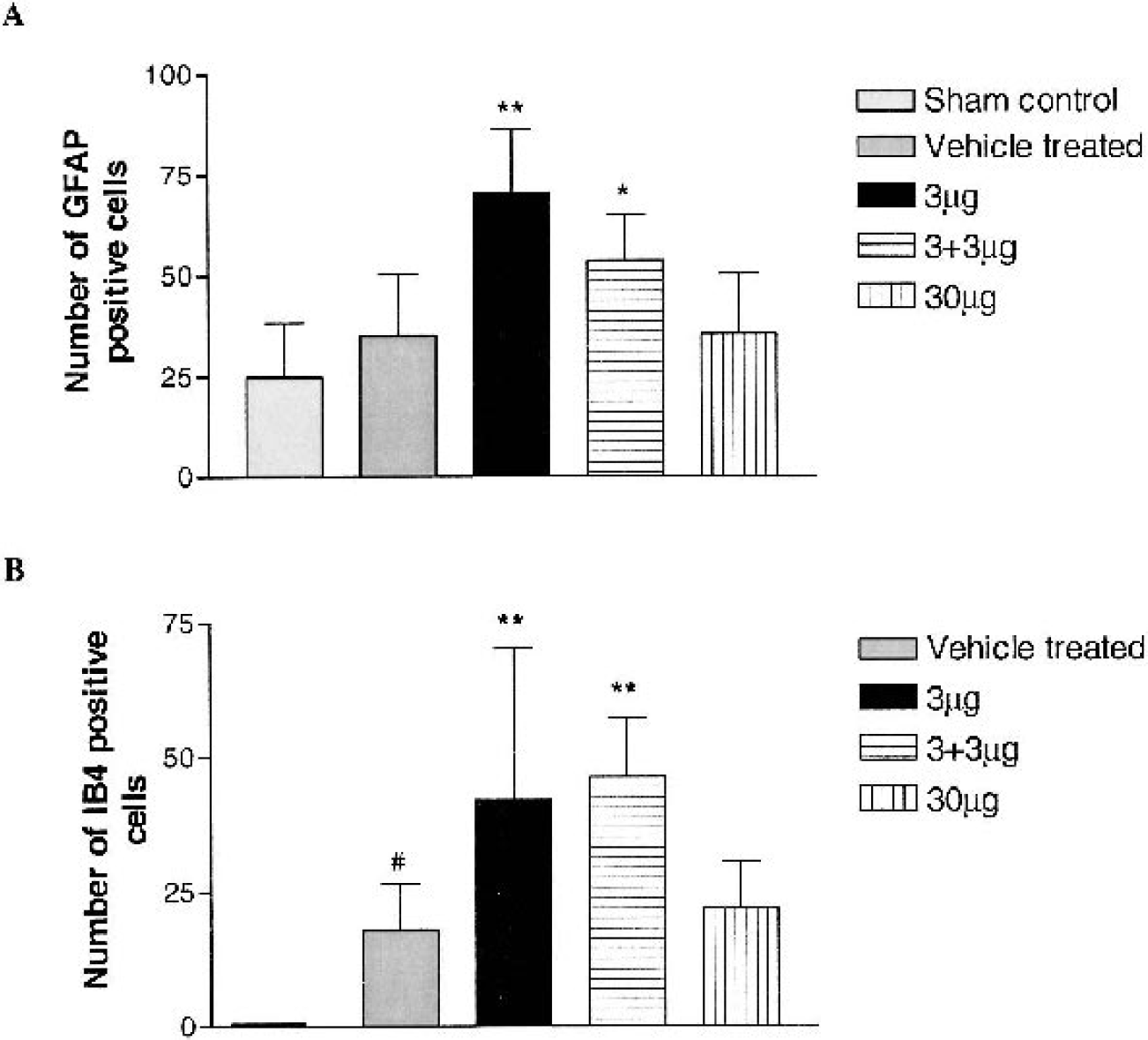
The number of glial fibrillary acidic protein (GFAP) and isolectin B-4 (IB-4) immunopositive cells were assessed in the intragyral cerebral white matter tracts.
Caspase-3 activation
In the vehicle-treated group, caspase-3p17 staining was distributed mainly within the white matter tracts, including the parasagittal cortex, corona radiata, and periventricular areas. The density of caspase-3 staining varied between cells due to the timing of cleavage of caspase-3. Cells with newly cleaved caspase-3 showed morphologically dense caspase-3p17 staining evenly distributed within the cytoplasm, with coarse granular fragments around the cells (Fig. 3). Thionine counterstaining showed condensed chromatin fragments within the caspase-3p17-positive cells (Fig. 3, arrow).
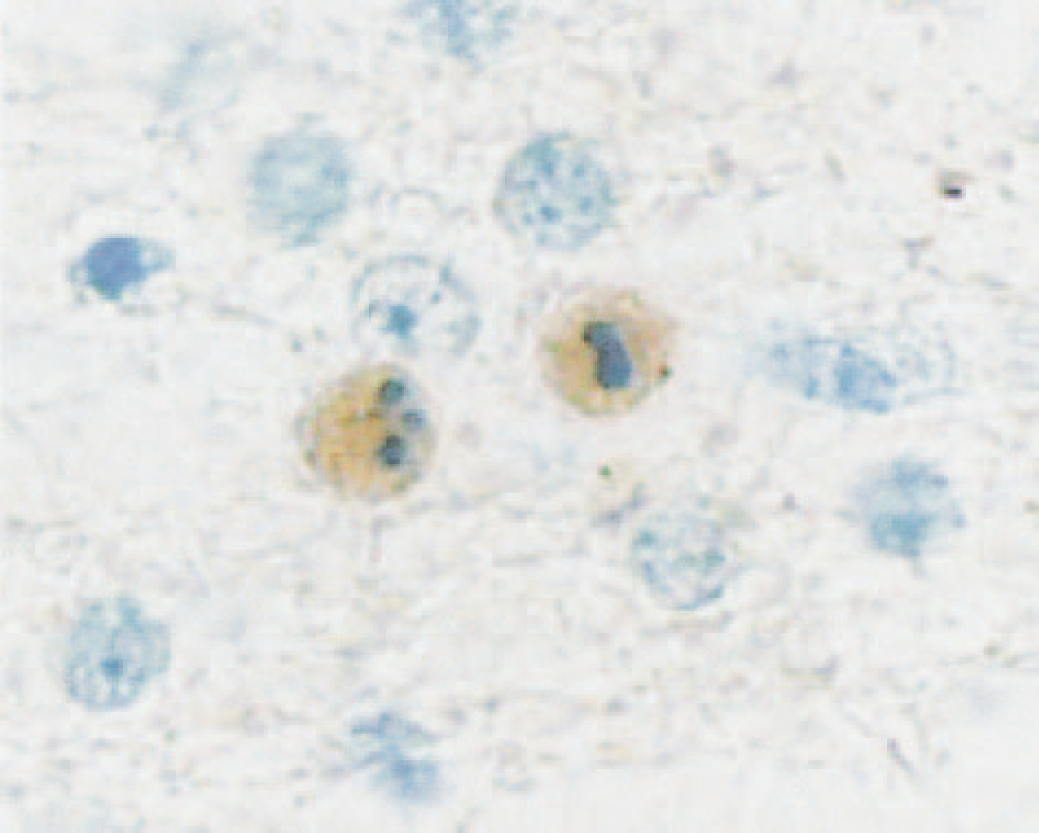
Photograph shows caspase-3-positive staining in the intragyral white matter tracts after ischemic injury. Thionine counterstaining showed a condensed nucleus within the cells, which were caspase-3p17 positive cytoplasm (×100).
In the sham control group, few caspase-3p17 positive cells were scattered within the white matter tracts. The number of caspase-3p17 positive cells significantly increased in the vehicle-treated group (70.1 ± 25.8, n = 8) compared to the sham control group (6.6 ± 2.7, n = 5, P < 0.001, Fig. 4A). A single dose of IGF-1 (3 μg, 25.4 ± 18.1, n = 9) or 3 + 3 μg (41.4 ± 33.8, n = 7) treatment significantly reduced caspase-3 activation (P < 0.001 and P < 0.05, respectively, Fig. 4A) compared to the vehicle-treated group (70.1 ± 25.8, n = 8). Similarly, 30 μg IGF treatment had no effect on caspase-3 activation. A similar peripheral distribution of PLP mRNA was seen in fluorescent staining (Fig. 5, green). Among them, fewer PLP positive cells had a morphology similar to that of caspase-3-positive staining (Fig. 5, red, arrows), with more accumulated cytoplasm staining around the condensed nucleus. Double labeling showed that the caspase-3-positive cells were co-localized with PLP mRNA containing (Fig. 5, arrows).
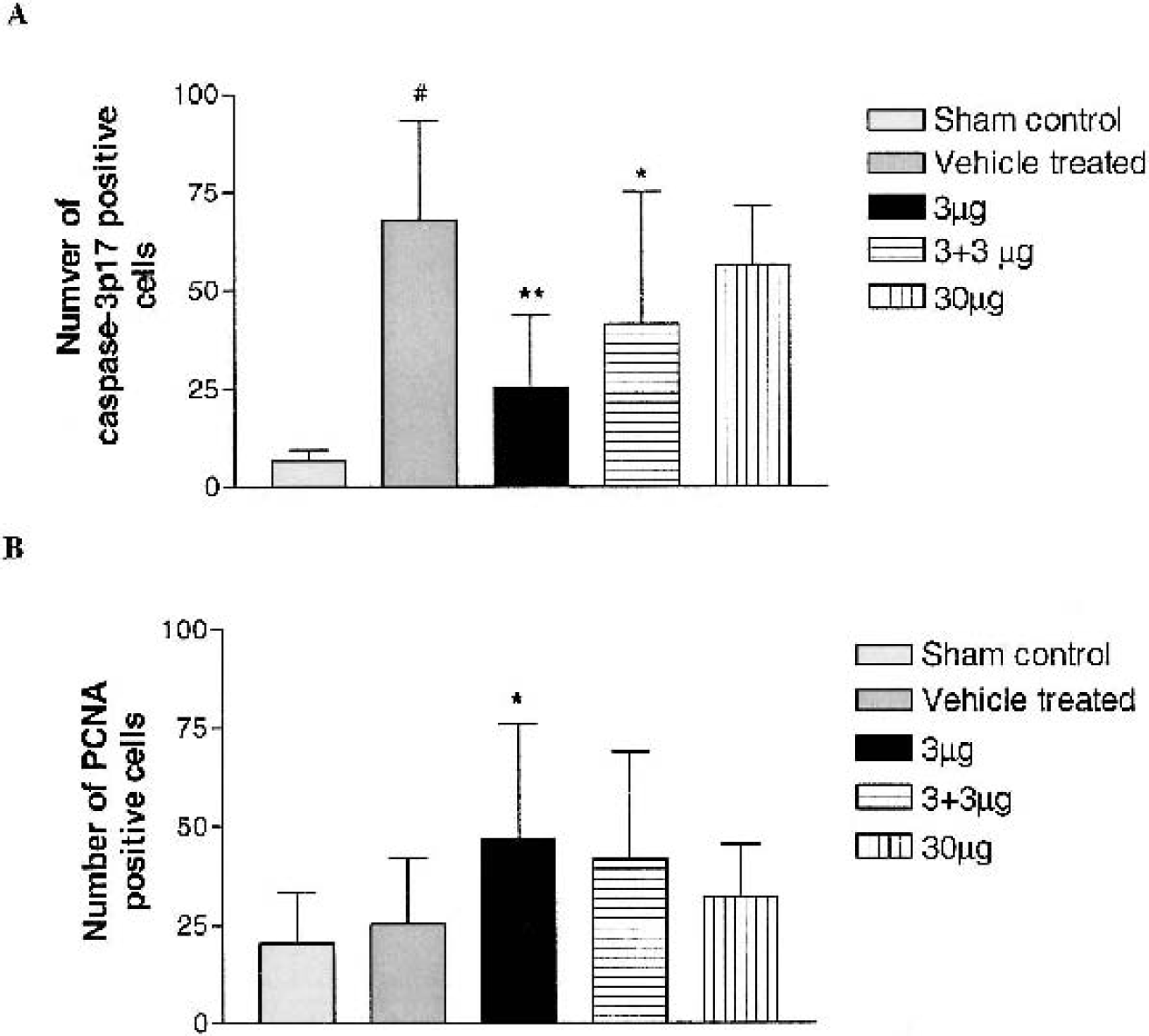
Numbers of caspase-3p17 positive cells assessed in the intragyral cerebral white matter tracts
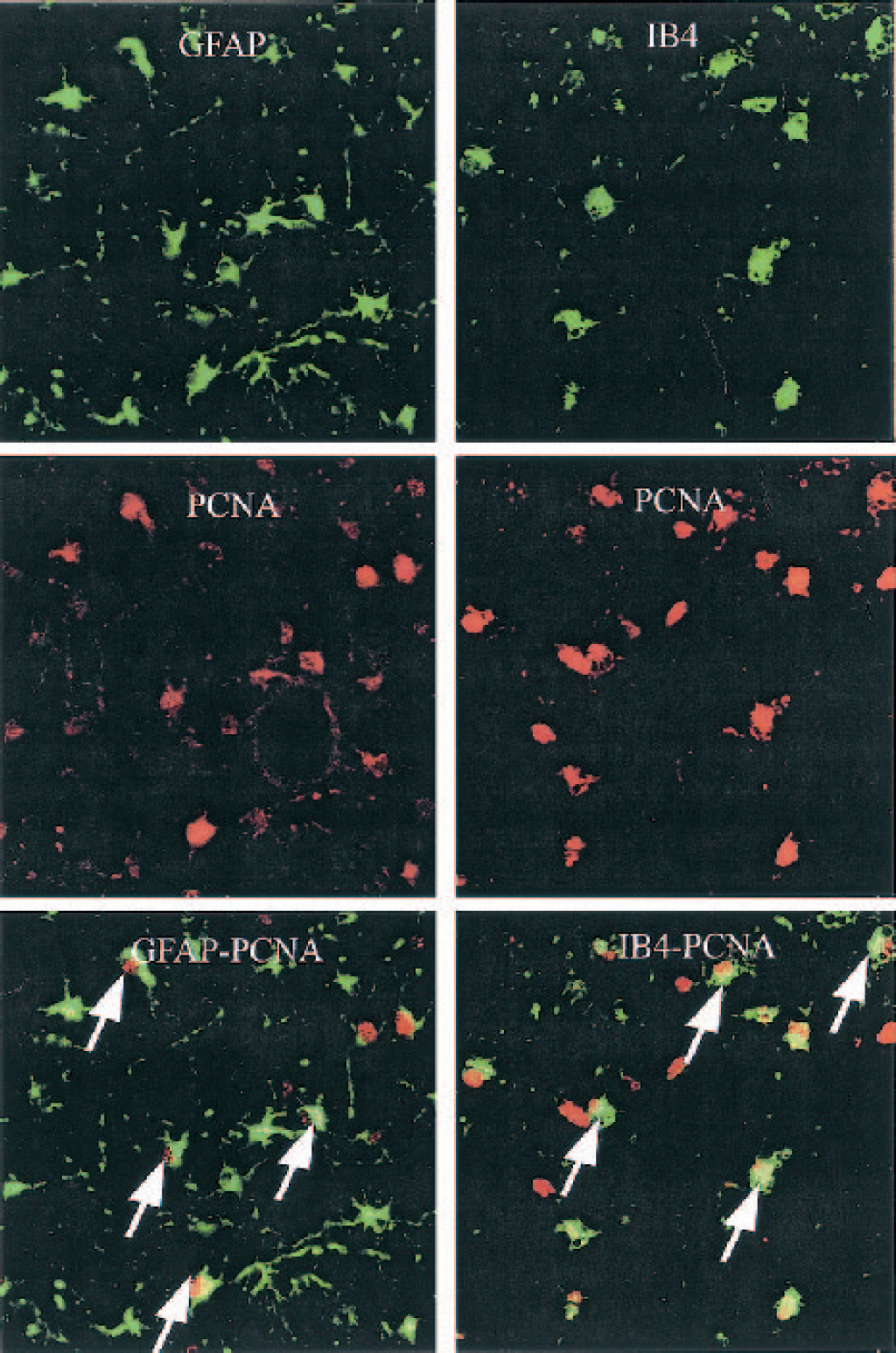
Double labeling of proliferating cell nuclear antigen (PCNA) and glial phenotypes. After ischemic injury, both GFAP and isolectin B4 staining located to both cytoplasm and processes. The confocal fluorescence photographs from intragyral white matter tract showed that PCNA immunopositive cells (red) were co-localized with both GFAP (green, left column, arrows) and IB-4 (green, right column, arrows) immunopositive cells.
PCNA immunoreactivity
In the sham control group only a few PCNA stained cells were detected in the white matter tracts and the staining was found in the nuclei. There was no increase in the number of PCNA positive cells in the vehicle-treated group (25.25 ± 16.71, n = 8) compared to the sham controls (20.33 ± 12.93, n = 5, Fig. 4B). Treatment with 3 μg IGF-1 (47.30 ± 29.07, n = 9) significantly increased the number of PCNA containing cells compared to the vehicle-treated group (25.25 ± 16.71, n = 8, Fig. 4B, P < 0.05). There was no significant difference between either 3 + 3 μg or 30 μg IGF-1-treated and vehicle-treated groups. Double labeling showed that PCNA-positive staining (Figs. 5 and 6, red) was co-localized with GFAP (Fig. 6, green), isolectin B4-containing cells (Fig. 6, green), and PLP mRNA-containing cells (Fig. 5, green).
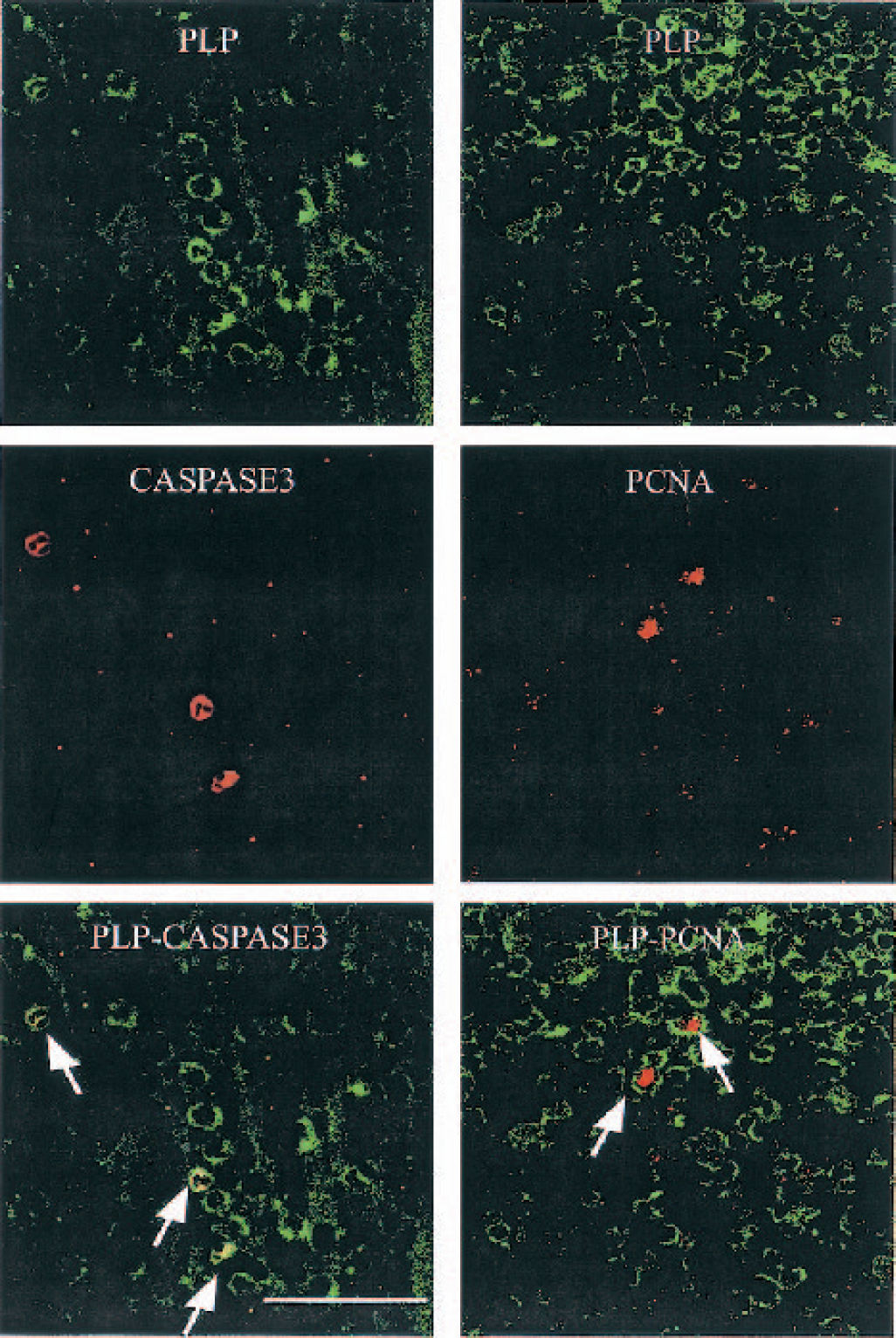
Double-labeling of proteolipid protein (PLP) with caspase-3 and proliferating cell nuclear antigen (PCNA). PLP mRNA-positive staining was peripherally distributed in cytoplasm. The confocal fluorescence photographs from the intragyral white matter tract showed that PLP mRNA (green) was co-localized with caspase-3 (red, left column, arrows). PCNA (red, right column, arrows)-positive cells were located within the PLP positive cells. Scale bar = 40 μm for both Figs 5 and 6.
DISCUSSION
The present study demonstrates that the effects of centrally administered IGF-1 on white matter recovery after severe cerebral ischemia are dose dependent and closely associated with suppression of caspase-3 activation in oligodendrocytes and promotion of glial proliferation in all phenotypes. These data suggest that both antiapoptotic effects and mitogenic effects contribute to the recovery of injured white matter after IGF-1 treatment.
Oligodendrocytes are the myelinating glia in the CNS. Proteolipid-protein gene expression has been used as a sensitive marker to detect bioactive oligodendrocytes at the translation stage of myelination. In vitro and in vivo evidence have shown that both mature and immature oligodendrocytes are vulnerable to hypoxic–ischemic injury (Petito et al., 1998; Back et al., 2002; Skoff et al., 2001). Using the same experimental model, we have previously reported a bell-shaped dose response of IGF-1 for neuronal rescue (Johnston et al., 1996), with loss of neuroprotection at higher doses. Similarly, the present study confirmed that high-dose (30 μg) IGF-1 failed to prevent ischemia-induced demyelination. The loss of treatment effect at the high dose may be due to down-regulation of IGF-1 receptors secondary to excessive free IGF-1 levels (Peruzzi et al., 1999). Although the protective effect of IGF-1 on preoligodendrocyte survival was sustained for at least 48 h in vitro, with no absolute reduction in IGF-1R levels; however, it is striking that the level of the IGF-1R was lower in the IGF-1-treated group after 48 h compared with controls, consistent with the hypothesis that free IGF-1 can modulate receptor expression (Ness and Wood, 2002). IGF-binding proteins (IGFBPs) can either inhibit or enhance IGF-1 bioactivity. IGFBPs, particularly IGFBP-2, have been identified as playing a critical role in mediating the neuroprotective action of IGF-1 after hypoxic–ischemic injury (Guan et al., 1996). A major role for IGFBPs has been suggested to be translocation of exogenous IGF-1 to the site of injury via white matter tracts (Guan et al., 2000). However, excessive free IGF-1 can downregulate the bioactivity of IGFBPs (Baxter, 1993); thus, reduced IGFBP activity could also contribute to the lack of effect of high-dose IGF-1.
The half-life of the IGF-1/IGFBPs complex is approximately 12 to 15 hours in plasma (Guler et al., 1989). A second dose of IGF-1 (3 μg) infused for an additional 24 h would be expected to maintain IGF-1 levels in CSF for longer. However, in the current study this extended infusion with IGF-1 showed no further improvement compared with a single dose, and indeed in some cases tended to be less effective. The window of opportunity for IGF-1 on neuroprotection has been well documented to be limited to a few hours after hypoxic–ischemic injury in adult rats (Guan et al., 2000). These observations are consistent with the hypothesis that IGF-1 is acting primarily by suppressing early, presumptively apoptotic, postischemic events after injury (Mason et al., 2000).
Apoptosis has been suggested to be relatively more important in ischemic brain damage in the neonate than in the adult, with caspase-3 activation believed to be a critical pathway in white matter damage, particularly in the cleaved 17-kd subunit form (Hu et al., 2000). Figure 3 demonstrates that the majority of caspase-3-positive cells had a clearly apoptotic morphology, including nuclear condensation, and were co-localized exclusively with oligodendrocytes (Fig. 5). Furthermore, in the vehicle-treated group, ischemia induced loss of myelinating oligodendrocytes, labeled by PLP mRNA expression was statistically associated with increased activation of caspase-3. These data indicate that ischemia-associated loss of PLP activity is at least in part related to caspase-3-activated apoptosis. However, quantitatively, caspase-3 expression after 4 days' recovery can only account for a minority of cell loss, with a relatively low density of caspase-positive cells observed. Speculatively, this discrepancy could be due to the timing of histologic evaluation, with activation of caspases occurring much earlier after ischemia. In addition to caspase-mediated cell death, noncaspase-mediated apoptotic pathways, such as apoptosis-inducing factor, may be important, as well as necrotic processes (Leist and Jaattela, 2001). Often, more than one of these three pathways seems to be activated simultaneously, and cell fate is then determined by the relative speed of each process (Leist and Jaattela, 2001).
We have previously reported that IGF-1 prevents ischemia-induced demyelination, with an increase in number of bioactive oligodendroglia (Guan et al., 2001), but were unable to distinguish the antiapoptotic and mitogenic effects of IGF-1. We now report that low-dose IGF-1 treatment inhibited the activation of caspase-3 in a dose-related manner similar to enhancement of bioactive oligodendroglia and myelin density. These data indicate that, at least in part, IGF-1 enhances oligodendroglial survival through inhibition of caspase-3. This is consistent with the observations that IGF-1 prevents hypoxia-induced neuronal death by inhibiting caspase-3-like activity in vitro (Tamatani et al., 1998) and attenuated the NMDA receptor–induced apoptotic cell death and associated caspase-3 activation (Takadera et al., 1999).
In addition, in the present study there was a positive correlation between cell proliferation and number of surviving oligodendrocytes (Figs. 1B and 4B). Both the effects on cell proliferation and on the numbers of oligodendrocytes (PLP-positive cells) fell when the dose of IGF-1 was increased. These data have several implications. First, double labeling in the low-dose IGF-1-treated group demonstrated that PCNA immunostaining was seen clearly within the cells that also show peripherally distributed PLP mRNA. These data confirm that promotion of regeneration of oligodendroglia by IGF-1 may be a significant factor in recovery of white matter after ischemia, since proliferating oligodendrocytes contribute to remyelination (Fig. 5) (Ludwin, 1988).
Second, IGF-1 also increased proliferation in astrocytes and microglia. This effect also strongly paralleled the effects of IGF-1 on oligodendrocytes, suggesting that this effect is not simply a nonspecific mitogenic effect. GFAP expression is essential for normal white matter architecture and blood–brain barrier integrity, and its absence can lead to late-onset central nervous system demyelination (Liedtka et al., 1996). It is well established that astrocytes play an important role in supporting neuronal and glial survival, including oligodendroglia and microglia by providing neurotrophic factors after hypoxic–ischemic insults (Miyazaki et al., 2001; Nawashiro et al., 2000; O'Donnell et al., 2002). Supporting networks of glial–glial interactions have been previously suggested to improve oligodendroglial survival (Corley et al., 2001; Dougherty et al., 2000). Traditionally, microglia have been considered to have a relatively negative influence on neural injury and recovery, opposite to that of astrocytes (Giulian et al., 1993). However, our data suggest that increased numbers of both active astrocyte and microglia are associated with better white matter recovery. There are some data to show that microglial activation can support a proregenerative environment in injured central nervous system through the release of cytokines, growth factors, and extracellular matrix molecules, which are critical for postinjury tissue repair in the central nervous system (Rabchevsky and Streit, 1998; Nagata et al., 1993; Nakajima et al., 1992). Speculatively, it may be that an earlier increase in extracellular IGF-1 levels may alter the balance of positive and negative effects of microglia. Thus, reactive glial cells appear to have a critical, although poorly understood role in white matter protection by IGF-1.
Finally, in addition to its role in DNA replication, PCNA is also required for DNA synthesis and nucleotide excision DNA repair. It has been shown that PCNA is expressed in surviving cells of damaged brain regions after a transient cerebral ischemia (Ju et al., 2000). This PCNA expression appears to be essential for neuronal survival (Ju et al., 2000), because impaired or insufficient function of PCNA results in a decreased DNA repair capacity and accumulation of DNA lesions, ultimately leading to cell death (Tomasevic et al., 1998). Given the close parallelism of the upregulation by IGF-1 of PCNA and the three glial phenotypes examined, the possibility arises that DNA repair in glial cells may also contribute to white matter recovery from ischemic injury.
CONCLUSION
IGF-1 prevented white matter damage from ischemic injury in a dose-dependent and time-of-deliverydependent manner. The mechanisms of postischemic white matter protection by exogenous IGF-1 appear to include suppression of late onset oligodendrocyte apoptosis, promotion of oligodendrocyte regeneration, and increased proliferation of reactive glia.
Footnotes
Acknowledgments:
We thank the Chiron Corporation, U.S.A., for the gift of IGF-1 and the Children's Hospital of Fudan University, Shanghai, China for support of Dr. Yun Cao.