
Abstract
Mild hypothermia is a robust neuroprotective treatment for stroke. Understanding the mechanisms underlying hypothermia's benefits will lead to more effective treatments to prevent stroke damage. Delta protein kinase C (δPKC) is a kinase that has been strongly implicated in executing ischemic damage. We investigated the effects of hypothermia on δPKC activation, as determined by its subcellular translocation, proteolytic cleavage, and phosphorylation in a focal cerebral ischemia model. The amount of constitutively activated C-terminal catalytic fragment of δPKC (CF-δPKC) increased after stroke. Both hypothermia (30 C) and the caspase-3-specific inhibitor, Z-DQMD-FMK, blocked the accumulation of activated δPKC in the penumbra. Other hallmarks of δPKC activation, its translocation to the mitochondria, and nucleus were observed in the penumbra as early as 10 mins after reperfusion. These events were blocked by hypothermia. Hypothermia also blocked CF-δPKC increases in the mitochondria and nuclei. Conversely, a specific δPKC activator, ψδRACK, decreased the neuroprotective effect of hypothermia. Finally, δPKC activity may lead to mitochondrial injury and cytochrome c release, as the timing of cytochrome c release corresponded to the time course of δPKC translocation. Both cytochrome c release and δPKC translocation were blocked by hypothermia. In conclusion, hypothermia protects against ischemic damage in part by suppressing δPKC activation after stroke.
Introduction
The protein kinase C (PKC) family of serine/threonine protein kinases consists of at least 12 closely related isozymes, which mediate a variety of cellular functions including apoptosis (Gutcher et al, 2003). Delta protein kinase C is involved in apoptosis in various types of cells including neurons (Anantharam et al, 2002).
Delta protein kinase C can be activated by several means: translocation to membranes to become fully activated in the presence of cofactors; proteolytic cleavage (Kanthasamy et al, 2003) to a constitutively active C-terminal catalytic fragment (CF-δPKC) (Emoto et al, 1995); and phosphorylation (Kayali et al, 2002). Delta protein kinase C is a substrate for caspase-3, an enzyme that plays a key role in apoptosis, which proteolytically cleaves δPKC to release the C-terminal catalytic fragment of δPKC (CF-δPKC) (Emoto et al, 1995). The CF-δPKC is a constitutively active enzyme, because it is freed from the autoinhibitory constraints imposed by the N-terminal regulatory domain (Emoto et al, 1995). In addition, several conserved threonine or serine phosphorylation sites are thought to regulate the activity of δPKC and the levels of phosphorylated-δPKC (p-δPKC) at those sites were used as markers for δPKC activation (Kayali et al, 2002).
Delta protein kinase C has been implicated in mediating ischemic and reperfusion brain injuries. Delta protein kinase C mRNA increases after ischemia in CA1 neurons in gerbil and rat models of global ischemia (Savithiry and Kumar, 1994; Koponen et al, 2000). Delta protein kinase C mRNA and protein levels increase near an ischemic lesion after focal cerebral ischemia in rat (Miettinen et al, 1996). Furthermore, a single injection of the δPKC-specific inhibitor, δV1–1, up to 6 h after reperfusion reduces infarct size after transient cerebral ischemia (Bright et al, 2004). Infarct size after transient focal ischemia is smaller also in δPKC-null mice as compared with wild-type mice (Chou et al, 2004). These findings suggest that δPKC plays a detrimental role after stroke. However, the mechanism of the δPKC activation process after stroke remains largely unknown.
Mild or moderate hypothermia is neuroprotective against experimental stroke (Busto et al, 1989). The protective mechanisms of hypothermia have been attributed to preservation of ATP (Erecinska et al, 2003), and the inhibition of a large array of adverse events. Hypothermia also inhibits apoptotic pathways after stroke (Yenari et al, 2002; Zhao et al, 2005a). Additionally, we recently showed that hypothermia might protect against ischemic damage partly through the PI3K/Akt survival pathway (Zhao et al, 2005b). Hypothermia also inhibited subcellular translocation of several isozymes of the PKC family (α, β, γ) after global ischemia (Harada et al, 2002). However, the effect of hypothermia on proapoptotic δPKC activation has not been studied after stroke. Here, we report the effects of hypothermia on δPKC activation after focal cerebral ischemia.
Materials and methods
Focal Cerebral Ischemia
Experimental protocols were approved by the Stanford University Administrative Panel on Laboratory Animal Care. Focal cerebral ischemia was generated as described (Zhao et al, 2005b). Male Sprague–Dawley rats (350 to 450 g) were used. Anesthesia was induced by 5% isoflurane and maintained with 1.5% to 2.5% isoflurane during surgery. Core body temperatures were monitored with a rectal probe. The distal middle cerebral artery was exposed and cauterized above the rhinal fissure. The bilateral common carotid arteries (CCAs) were occluded for 1 h then released, while the distal middle cerebral artery remained occluded. Rats were divided into normothermic or hypothermic groups (or sham surgery controls). Core temperature in the normothermic group was maintained at 37°C throughout the surgery. The hypothermic group was maintained at a core temperature of 30°C by spraying 70% methanol onto the rat's body. Hypothermia was induced 10 mins before ischemia and was maintained for 1 h after ischemia onset. Temperature was returned to 37°C for 20 mins with a light and a heating pad manually. We have previously shown a high correlation between core temperature and brain temperature in hypothermic rats (Zhao et al, 2004).
Sample Preparations for Western Blot
Rats survived 10 mins, 1,4,8, 24, and 48 h (n = 3 to 6/each time point) after CCA release were killed with an overdose of isoflurane followed by intracardiac perfusion with normal saline. The brains were removed and tissue corresponding to the ischemic core and penumbra was dissected for Western blots as described (Zhao et al, 2005b). The ischemic penumbra, a viable ischemic region that will eventually evolve into infarction, was defined as the tissue saved by hypothermia 48 h after CCA release. The corresponding regions from sham surgery rats were also dissected as controls. Solutions for Western blots were prepared as detailed below.
For whole-cell extracts, brain tissues were homogenized in a glass homogenizer (Wheaton, Millville, NJ, USA) in seven volumes of cold cell lysis buffer (Cat #9803, Cell Signaling Technology, Beverly, MA, USA) containing essential protease and phosphatase inhibitors. Phenylmethylsulphonyl fluoride (1 mmol/L) was added before use according to the manufacturer's instructions. The homogenates were centrifuged at 14,000g for 10 mins at 4°C, and the supernatants were collected as whole-cell extracts according to the manufacturer's instructions.
Cytosolic and particulate (membrane) subcellular fractionation (Bright et al, 2004): Rat brains were homogenized in 7 volumes of homogenization buffer (20 mmol/L Tris-HCl (pH 7.5), 2 mmol/L ethylenediaminetetraacetic acid, 10 mmol/L ethyleneglycol tetraacetate, 250 mmol/L sucrose plus 0.7% protease and phosphatase inhibitor cocktails (Sigma)) and were centrifuged at 100.000 g for 30 mins at 4°C. The supernatants were collected as the cytosolic fraction. The pellets were resuspended in homogenization buffer containing 1% Triton X-100, and centrifuged at 100,000g for 30 mins at 4°C. The supernatants were collected as the membrane fraction.
Nuclear, cytosolic, and mitochondria subcellular fractionation (Yenari et al, 2002; Zhao et al, 2005a): Brains were removed after 10 mins to 48 h after CCA release and were homogenized in a glass homogenizer in 7 volumes of cold suspension buffer (20 mmol/L HEPES-KOH (pH 7.5), 250 mmol/L sucrose, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L ethylenediaminetetraacetic acid, and 1 mmol/L ethyleneglycol tetraacetate plus 0.7% protease and phosphatase inhibitor cocktails (Sigma)). The homogenates were centrifuged at 750g for 10 mins at 4°C and then at 10,000g for 20 mins at 4°C. The 750g pellets were used to obtain the crude nuclear fraction and this fraction was purified using a nuclear extraction kit (CelLytic NuCLEAR extraction kit, Sigma), according to the manufacturer's instructions. The 10,000g pellets were used to obtain the mitochondrial fraction. The supernatant was taken off as the cytosolic fraction.
Western Blots
Western blots were performed as described (Zhao et al, 2005b). Protein concentration was determined using Bradford protein assay kit (Bio-Rad Laboratories Inc., Hercules, CA, USA). Sample (20 μg) was mixed with 5× sodium dodecyl sulfate buffer, boiled for 5 mins, and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis for 1 h. Protein bands were transferred from the gel to a polyvinylidene fluoride membrane (Hybond-P, Amersham Biosciences, Arlington Heights, IL, USA) for 1 h. Membranes were blocked with 5% milk in phosphate-buffered saline containing 0.1% Tween-20 and then probed for δPKC (C-17, 1:5000, rabbit polyclonal, Santa Cruz Biotechnologies, Santa Cruz, CA, USA), p-δPKC (Thr505) (1:1000, rabbit polyclonal, Cell Signaling technology), p-δPKC (Ser643) (sc-18370-R, 1:200, rabbit polyclonal, Santa Cruz Biotechnologies), or cytochrome c (1:1000, rabbit polyclonal, Cell Signal Technology) by incubating in the primary antibody for 1 h followed by a horseradish peroxidase-conjugated secondary anti-rabbit or anti-mouse IgG antibody (1:2000, Cell Signaling Technology) for 1 h at room temperature. Protein bands were detected using an enhanced chemiluminescence system (Western blotting detection reagent, Amersham Biosciences) and exposed to BioMax film (Kodak, Rochester, NY, USA). Films were scanned with a photoscanner and analyzed using ImageJ (US National Institute of Health, Bethesda, MD, USA). Densities of protein bands were measured to assess membrane translocation of δPKC, CF-δPKC, or p-δPKC (Ser643) as described (Bright et al, 2004). To confirm even loading of proteins, membranes were stripped and probed for β-actin (AC-15, 1:40,000, mouse monoclonal, Sigma). To verify accurate separation of subcellular fractionation, membranes for mitochondrial and nuclear proteins were stripped and probed for the mitochondrial marker COX IV (20E8, 1 μg/mL, mouse monoclonal, Molecular Probes, Eugene, OR, USA) or the nuclear maker TFIID (N-12, 1:200, rabbit polyclonal, Santa Cruz Biotechnologies), respectively.
Caspase-3 Inhibitor Injection and Delta Protein Kinase C Cleavage Assay
A cell-permeable caspase-3-specific inhibitor, Z-DQMDFMK (Caspase-3 inhibitor V, Calbiochem, La Jolla, CA, USA), was dissolved in dimethyl sulfoxide and phosphate-buffered saline (Z-DQMD-FMK, 0.3 μg/μL in 1% dimethyl sulfoxide in phosphate-buffered saline; vehicle, 1% dimethyl sulfoxide in phosphate-buffered saline). Rats were anesthetized and placed in stereotactic frames. The drug solution (1.5 μg) or the vehicle was injected into the ventricular space ipsilateral to the ischemia (5 μL, from Bregma: 0.9 mm posterior, 1.5 mm lateral, 3.5 mm deep) at 2 h after CCA release. At 24 h after CCA release, brains were removed and whole-cell extracts were prepared for CF-δPKC detection by Western blots.
ψδRACK Delivery and Infarct Size Measurement
Cell membrane-permeating TAT47–57 carrier peptide (Asoh et al, 2002) and TAT47–57 conjugated δPKC activator peptide, ψδRACK, were synthesized as described (Chen et al, 2001). ψδRACK mimics the δPKC-specific receptors for activated C-kinase (RACK) binding site and causes the conformational change in δPKC, allowing it to translocate to RACK. These peptides have been shown to be delivered across the blood–brain barrier (Bright et al, 2004). ψδRACK or TAT peptides (0.2 mg/kg) were delivered intraperitoneally to the hypothermic rats immediately after CCA release. Rat brains were harvested 24 h later and sliced into 3 mm coronal sections, and incubated in phosphate-buffered saline containing 2% 2,3,5-triphenyl tetrazolium chloride (Sigma) at 37°C for 20 mins. Infarct cortex was measured and normalized to the non-ischemic contralateral cortex and expressed as percentages.
Immunofluorescence Staining and Confocal Microscopy
Free-floating immunostaining was performed as described (Zhao et al, 2005b). Brain slices of 50 μm were prepared on a vibratome and stored in anti-freeze solution at −20°C. Slices were incubated in the solutions of primary antibodies at 4°C overnight. Concentrations of primary antibodies were follows: δPKC (1:200), p-δPKC (Ser643) (1:200), p-δPKC (Thr505) (1:200) and MAP2 (HM-2, 1:500, mouse monoclonal, Sigma). An MAP2 antibody was stained to determine the ischemic core and penumbra as described (Zhao et al, 2005b). Sections were incubated for 2 h at room temperature in the solutions of fluorescein isothiocyanate or Cy3-conjugated secondary anti-rabbit or anti-mouse IgG antibodies (1:200, Jackson ImmunoResearch Laboratories, West Grove, PA, USA), and mounted with Vectashield DAPI (Vector Laboratories, Burlingame, CA, USA). The sections were examined under a confocal laser-scanning microscope (LSM510, Carl Zeiss, Oberkochen, Germany).
Statistical Analyses
Differences in protein bands from Western blots at the same temperature were analyzed using one-way analysis of variance (ANOVA) followed by Tukey's post hoc test. Two-way ANOVA followed by Tukey's post hoc test was used to determine the protective effect of hypothermia on optical densities of protein bands. To examine the effect of caspase-3-specific inhibitor on CF-ψδPKC or the effect of ψδRACK on infarct size, one-way ANOVA followed by Tukey's post hoc test was performed. All tests were considered statistically significant at P-values <0.05. Data are presented as means±s.e.m.
Results
Stroke Causes Proteolytic Cleavage of Delta Protein Kinase C, which is Blocked by Hypothermia
Our previous work had confirmed that moderate hypothermia (30°C) reduced infarct size by ∼80% compared with normothermia (37°C) after focal ischemia caused by 1 h of transient bilateral CCA occlusion plus permanent middle cerebral artery occlusion (Zhao et al, 2005b). In this study, we further investigated δPKC activation after stroke and the protective effects of hypothermia on δPKC activation employing the same stroke model.
We first examined proteolytic activation of δPKC (Kanthasamy et al, 2003). The results of Western blot analyses using a δPKC antibody that recognizes the C-terminus of δPKC indicated that δPKC levels decreased in the whole-cell extracts. At 4 and 24 h after CCA release, full-length δPKC decreased more in the ischemic core as compared with the penumbra (Figures 1A and 1C). Concomitant with such changes, the levels of the CF-δPKC increased slightly at 10 mins, but markedly at 4 and 24 h in the ischemic core. Lesser increases were observed in the penumbra (Figures 1A and 1D). These results suggest that δPKC was cleaved after stroke. Although hypothermia did not markedly affect the amount of the full-length δPKC, it significantly reduced the levels of the activated δPKC cleavage product in the penumbra but not in the ischemic core (Figures 1B and 1D).
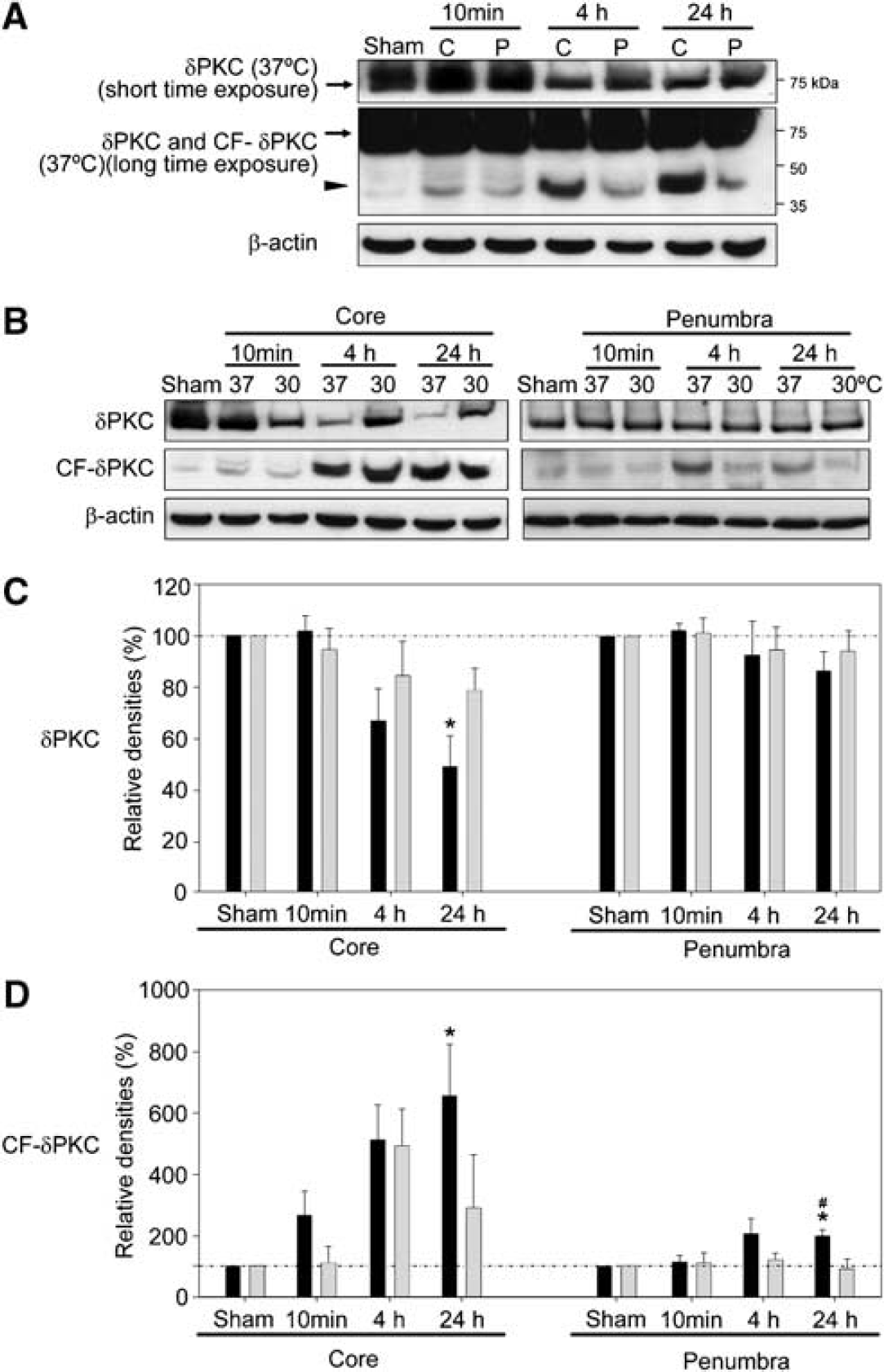
Hypothermia suppresses proteolytic cleavage of δPKC after stroke. (
Proteolytic Cleavage of Delta Protein Kinase C is Suppressed by a Caspase-3-Specific Inhibitor
To investigate whether δPKC cleavage after stroke is caused by caspase-3 activation, we examined the effect of a cell-permeable caspase-3-specific inhibitor, Z-DQMD-FMK, on CF-δPKC generation (Figure 2A). In the penumbra at 24 h after CCA release, the caspase-3 inhibitor reduced δPKC cleavage by ∼40%, as compared with vehicle (P < 0.01) (Figure 2B). In the ischemic core, the caspase-3-specific inhibitor did not significantly reduce the δPKC cleavage.
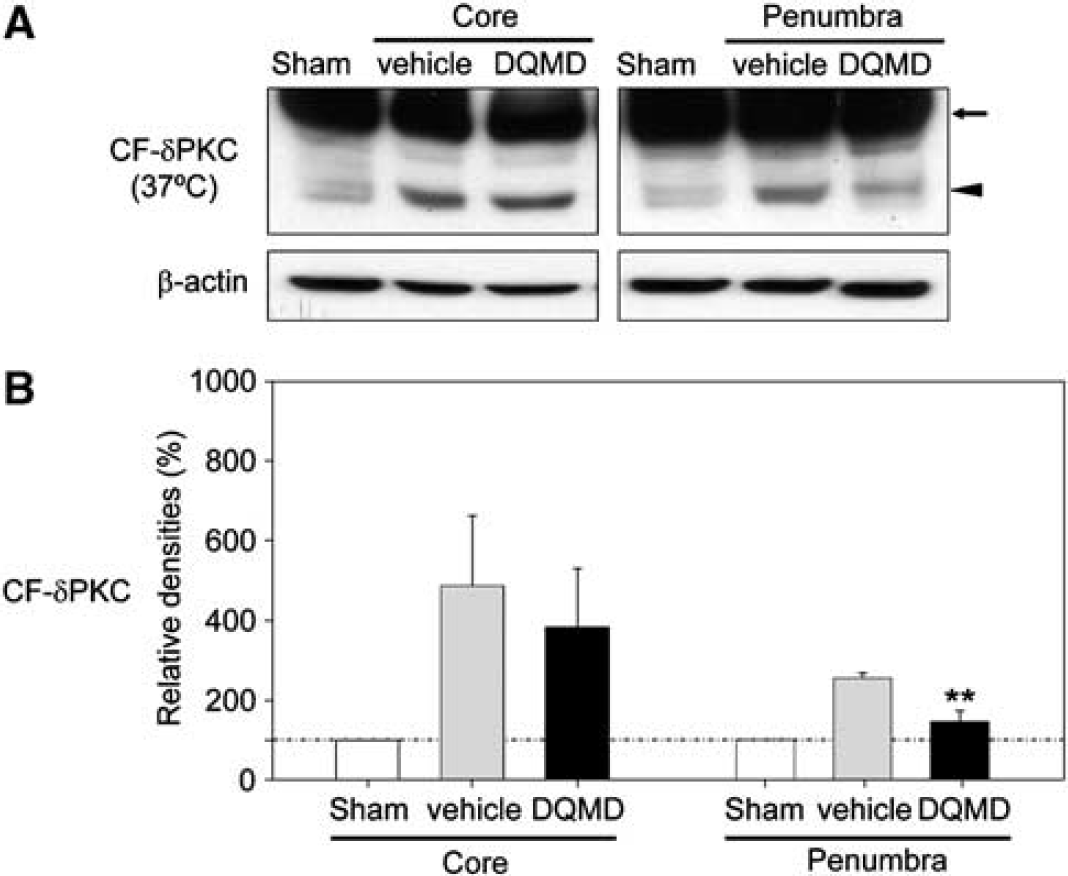
Attenuation of δPKC cleavage by a caspase-3-specific inhibitor. (
Hypothermia Attenuates Dephosphorylation of Phosphorylated-Delta Protein Kinase C (Thr505) after Stroke
Delta protein kinase C phosphorylation can also affect its catalytic activity (Kayali et al, 2002). While levels of p-δPKC (Ser643) did not change after stroke, levels of p-δPKC (Thr505) dramatically decreased in the ischemic core and moderately decreased in the penumbra (Figures 3A, 3C and 3D). Hypothermia attenuated dephosphorylation of p-δPKC (Thr505) at 4 h after CCA release in both the ischemic core and penumbra (Figures 3B and 3C). The levels of p-δPKC (Ser643) did not change significantly after stroke at either temperature (Figures 3B and 3D).
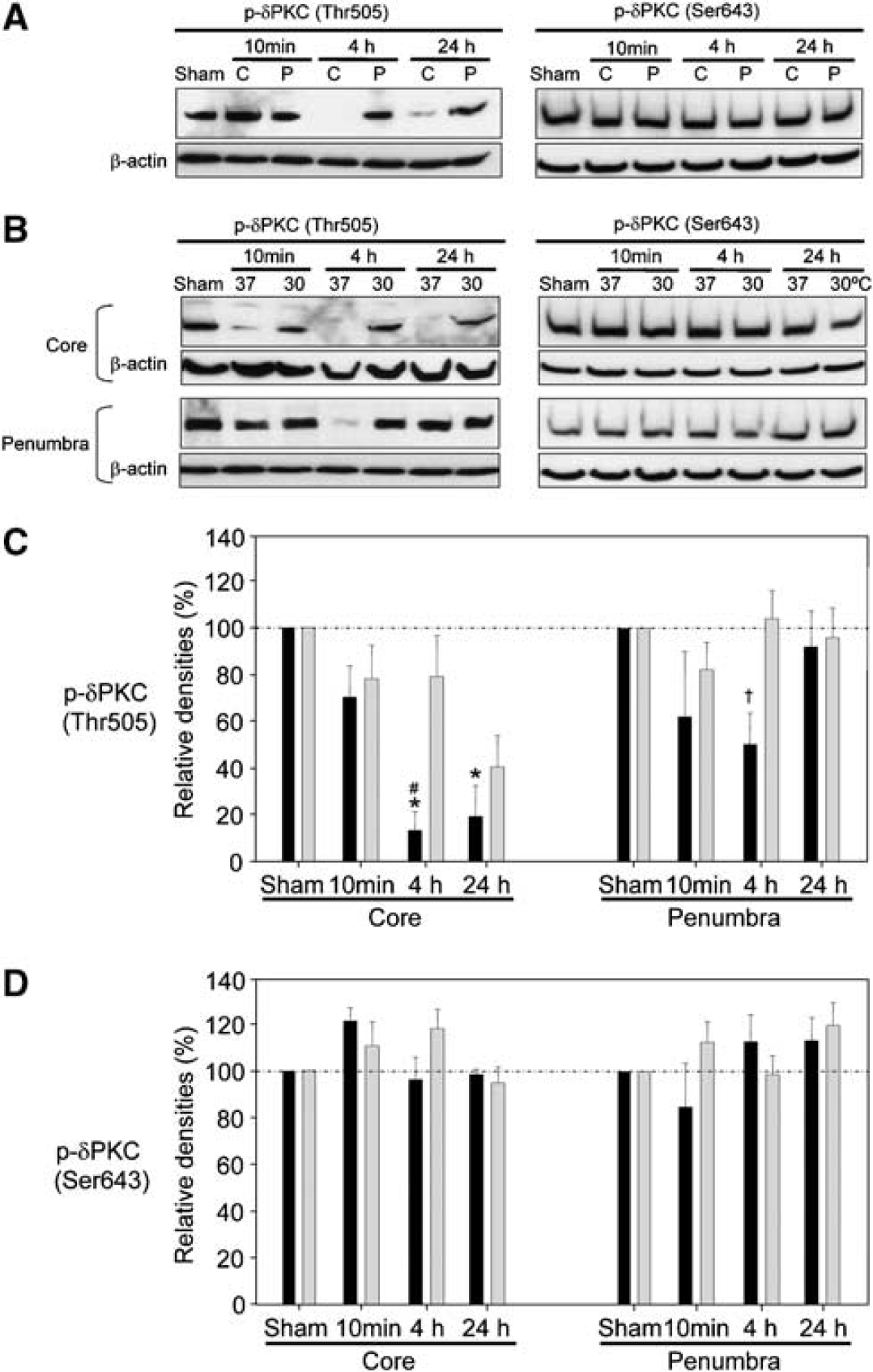
Hypothermia blocks the decrease in p-δPKC (Thr505) after stroke, but p-δPKC (Ser643) does not change after stroke in normothermic rats. (
Stroke Causes Membrane Translocation of Delta Protein Kinase C and Phosphorylated-Delta Protein Kinase C (Ser643) in the Penumbra, but not in the Ischemic Core; Hypothermia Prevents this Membrane Translocation
To investigate membrane translocation of δPKC, another indication of PKC activation (Kraft and Anderson, 1983), Western blots were performed to assess protein levels of δPKC and CF-δPKC in the cytosolic and membrane subcellular fractions after stroke. In sham-operated brains, δPKC was observed in both the cytosolic and membrane fractions (Figure 4A). The amount of membrane-associated δPKC increased at 10 mins and 4 h in the penumbra from normothermic rats subjected to stroke, concomitant with the decrease in δPKC levels in the cytosolic fraction as compared with sham rats (Figures 4A and 4D). This confirms our previous observation that δPKC begins to translocate from the cytoplasm to the plasma membrane immediately after stroke (Bright et al, 2004). In contrast to the penumbra, the amount of δPKC in the membrane fraction did not significantly increase in the ischemic core, although δPKC in the cytosolic fraction decreased (Figures 4A and 4C), possibly because of its proteolytic cleavage (Figures 4A and 4E). In the penumbra, hypothermia blocked both the decrease in cytosolic δPKC and the increase in membraneous δPKC (Figures 4B and 4D). Thus, hypothermia attenuated the membrane translocation of both δPKC and the constitutively active CF-δPKC in the penumbra after stroke. However, hypothermia had no effect on the post-stroke membrane translocation of CF-δPKC in the ischemic core (Figure 4E).
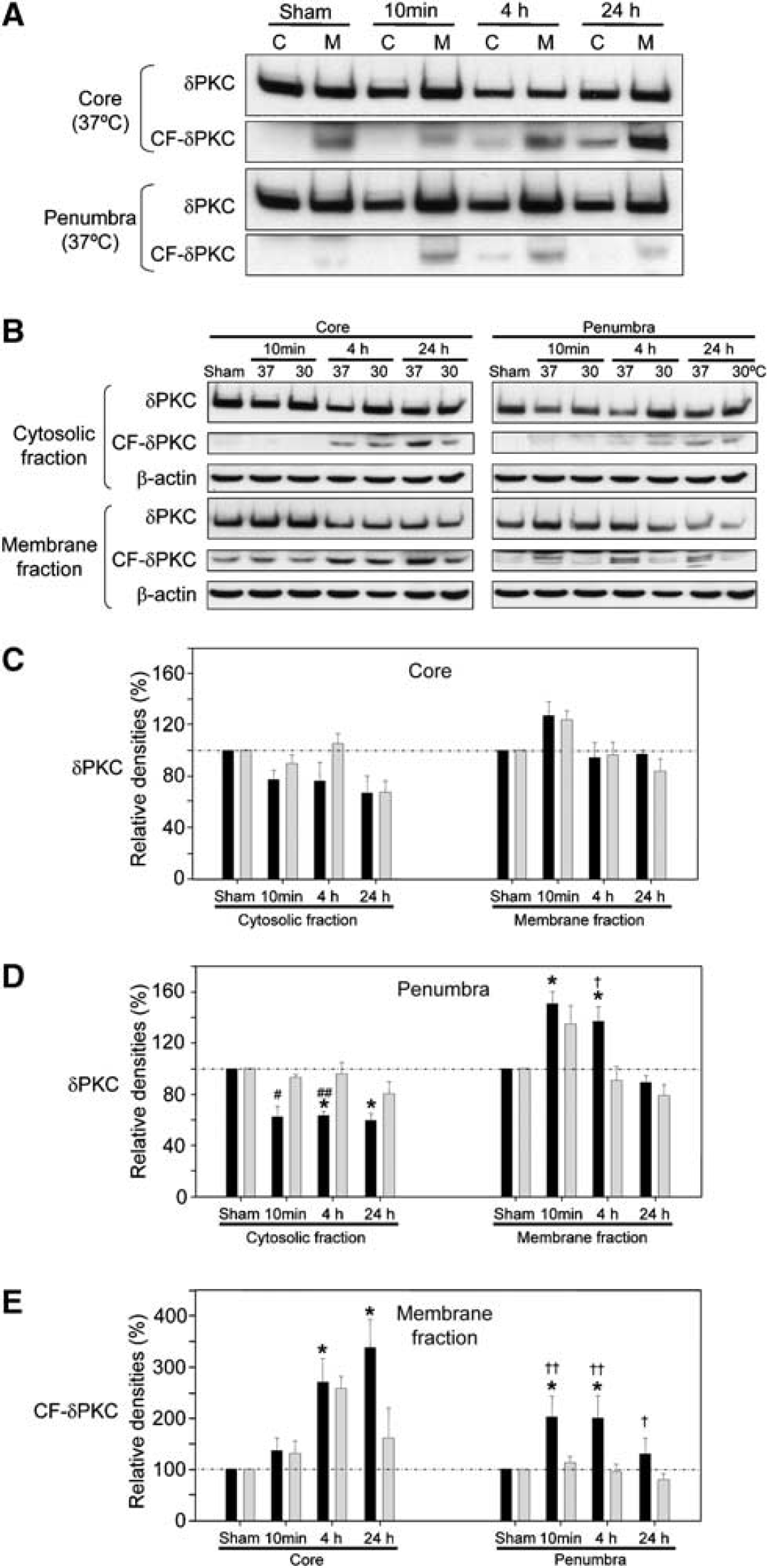
Hypothermia blocks membrane translocation of δPKC and CF-δPKC in the penumbra. (
We also investigated membrane translocation of p-δPKC (Ser643) after stroke. Similar to δPKC, stroke-induced membrane translocation of p-δPKC (Ser643) in the penumbra was inhibited by hypothermia, but not in the ischemic core (data not shown).
Hypothermia Suppresses Mitochondrial Delta Protein Kinase C and Phosphorylated Delta Protein Kinase C (Ser643) Translocation
As we found in cardiac ischemia (Murriel et al, 2004), δPKC translocated to the mitochondrial fraction starting at 10 mins after termination of the ischemic insult in both the ischemic core and penumbra (Figures 5A and 5B). In addition, an increase in mitochondrial CF-δPKC was also observed at 4 and 24 h, being more marked in the ischemic core and less in the penumbra (Figures 5A and 5C). Hypothermia suppressed mitochondrial translocation of δPKC in the penumbra, but not in the ischemic core (Figures 5A and 5B). Hypothermia also suppressed the increase in mitochondrial CF-δPKC in both the ischemic core and penumbra, even though a significant increase in mitochondrial CF-δPKC was still observed in the ischemic core at 24 h in hypothermic rats (Figures 5A and 5C). Mitochondrial translocation of p-δPKC (Ser643) was also observed in both the ischemic core and penumbra, which was suppressed by hypothermia (Figures 5A and 5D).
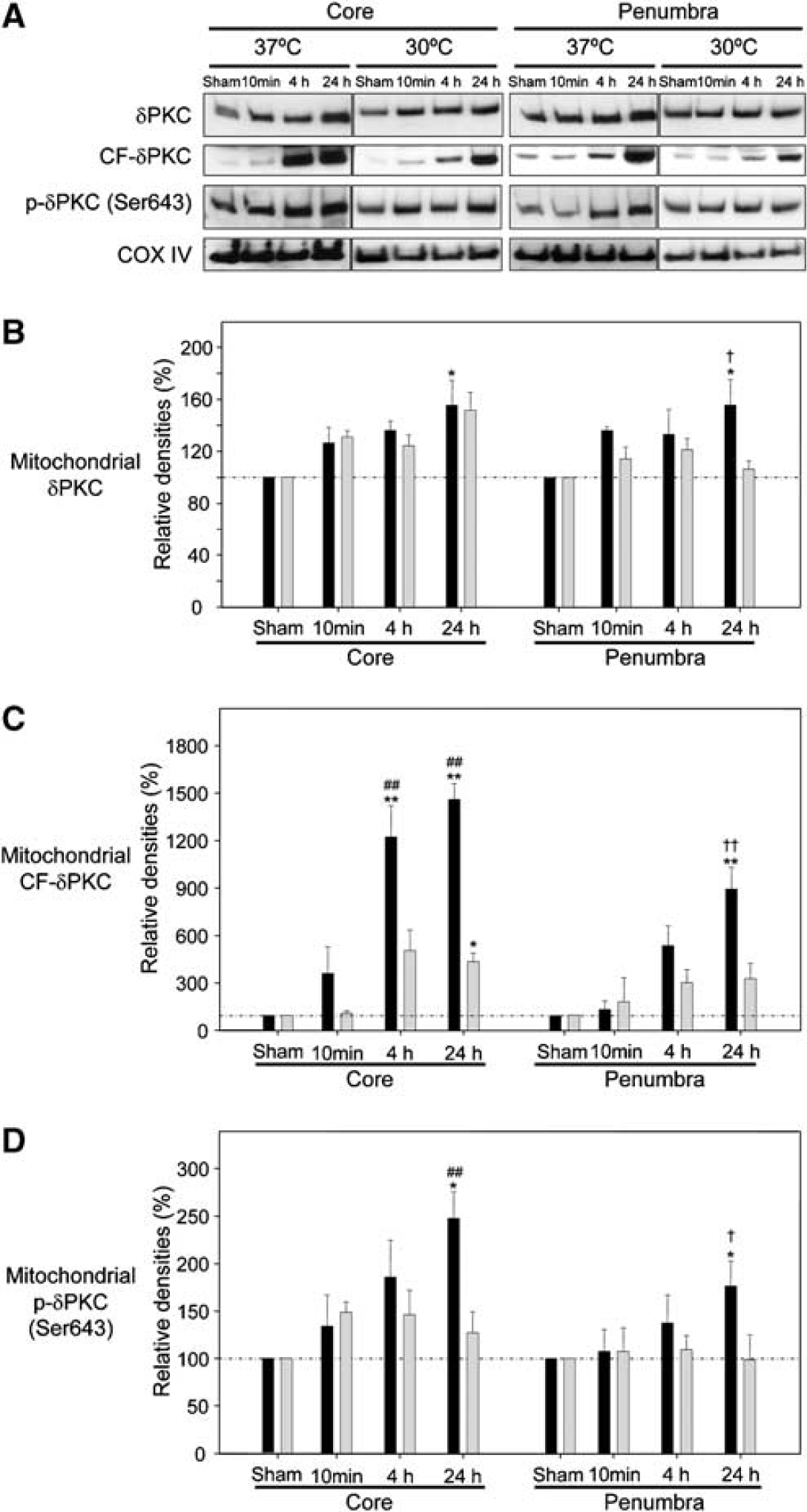
Hypothermia suppresses mitochondrial translocation of δPKC. (
Nuclear Translocation of Delta Protein Kinase C, C-terminal Catalytic fragment-Delta Protein Kinase C, and Phosphorylated Delta Protein Kinase C (Ser643) in the Penumbra after Stroke is Reduced by Hypothermia
Some δPKC and p-δPKC (Ser643), but not the CF-δPKC, was observed in the nuclear fraction of sham-operated brains (Figure 6A). At 24 h after CCA release, nuclear levels of δPKC (Figures 6A and 6B) and p-δPKC (Ser643) (Figures 6A and 6C) significantly increased, and CF-δPKC (Figures 6A and 6F) was detected in the nuclear fraction of both the ischemic core and penumbra. Hypothermia attenuated these increases in the penumbra but not in the ischemic core (Figures 6D–6H).
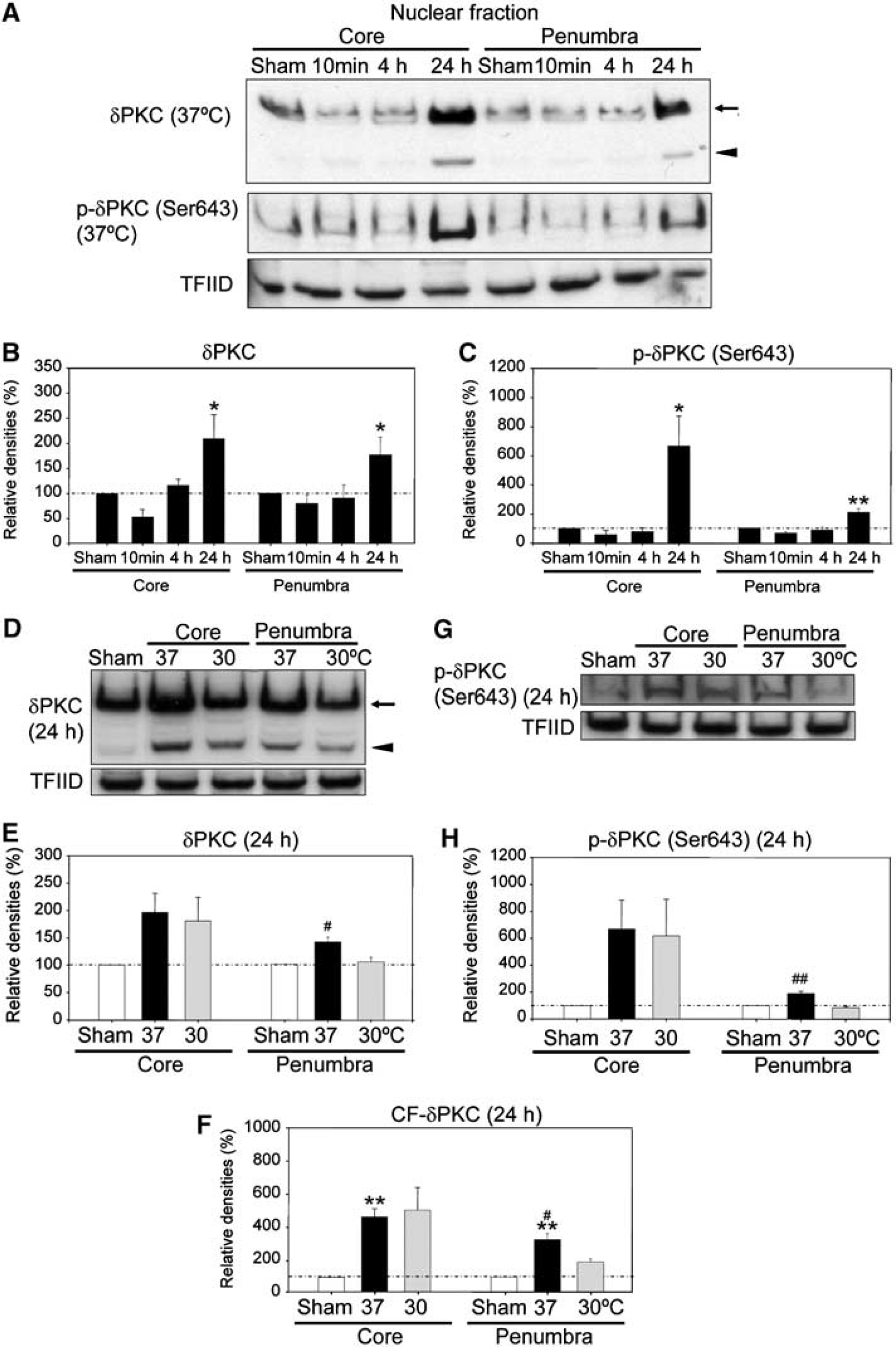
Hypothermia blocks nuclear translocation of δPKC. (
To confirm nuclear translocation of δPKC at 24 h, we performed confocal immunofluorescence microscopy (Figure 7). In sham-operated brains, δPKC was observed mainly in the cytoplasm and axons of the neurons. After stroke, δPKC translocated to the nuclei of neurons in the ischemic core and penumbra; this translocation was suppressed by hypothermia.
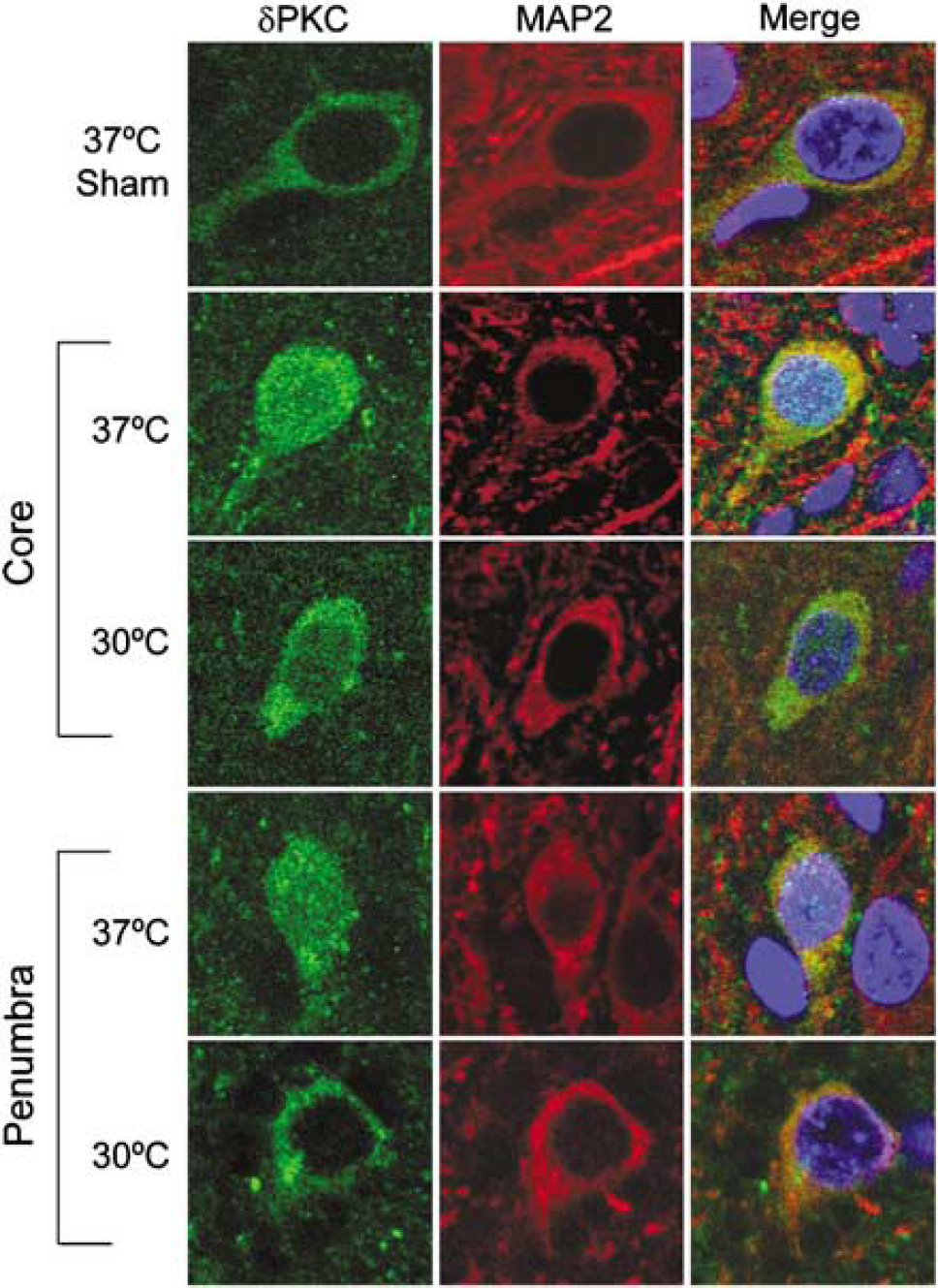
Subcellular localization of δPKC with and without ischemia. Triple staining of δPKC (green), microtubule-associated protein 2 (MAP2; red) and DAPI (blue). Hypothermia attenuated nuclear translocation of δPKC after stroke in the ischemic core and penumbra. Scale bar, 10 μm.
Cytochrome c is Released after Stroke and Hypothermia Blocks Cytochrome c Release During Early Reperfusion
Mitochondrial translocation of δPKC and CF-δPKC might cause mitochondrial injury leading to caspase-3 activation and apoptosis. Thus, the time course of cytochrome c release from the mitochondria to the cytoplasm was determined. Cytosolic cytochrome c levels significantly increased at 10 mins after CCA release in the ischemic core and penumbra, and again at 48 h in the core and at 24 h in the penumbra (Figures 8A and 8B). Hypothermia completely blocked cytochrome c release at 10 mins in the penumbra, but not in the ischemic core (Figures 8C and 8D).
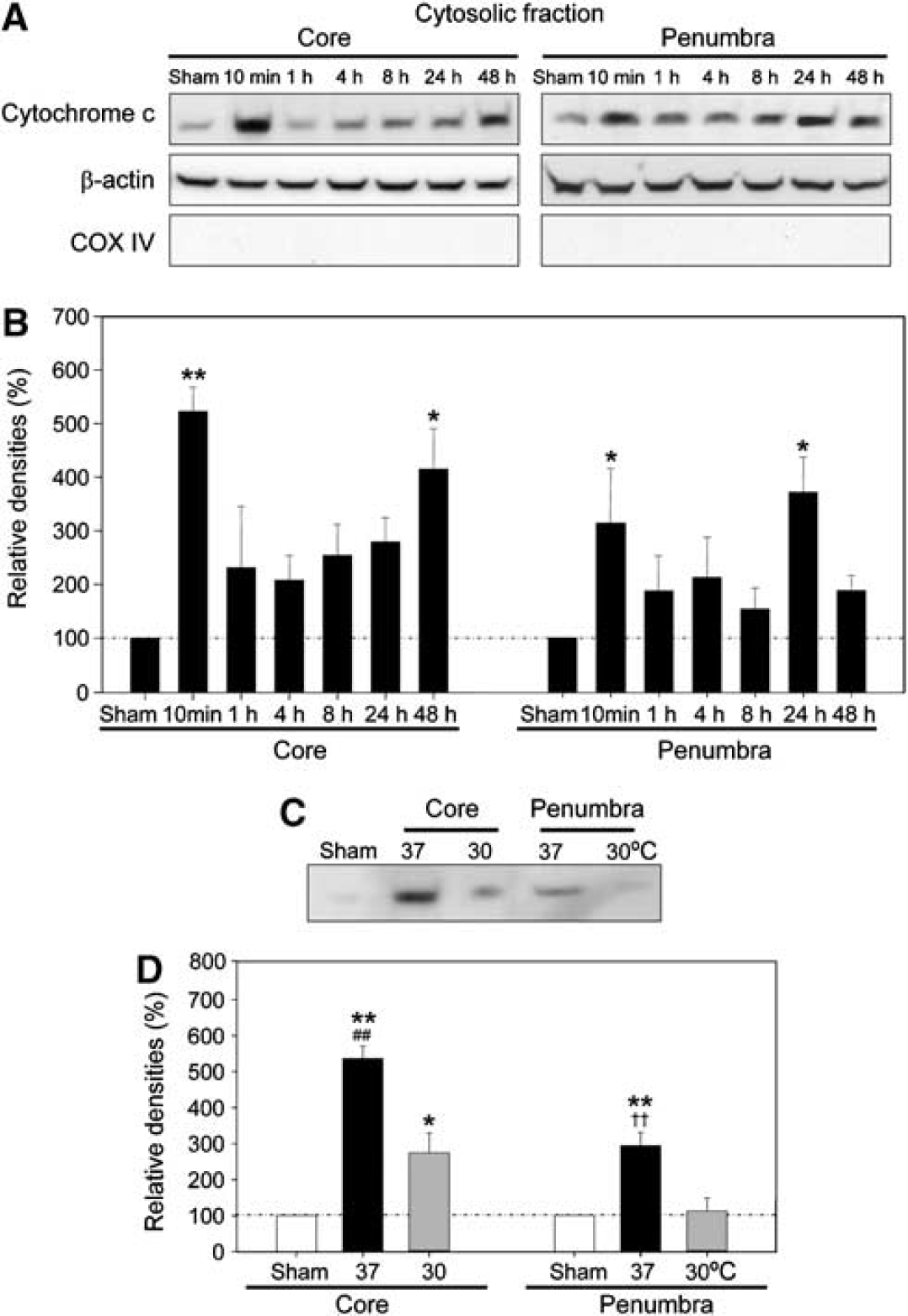
Biphasic release of cytochrome c after stroke. (
The Delta Protein Kinase C Activator, ψδRACK, Partially Reverses the Protective Effect of Hypothermia
To provide direct evidence that δPKC inhibition contributes to the hypothermia-induced protective effect, we examined the effect of δPKC activation on infarct size in hypothermic rats (Figure 9A). Intraperitoneal injection of the δPKC-selective activator, ψδRACK (Chen et al, 2001), after CCA release partially reversed the protective effect of hypothermia at 24 h (Figure 9B).
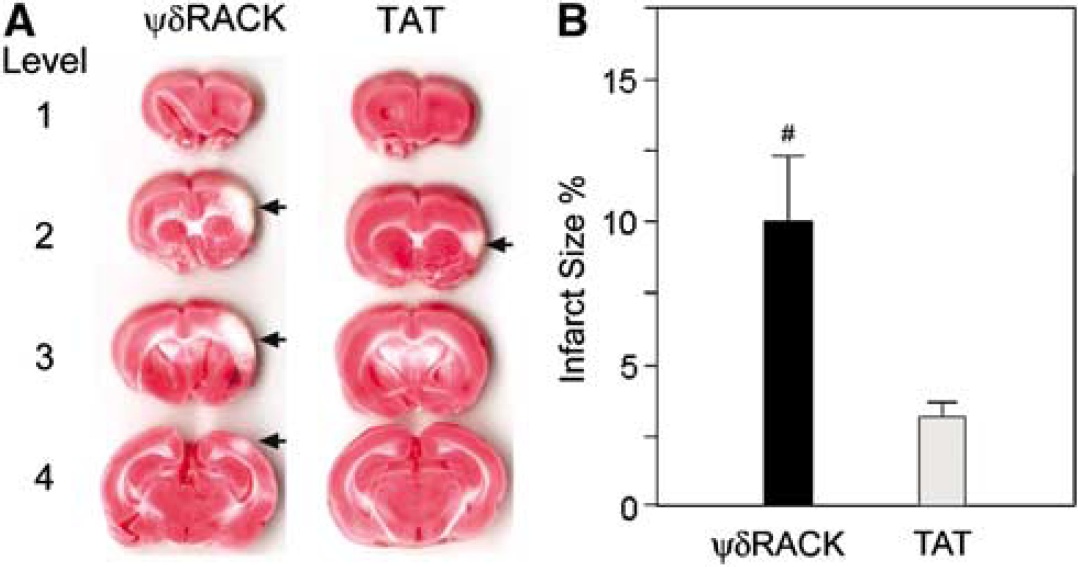
Delta protein kinase C-selective activator partially reverses the protective effect of hypothermia. (
Discussion
We had recently shown that inhibition of δPKC results in decreased infarct size after transient cerebral ischemia (Bright et al, 2004). In the present study, we provide evidence for the first time that hypothermia exerts a protective effect in the penumbra by suppressing δPKC activity in a rat focal cerebral ischemia model. Several novel observations were made. First, we showed that after focal cerebral ischemia, δPKC translocated to the mitochondria and nuclei, and that hypothermia attenuated such subcellular translocation in the penumbra but not in the ischemic core. Second, we showed that δPKC is cleaved by caspase-3 to generate a catalytic fragment after focal ischemia, and that the resultant CF-δPKC increased in the membrane fraction, mitochondria, and nuclei; these effects were suppressed by hypothermia in the penumbra. We have shown δPKC activation via its subcellular translocation and proteolytic cleavage, although it is not known which mechanism is critical. However, it is clear that proteolytic cleavage induces irreversible activation. Third, we observed cytochrome c release as early as 10 mins after CCA release, and this was also inhibited by hypothermia. We also found an increase in cytochrome c release in ischemic core at 48 h of reperfusion, which might be derived from remaining dead cells or cell debris in the ischemic tissues. However, the exact mechanism of this increased cytochorme c is not clear. Most importantly, the specific δPKC activator, ψδRACK, blocked the protective effect of hypothermia, providing further evidence that δPKC inhibition contributes to the protective effect of hypothermia.
Proteolytic Cleavage of Delta Protein Kinase C after Stroke is Suppressed by Hypothermia in the Penumbra
Delta protein kinase C activation by caspase-3-mediated proteolytic cleavage occurs in response to a variety of stimuli, including DNA-damaging agents, Fas ligand, and ultraviolet-irradiation. Delta protein kinase C cleavage has also been shown in neuronal cells after exposure to neurotoxic agents (Anantharam et al, 2002). When CF-δPKC is over-expressed in cultured cells, it rapidly induces apoptosis (Raval et al, 2005).
Few reports have investigated CF-δPKC generation after stroke. A recent study showed that δPKC is activated via caspase-3-mediated cleavage in hippocampal CA1 neurons after cardiac arrest in rats (Raval et al, 2005). We found a similar phenomenon after focal ischemia in this study. Full-length δPKC decreased after stroke because of its proteolytic cleavage, which differs from findings in another report (Miettinen et al, 1996) that used a different model of focal cerebral ischemia. C-terminal catalytic fragment-δPKC increased markedly in the ischemic core and moderately so in the penumbra after stroke. C-terminal catalytic fragment-δPKC accumulation in the penumbra was prevented by a caspase-3 inhibitor, which suggests that its cleavage might be regulated by caspase-3 activity. Delta protein kinase C cleavage after stroke in the penumbra was prevented by hypothermia, but not in the ischemic core, where hypothermia does not prevent tissue damage. We previously found that caspase-3 activity after global ischemia is markedly attenuated by hypothermia (Zhao et al, 2005a), which is consistent with our hypothesis that hypothermia inhibits δPKC cleavage by attenuating caspase-3 activity.
Hypothermia Reduces Subcellular Translocation of Delta Protein Kinase C to the Membrane Fraction and Mitochondria in the Penumbra
Membrane translocation of δPKC occurred as early as 10 mins after CCA release, which is consistent with our previous report using a suture middle cerebral artery occlusion model (Bright et al, 2004). This suggests that δPKC activation via its membrane translocation starts immediately after reperfusion after focal ischemia, although δPKC kinase activity should be measured in the future. We did not see membrane translocation of δPKC in the ischemic core at 24 h, perhaps because of δPKC proteolysis.
The membrane fraction is derived from several organelles such as mitochondria and nuclei. We showed that δPKC started to translocate to mitochondrial fraction at 10 mins after CCA release, and that mitochondrial fraction of CF-δPKC increased after several hours. Mitochondrial translocation of δPKC in response to diverse stimuli has been reported in several oxidative stress models (Churchill et al, 2005). In addition, accumulation of CF-δPKC in the mitochondria is responsible for mitochondrial damage induced by a proteasome inhibitor (Durrant et al, 2004). Furthermore, mitochondrial δPKC triggers cytochrome c release via loss of mitochondrial membrane potential, and thereby activation of caspase-3 (Majumder et al, 2000). Interestingly, release of cytochrome c from the mitochondria was seen as early as 10 mins after CCA release in the current study, which corresponded to the time course of δPKC translocation. Whether, δPKC translocation to the mitochondria triggers cytochrome c release and CF-δPKC formation in the ischemic brain, as in the ischemic heart (Murriel et al, 2004), remains to be determined. Nonetheless, our data are consistent with δPKC translocation to the mitochondria as the trigger for activation of caspase-3 and further δPKC cleavage (Figure 10).
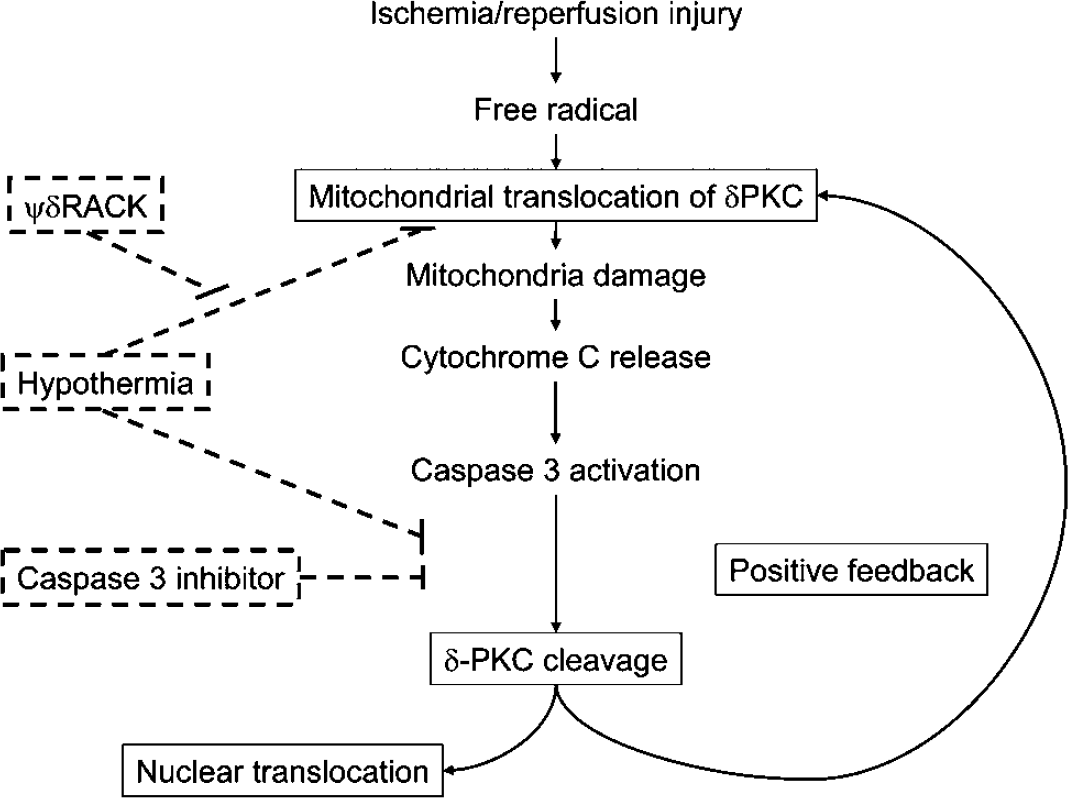
Hypothesis for δPKC activation after stroke and the effects of hypothermia. Free radical generation after ischemic and reperfusion injury causes immediate mitochondrial translocation of δPKC, resulting in mitochondrial damage. Mitochondrial damage promotes cytochrome c-dependent caspase-3 activation leading to CF-δPKC production. A recent study suggests that caspase-3 function in the early stage of apoptosis, which causes mitochondrial cytochrome c release (Lakhani et al, 2006). We also hypothesize that caspase-3 activation is responsible for δPKC cleavage. C-terminal catalytic fragment-δPKC feeds back onto the mitochondria, which further activates caspase-3, therefore leading to more δPKC cleavage (positive feedback). Delta protein kinase C and CF-δPKC also translocates into the nuclei and might cause changes in nuclear functions. Hypothermia prevents ischemic damage by attenuating subcellular translocation and proteolytic cleavage of δPKC.
Our previous study showed that hypothermia attenuates cytochrome c release after stroke using a rat transient focal cerebral ischemia model (Yenari et al, 2002), suggesting that the neuroprotective effect of hypothermia might be a result of maintaining mitochondrial integrity. One potential mechanism for this protection may be through reduced mitochondrial translocation of δPKC.
Hypothermia Inhibits Nuclear Translocation of Delta Protein Kinase C and C-terminal Catalytic Fragment-Delta Protein Kinase C in the Penumbra
Nuclear translocation of δPKC causes apoptosis in various models, which include glioma cells treated with etoposide (Blass et al, 2002) and cytokine-deprived T-cells (Scheel-Toellner et al, 1999). Overexpression of CF-δPKC in cultured cells results in its nuclear translocation and apoptosis. Delta protein kinase C cleavage by caspase-3 not only generates CF-δPKC but also facilitates nuclear transport, mediated by a nuclear localization signal in the C-terminus of δPKC (DeVries et al, 2002). Consistent with these previous reports, we observed δPKC and CF-δPKC translocated into the nuclei after stroke, and hypothermia inhibits such translocation in the penumbra but not in the ischemic core. Hypothermia may exert a protective effect by blocking nuclear translocation of δPKC in the penumbra, but we do no yet have functional evidence to support this idea.
Delta protein kinase C and CF-δPKC phosphorylate nuclear proteins, leading to apoptosis in cultured cells. Delta protein kinase C phosphorylates lamin B, a nuclear structural protein, causing the disassembly of the nuclear lamina (Cross et al, 2000). C-terminal catalytic fragment-δPKC also phosphorylates and suppresses DNA-dependent protein kinase, an enzyme that plays an essential role in the repair of DNA double-strand breaks (Bharti et al, 1998). Furthermore, CF-δPKC phosphorylates p73β, a homologue of the p53 tumor suppressor, stimulating p73β-mediated apoptosis (Ren et al, 2002). The critical δPKC and CF-δPKC nuclear substrates in the ischemic brain are currently unknown.
Hypothermia Blocks Dephosphorylation of Phosphorylated-Delta Protein Kinase C (Thr505) after Stroke
Phosphorylation of several conserved serine and/or threonine residues regulates the activity of PKC isozymes. In particular, phosphorylation of threonine residue in the ‘activation-loop’ is considered to be essential for the activation of several PKC isozymes. However, the role of phosphorylation of the corresponding threonine residue Thr505, in δPKC is still controversial (Le Good et al, 1998; Stempka et al, 1999). In addition to the serine/threonine phosphorylation, tyrosine phosphorylation might modulate δPKC activity. Nevertheless, it is not known how cerebral ischemia affects phosphorylation of δPKC. We observed that total levels of p-δPKC (Ser643) did not change after stroke, and that p-δPKC (Thr505) markedly decreased in the ischemic core, but only mildly so in the penumbra. Hypothermia did not change the level of p-δPKC (Ser643) after stroke, but it attenuated the decreases in p-δPKC (Thr505). Thus, the phosphorylation levels may not reflect δPKC activity.
The mechanisms underlying the decrease in p-δPKC (Thr505) after stroke and its inhibition by hypothermia is unknown. Previously, using the same model, we found that PDK1 was phosphorylated (p-PDK1). The levels of P-PDK1 decreased after stroke, and hypothermia suppressed the decrease in p-PDK1 (Zhao et al, 2005b). PDK1 has been shown to phosphorylate Thr505 in δPKC (Hodgkinson and Sale, 2002). It is possible that the decrease in p-δPKC (Thr505) might be caused in part by decreased activity of PDK1 after stroke. Taken together, our studies are consistent with δPKC playing critical roles in ischemic–reperfusion injury by participating in the caspase-dependent apoptotic pathway and the PI3K/Akt survival pathway.
Delta Protein Kinase C as Therapeutic Target for a Focal Cerebral Ischemia
We had previously shown that administration of the δPKC-specific inhibitor peptide, δV1-1, significantly reduced infarct size when delivered over an extended time of reperfusion after a 2-h middle cerebral artery occlusion (Bright et al, 2004). Here we further implicate δPKC as a key player in ischemic damage by demonstrating that the specific δPKC activator, ψδRACK, partially reversed the protective effect of hypothermia. ψδRACK did not completely block the neuroprotective effect of hypothermia, but this could be because of incomplete efficacy of the peptide to activate δPKC in vivo. Although we have mainly discussed the activation of δPKC in neurons, a recent study using δPKC-null mice identified a critical role for neutrophil δPKC, as a mediator of inflammation and oxidative stress during ischemic and reperfusion injury (Chou et al, 2004). In summary, these findings suggest that δPKC is a promising therapeutic target against stroke.
Footnotes
Acknowledgements
The authors thank Dr Bruce Schaar for editing, Beth Hoyte for preparation of the figures, and David Kunis for technical assistance.
Dr Mochly-Rosen is the founder of KAI Pharmaceuticals, Inc., a company that plans to bring PKC regulators to the clinic. However, none of the work described here is based on or supported by the company.