
Abstract
Mononuclear phagocytes are a population of multi-phenotypic cells and have dual roles in brain destruction/reconstruction. The phenotype-specific roles of microglia/macrophages in traumatic brain injury (TBI) are, however, poorly characterized. In the present study, TBI was induced in mice by a controlled cortical impact (CCI) and animals were killed at 1 to 14 days post injury. Real-time polymerase chain reaction (RT–PCR) and immunofluorescence staining for M1 and M2 markers were performed to characterize phenotypic changes of microglia/macrophages in both gray and white matter. We found that the number of M1-like phagocytes increased in cortex, striatum and corpus callosum (CC) during the first week and remained elevated until at least 14 days after TBI. In contrast, M2-like microglia/macrophages peaked at 5 days, but decreased rapidly thereafter. Notably, the severity of white matter injury (WMI), manifested by immunohistochemical staining for neurofilament SMI-32, was strongly correlated with the number of M1-like phagocytes.
INTRODUCTION
Traumatic brain injury (TBI) is a neurologic disorder with growing prevalence among young adults. Traumatic brain injury poses a significant financial burden to society as well as physical and emotional burden to victims and caregivers. Neurologic morbidities after TBI are common and include cognitive impairments, dementia, epilepsy, and depression.1, 2, 3, 4 The impact of TBI on brain tissue has been studied for many decades; from these studies, TBI is known to damage gray matter and elicit neuronal death. However, TBI can also elicit severe white matter injury (WMI) that correlates with long-term deficits in motor and cognitive function.5, 6 Complications after TBI involve more than the initial tissue damage induced directly by mechanical trauma.1, 7, 8 Delayed secondary events, such as ischemia, lipid degradation, free radical formation, excitotoxicity, and protease release can be equally devastating.9, 10, 11 For example, secondary events can lead to demyelination, axonal degeneration, neuronal death, cavitation, and glial scarring around the area of the initial damage.9, 10, 11 Inflammation, including the activation of resident microglial cells, is thought to have an important role in these secondary changes.3, 9, 10, 12
Microglia/macrophages are the primary mediators of the immune defense system in the central nervous system (CNS) and are integral to subsequent inflammatory responses.12, 13 Resident microglia and peripheral macrophages are rapidly mobilized to the site of injury and initiate the release of effector molecules and recruitment of other immune cells.14, 15 An increasing number of studies now agree that microglia/macrophages are highly plastic cells that can assume diverse phenotypes and engage different functional programs in response to specific microenvironmental signals.13, 15 The dual roles of distinctly polarized microglia/macrophage populations have been reported in several CNS disorders, including stroke, 16 multiple sclerosis, 17 and spinal cord injury. 18 However, the specific roles of polarized microglia/macrophages in the pathophysiology of TBI have not been explored.
The goal of the present study was to analyze the temporal kinetics of microglia/macrophage polarization after TBI using a well-established murine model of controlled cortical impact (CCI). Based on our previous observations in ischemic injury models,
16
we tested the hypothesis that microglia/macrophages respond dynamically to mechanical trauma, switching from a transient M2 phenotype to a sustained M1 phenotype. In support of this hypothesis, we found that microglia/macrophages expressed the early M2 phenotype after TBI, but that this was gradually replaced by the M1 phenotype at the site of injury. Notably, the severity of WMI was strongly correlated with activation of the M1 phenotype.
MATERIALS AND METHODS
Animals
Young male C57BL/6J mice (10- to 12-week-old) were obtained from Jackson Laboratory (Bar Harbor, Maine, USA). Animals were housed in groups of four per cage in a temperature- and humidity-controlled animal facility with a 12-hour light–dark cycle. Food and water were available
Traumatic Brain Injury
Traumatic brain injury was induced in C57/BL6 mice by a CCI, as described previously. 11 Briefly, all animals were randomly assigned into experimental groups through the use of a lottery drawing box. Mice were anesthetized with 1.5% isoflurane in a 30% O2/68.5% N2O mixture under spontaneous breathing conditions. Using aseptic techniques and principles consistent with IACUC surgical policy, an approximately 4-mm craniotomy was performed over the right parietotemporal cortex using a motorized drill. The CCI was centered 2 mm lateral to the midline and 1.0 mm anterior to the bregma and produced with a pneumatically driven CCI device (Precision Systems and Instrumentation, Fairfax, VA, USA) using a 3 mm flat-tip impounder (velocity, 3.5 m/second; duration, 150 milliseconds; depth, 1.5 mm). Immediately after injury, the bone flap was replaced and sealed with Koldmount cement (Vernon Benshoff, Albany, NY, USA), and the scalp was sutured shut. Rectal temperature was controlled at 37°C±0.5°C during surgery and up to 30 minutes after TBI using a temperature-regulated heating pad. Sham animals were subjected to all aspects of the protocol (surgery, anesthesia, craniotomy, recovery) except for CCI.
Immunohistochemistry and Cell Counting
After killing by an anesthetic overdose and perfusion with cold saline followed by formaldehyde (4% in phosphate-buffered saline), brains were removed and cryoprotected in 30% sucrose in phosphate-buffered saline. Serial sections (25-
Fluorescence Quantification
All fluorescence values were generated using confocal microscopy (FV1000, Olympus, Tokyo, Japan) and analyzed semi-quantitatively with ImageJ software. Fluorescence intensity of SMI-32 in the CC area affected by CCI was contrasted with the CC of sham animals. The region of interest for the CC area was drawn close to the lesion cavity, whereas a mirror image of this outline was drawn over the contralateral CC in the contralateral hemisphere. Similar regions of interest were drawn in sham animals in the same anatomic regions. The analysis was done by an investigator masked to experimental group.
Real-Time Polymerase Chain Reaction
Total RNA was isolated from sham and injured brains using the RNeasy Mini Kit (Qiagen, Germantown, MD, USA) according to the manufacturer's instructions. Five micrograms of RNA was used to synthesize the first strand of cDNA using the Superscript First-Strand Synthesis System (Invitrogen, Grand Island, NY, USA) for reverse-transcription polymerase chain reaction (RT–PCR). Polymerase chain reaction was performed with the Opticon 2 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA), and SYBR gene PCR Master Mix (Invitrogen) as described previously. 16 The cycle time values were normalized to glyceraldehyde 3-phosphate dehydrogenase messenger RNA levels in the same sample. The expression levels of the messenger RNA were then reported as fold changes versus sham controls. The sequences of the primer pairs for M1 phenotype genes are as follows (in pairs, sense, and antisense): (1) iNOS: CAAGCACCTTGGAAGAGGAG and 5′-AAGGCCAAACACAGCATACC-3′; (2) CD32: 5′-AATCCTGCCGTTCCTACTGATC-3′ and 5′-GTGTCACCGTGTCTTCCTTGAG-3′; (3) CD16: 5′-TTTGGACACCCAGATGTTTCAG-3′ and 5′-GTCTTCCTTGAGCACCTGGATC-3′; (4) CD86: 5′-GACCGTTGTGTGTGTTCTGG-3′ and 5′-GATGAGCAGCATCACAAGGA-3′; (5) CD11b: 5′-CCAAGACGATCTCAGCATCA-3′ and 5′-TTCTGGCTTGCTGAATCCTT-3′. The sequences of the primer pairs for M2 phenotype genes are as follows: CD206: 5′-CAAGGAAGGTTGGCATTTGT-3′ and 5′-CCTTTCAGTCCTTTGCAAGC-3′; (2) Arg1: 5′-TCACCTGAGCTTTGATGTCG-3′ and 5′-CTGAAAGGAGCCCTGTCTTG-3′; (3) IL-10: 5′-CCAAGCCTTATCGGAAATGA-3′ and 5′-TTTTCACAGGGGAGAAATCG-3′; (4) CCL-22: 5′-CTGATGCAGGTCCCTATGGT-3′ and 5′-GCAGGATTTTGAGGTCCAGA-3′; (5) TGF-b: 5′-TGCGCTTGCAGAGATTAAAA-3′ and 5′-CGTCAAAAGACAGCCACTCA-3′; (6) Ym1/2: 5′-CAGGGTAATGAGTGGGTTGG-3′ and 5′-CACGGCACCTCCTAAATTGT-3′.
Primary Microglia and Oligodendrocyte Cultures
Primary microglia and oligodendrocytes were prepared from mixed glial cultures of 1-day-old mouse brains, as described previously.16, 19 Briefly, microglia were isolated by shaking flasks containing mixed glia for 1 hour at 180
Measurements of Cell Damage
Metabolic viability was assessed with the MTT method. The MTT solution (50
Phagocytosis Assay
For analysis of phagocytosis, microglia (1 × 105 cells/well) were plated into 8-well chamber slides (Nunc, ThermoScientific, Pittsburgh, PA, USA) as described previously. 16 Nile red fluorescent microspheres (Invitrogen) were solubilized to a concentration of 0.03% solids. Cells were incubated with or without microspheres for 3 hours. Cells were then rinsed with PBS and fixed in 4% paraformaldehyde. Alexa Fluor 488 phalloidin (Invitrogen) was added and incubated at room temperature in the dark for 1 hour. Images were captured with an Olympus confocal microscope.
Statistical Analyses
All values are presented as mean±standard error of the mean. Data with two groups were analyzed with the Student's
RESULTS
Dynamic Changes in Messenger RNA Expression of M1 and M2 Polarization Markers
Polarized microglia/macrophages can be distinguished by surface marker expression and cytokine/chemokine production. Real-time polymerase chain reaction was performed using total RNA extracted from ipsilateral striatum at 1, 3, 5, 7, and 14 days after TBI or from sham-operated brains (Figure 1A). Levels of M1-type genes (iNOS, CD11b, CD16, CD32, and CD86) gradually increased over time from day 3 onward and peaked at day 5 or 7 (Figure 1B). Furthermore, all but CD86 were maintained at relatively high levels for at least 14 days after TBI.
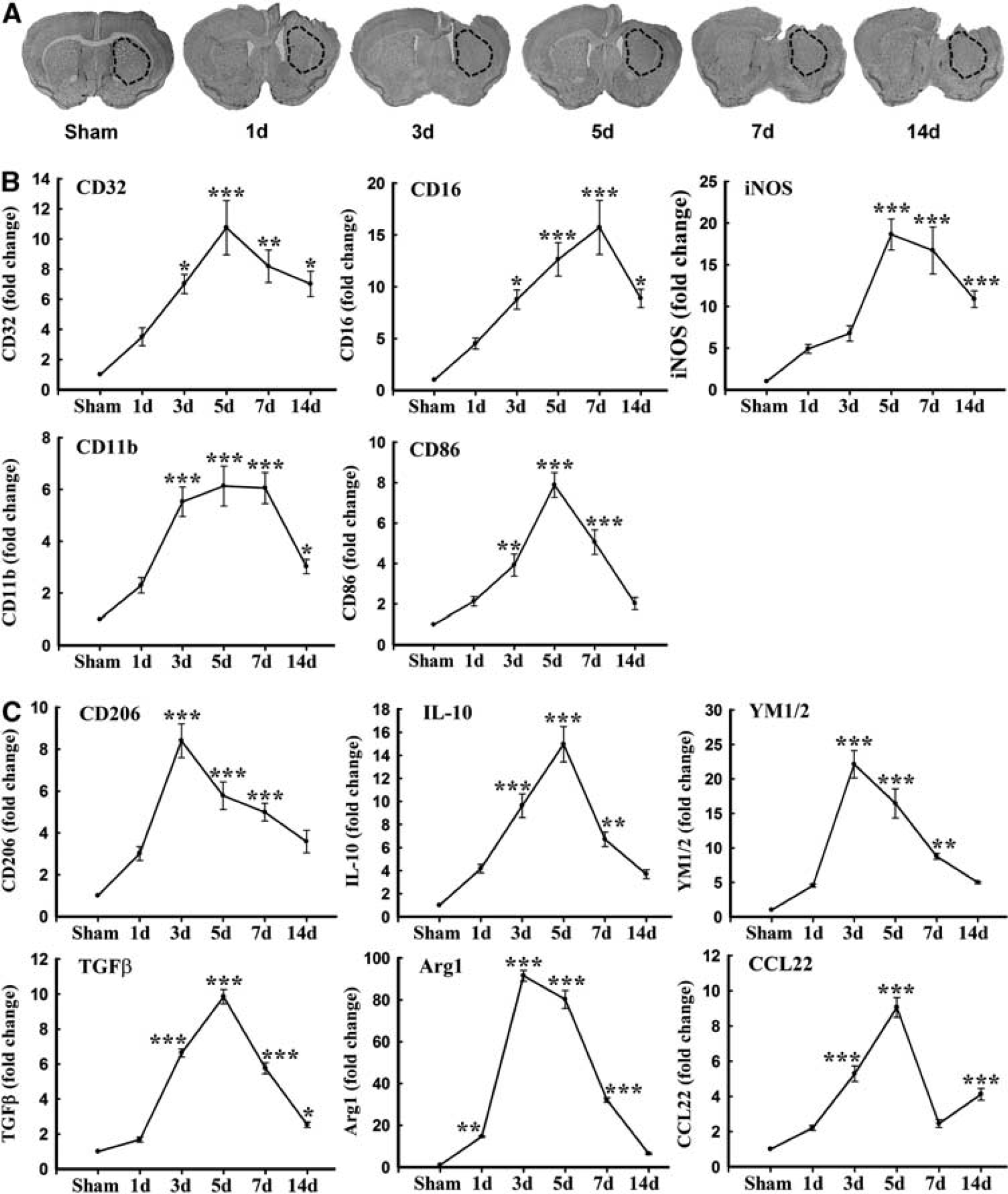
Traumatic brain injury (TBI) induced changes in messenger RNA (mRNA) expression of M1 and M2 polarization markers in injured tissue. (
In contrast to M1 markers, the messenger RNA expression of M2 markers, including CD206, Arg1, CCL-22, Ym1/2, IL-10, and transforming growth factor-
Dynamic Changes in Microglia/Macrophages with M1 or M2 Phenotypes
M1 and M2 signature genes are not only expressed in microglia/macrophages but also in other CNS cells or infiltrating immune cells. The real-time PCR data on striatal tissue therefore reflect a mixture of cell types. To specifically evaluate the polarization state of microglia/macrophages after TBI, representative M1-associated or M2-associated marker proteins were double labeled with the microglia/macrophage marker Iba1 in cortex, striatum, and CC near the lesion cavity, and in their corresponding contralateral regions (Figures 2A and 3A). The lack of a specific antibody for either microglia or macrophages precluded the distinction between local microglia and circulating macrophages recruited after injury into the brain. Thus, immunofluorescence for M1 and M2 markers in Iba1+ cells may reflect both microglia and macrophages.
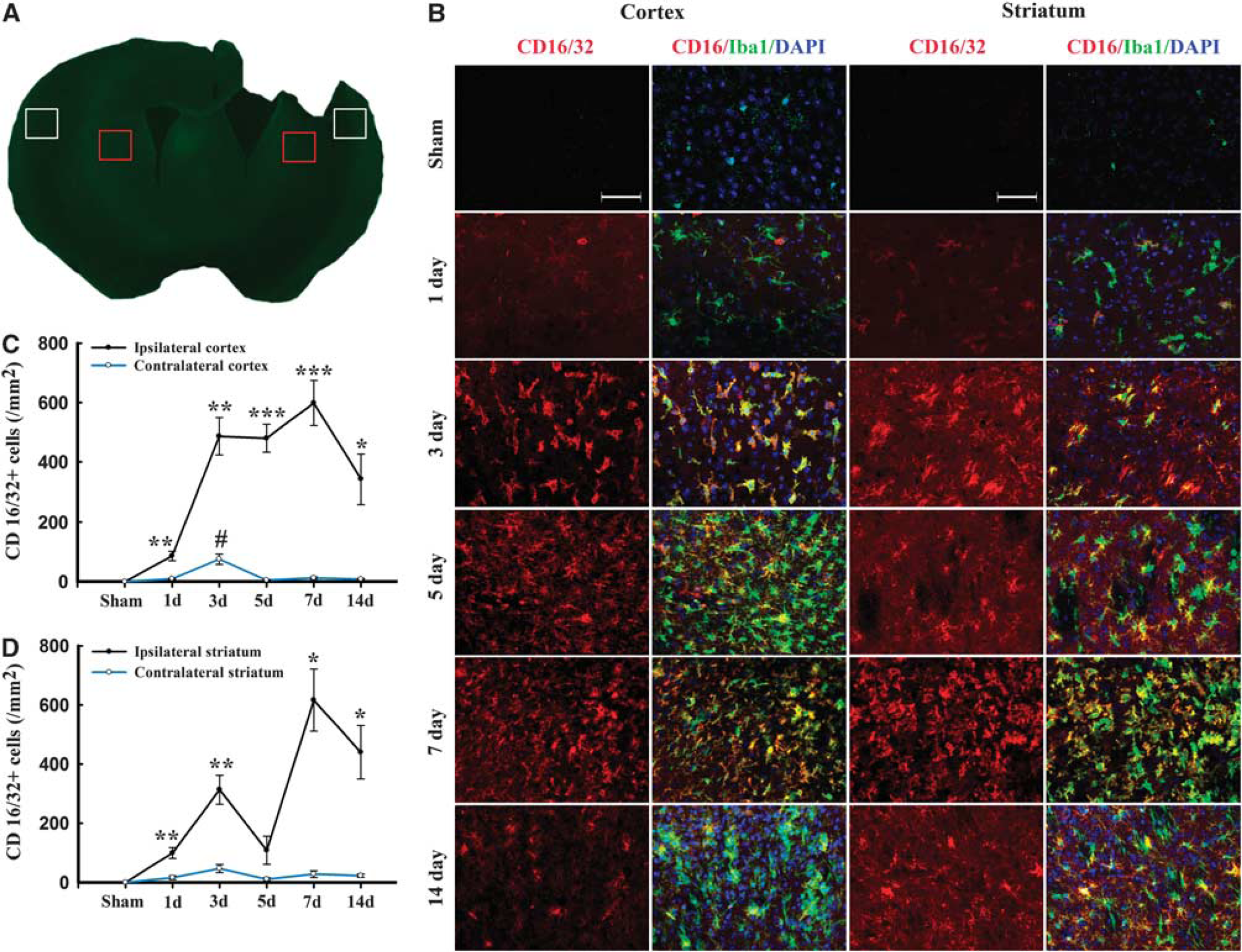
Temporal changes in microglia/macrophages expressing the M1 phenotype in cortex and striatum after traumatic brain injury (TBI). (
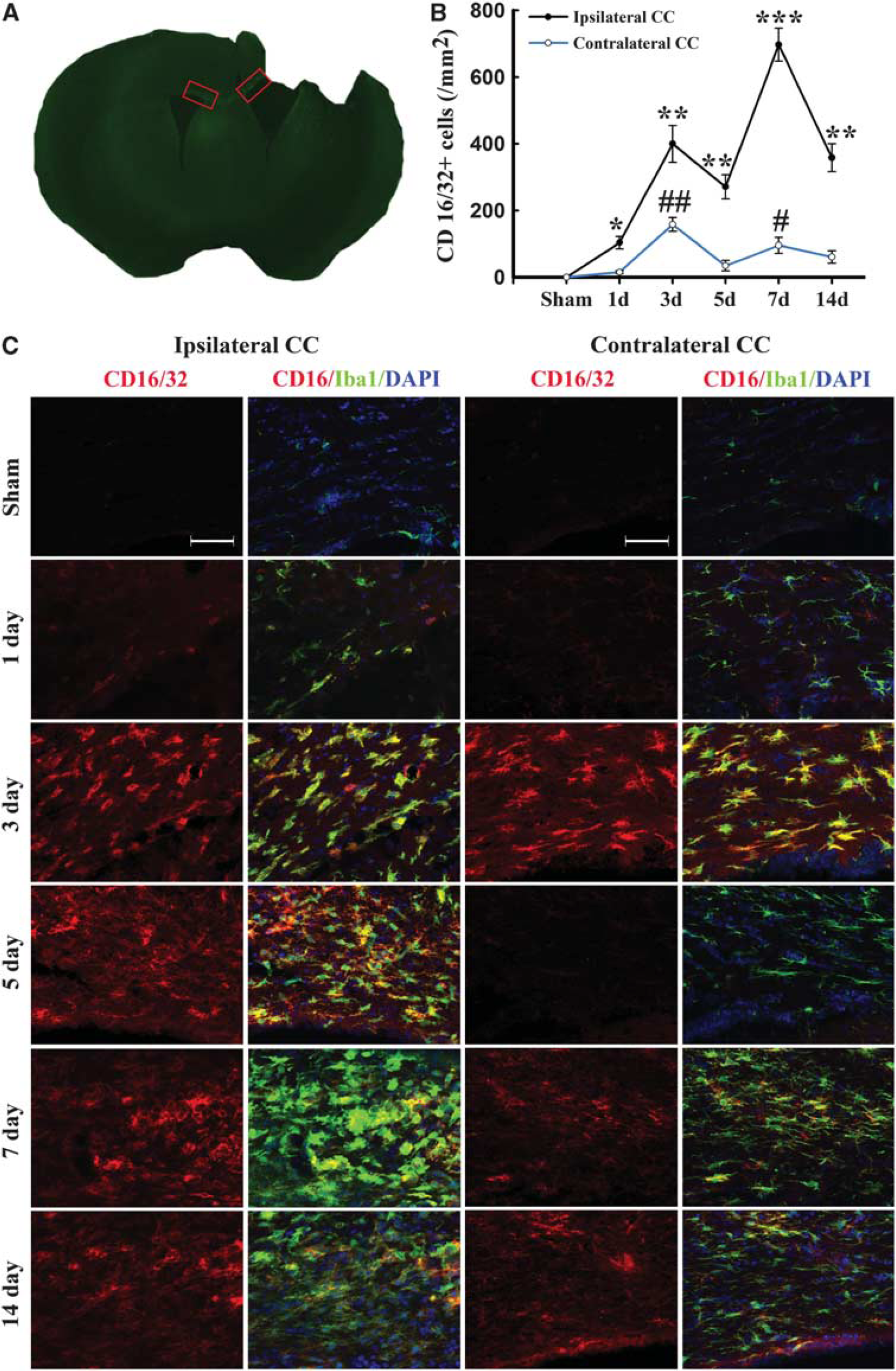
Temporal changes in microglia/macrophages expressing the M1 phenotype in corpus callosum (CC) after traumatic brain injury (TBI). (
Expression of the M1 marker CD16/32 slightly increased in cortex at day 1 after TBI and was significantly raised thereafter until day 14 (Figures 2B and C). This finding was consistent with the real-time PCR data. Interestingly, the number of CD16/32-positive microglia/macrophages in the contralateral cortex 3 days after TBI was higher than in sham-operated mice (Figure 2B). In the CC areas and ipsilateral striatum, CD16/32 expression peaked bimodally at 3 and 7 days after TBI (Figures 2B, 2D and 3B–C). CD16/32-positive cells were co-localized with Iba1, indicating that the majority of CD16/32 expression was associated with microglia/macrophages.
Immunofluorescence for the M2 marker CD206 increased significantly over control levels in the ipsilateral cortex and striatum at 3 days after TBI, peaking on day 5, and then decreasing back to baseline on day 14 (Figure 4). In contrast to the striatal and cortical expression of CD206, the ipsilateral CC area exhibited a peak in CD206 expression from day 3 onwards until day 5, followed by a gradual decrease to control levels (Figures 5A and C). Finally, CD206 was slightly increased after TBI in the contralateral CC, but only on day 3. Taken together, these findings reveal a temporal shift in microglial phenotype from transient M2 to sustained M1 in the 14 days after mechanical trauma.
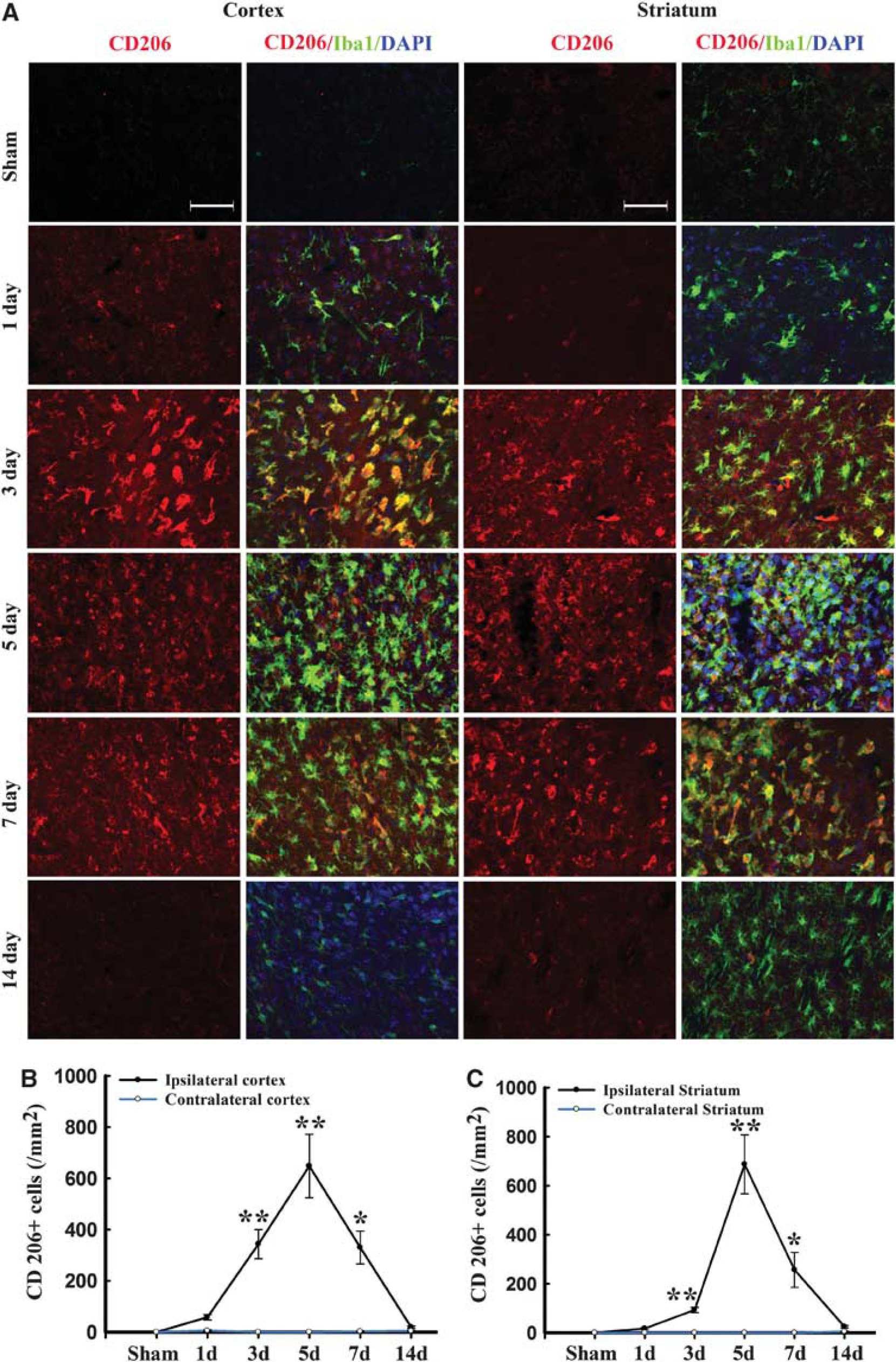
Temporal changes in microglia/macrophages expressing the M2 phenotype in cortex and striatum after traumatic brain injury (TBI). (
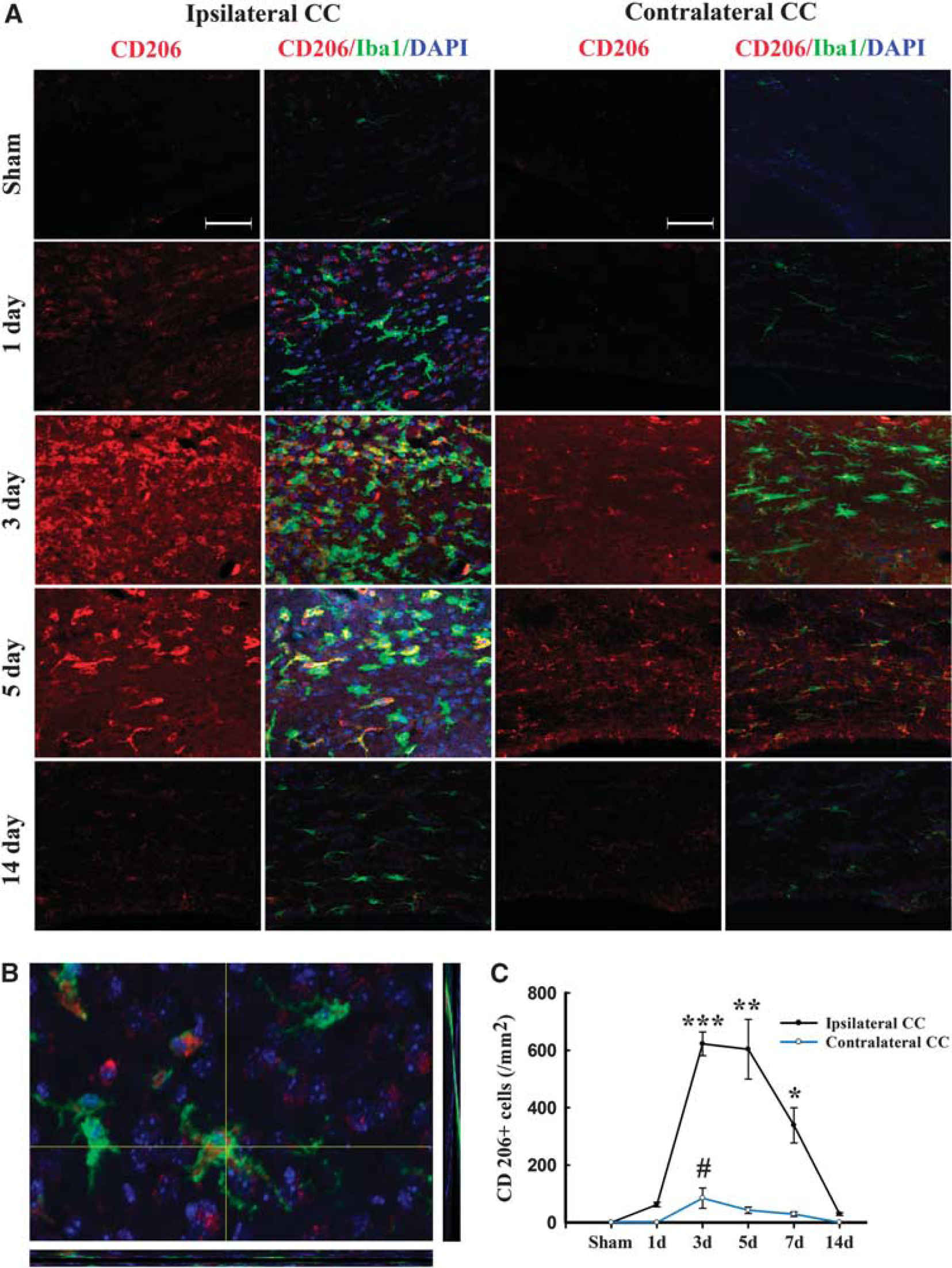
Temporal changes in microglia/macrophages expressing the M2 phenotype in corpus callosum (CC) after traumatic brain injury (TBI). (
White Matter Damage after Traumatic Brain Injury
To verify the presence of WMI in this TBI model, we performed co-immunostaining for MBP and non-phosphorylated neurofilament (SMI-32) in the CC area. Neurofilaments in healthy myelinated axons are heavily phosphorylated. Dephosphorylation of a neurofilament H epitope specifically recognized by the SMI-32 antibody, is a known marker of axonal damage. 20 As expected, SMI-32 immunoreactivity was rarely seen in normal-appearing white matter from sham-treated brains (Figure 6A). At 3 and 7 days after TBI, abundant SMI-32 immunoreactivity was detected within the CC (Figure 6B). The SMI-32 staining appeared as thin lines and small dots, reflecting the morphology of damaged axons (Figure 6A). We also found that MBP staining decreased early (day 3) after TBI and then increased by day 7, as compared with the contralateral hemisphere. It has been reported previously that the levels of MBP fragments change over time after TBI, with an early reduction (day 1 to 3) followed by dramatic increases by day 5 after TBI. 21 Thus our results are largely consistent with these previous findings and suggest that the late increase of MBP may represent an attempt by the brain to compensate for myelin loss from the initial injury. However, the anatomic distribution of MBP immunoreactivity remained irregular and discontinuous through day 7 after TBI (Figure 6A), suggesting ongoing impairments in axon myelination. Thus, the compensatory increases in MBP appear to be insufficient to fully restore the integrity of white matter after TBI.
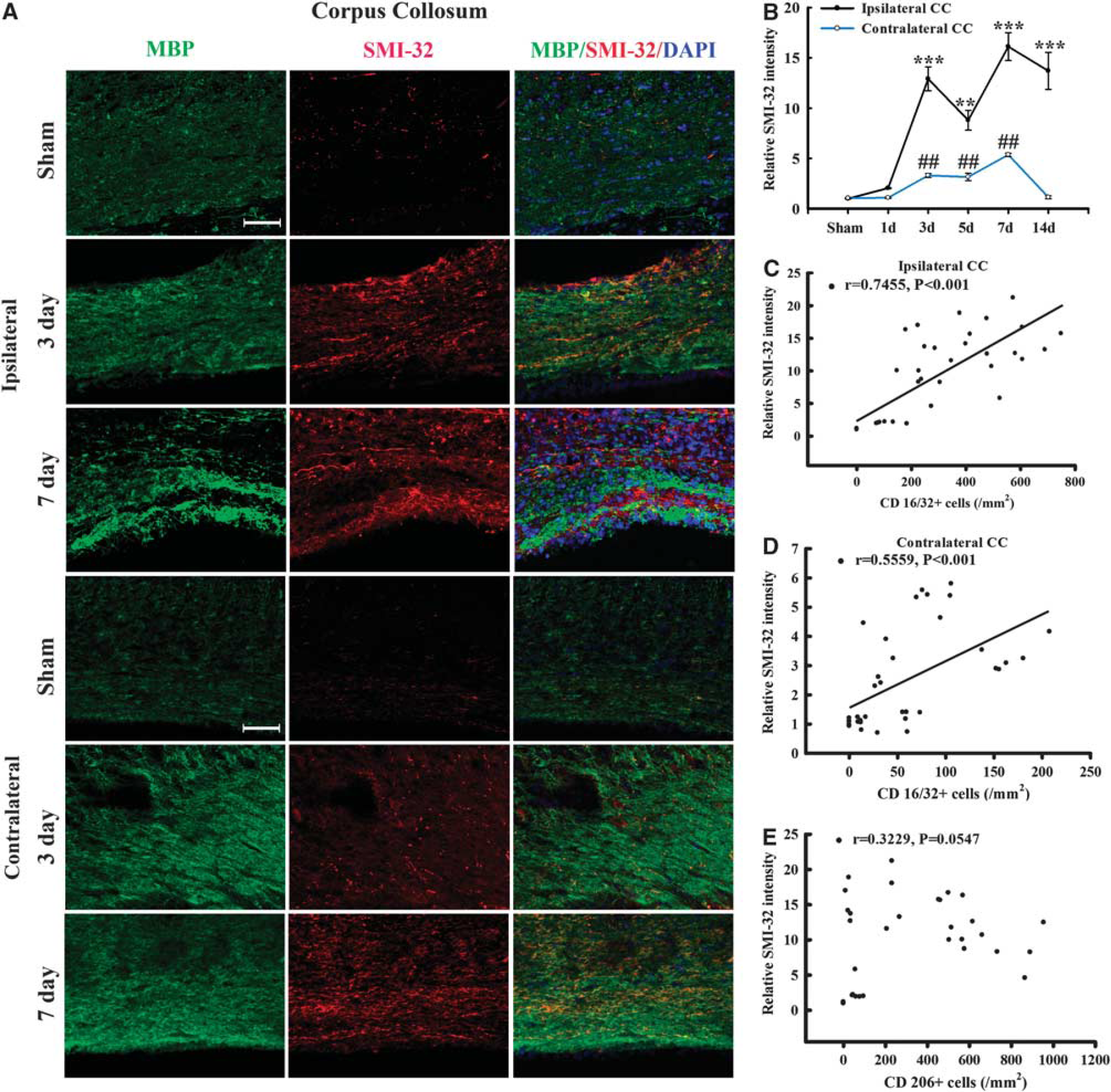
Temporal changes in white matter injury after traumatic brain injury (TBI). (
Remarkably, there was a direct positive correlation of SMI-32 intensity with the number of CD16/32+cells for all animals in both the ipsilateral and contralateral CC (all
Effect of M1 and M2 Microglia on Post-Oxygen Glucose-Deprived Oligodendrocyte Survival
Central nervous system mechanical trauma and ischemic or hemorrhagic stroke share common detrimental features such as reduced blood flow and energy failure, and all three insults directly result in cell death and tissue loss. These common features can be mimicked by exposing cells to hypoxic conditions in glucose-free medium
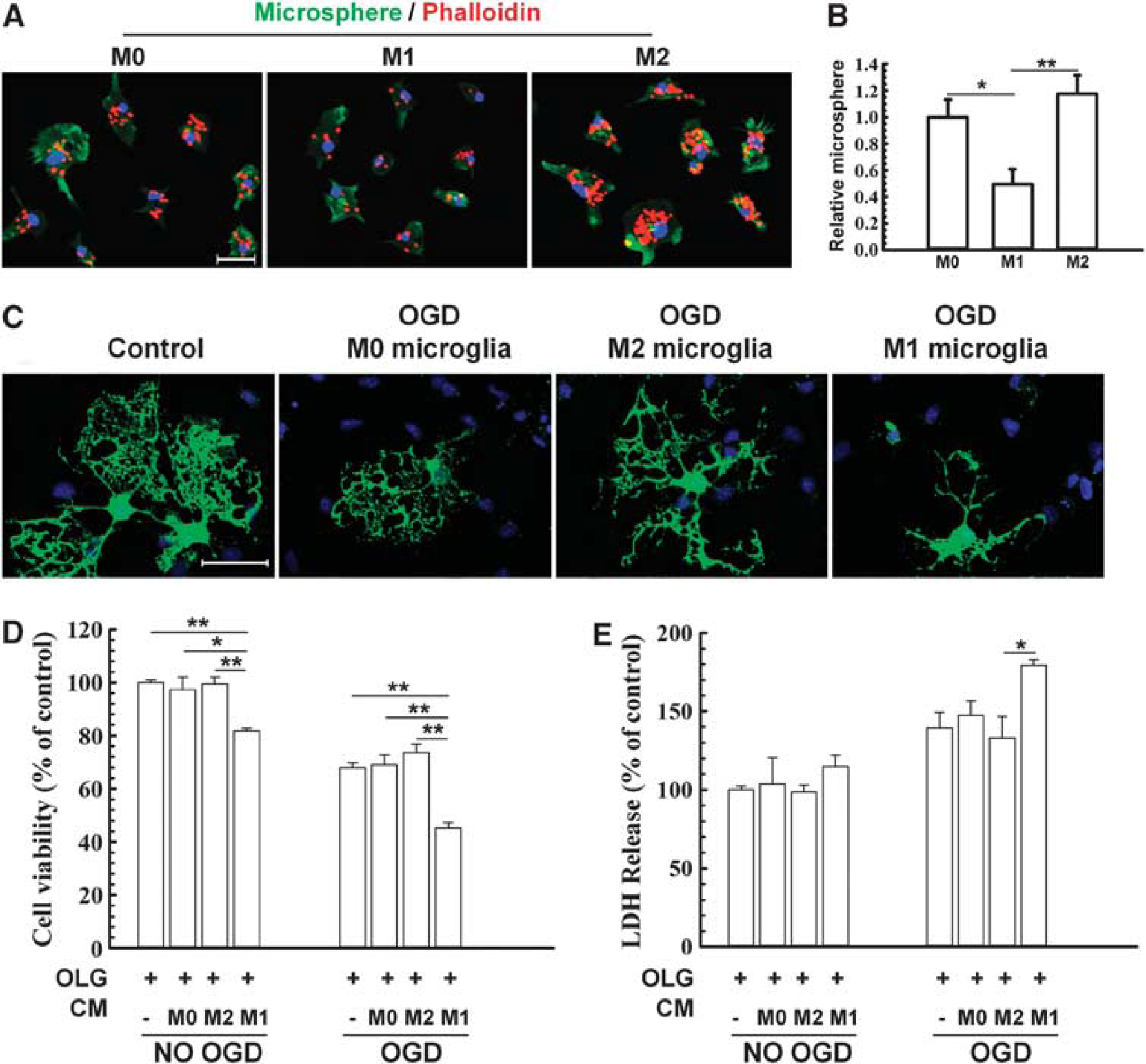
Effect of M1 and M2 microglia/macrophages on oxygen glucose deprivation (OGD)-induced injury in oligodendrocytes. (
Conditioned medium was collected from M1 polarized, M2 polarized, or non-polarized microglia and applied to non-OGD or post-OGD oligodendrocyte cultures (Figures 7C, D, and E). Myelin basic protein fluorescence was relatively stable in these mature oligodendrocytes in control conditions. However, OGD evoked rapid and severe loss of processes in media from unstimulated microglia (Figure 7C). This loss of processes was further exacerbated by M1 media but alleviated by M2 media (Figure 7C). Lactate dehydrogenase release and MTT assays both showed that the addition of M1 microglia-conditioned media reduced oligodendrocyte survival after OGD compared with all other groups (Figures 7D and E).
DISCUSSION
Although WMI is a frequent complication of TBI and an important determinant of cognitive impairment, the underlying pathophysiology remains poorly understood. 22 The present study characterizes the dynamic microglia/macrophage response to TBI and its relation to WMI. Microglia/macrophages are the primary mediators of the innate immune response to injury and disease in the CNS.13, 16 The functions of microglia in the injured CNS are under intense scrutiny, and recent research provides strong support for dual microglial roles, both beneficial and destructive, as well as differential activation of these immune cells into one of at least two phenotypes, M1 or M2.13, 18, 23 On one hand, microglia/macrophage activation is thought to benefit the injured brain by removing cellular debris and restoring tissue integrity.24, 25 However, a large number of studies have also shown that microglia release proinflammatory mediators that contribute to neuronal dysfunction and cell death.13, 26, 27 In other words, microglia can either promote delayed cell damage by generating proinflammatory cytokines or participate in regenerative processes by scavenging necrotic tissue.28, 29 Thus, microglial activation in white matter after cerebral trauma may reflect either or both of these processes. 30 An improved understanding of the dynamic equilibrium between beneficial and destructive microglia/macrophages may advance our knowledge of post-trauma recovery and help ameliorate WMI in trauma victims.
In the present study, we described the temporal kinetics of microglia/macrophage polarization in both gray and white matter after TBI. It is evident from our findings that the dynamic changes of microglia/macrophages are quite different in white and gray matter. For example, a clear M2–to-M1 transit is apparent in gray matter in response to TBI, whereas the changes in white matter seem to be bimodal. The majority of microglia/macrophages migrating or infiltrating into the lesioned white matter areas assume the M1 phenotype at day 3, as evidenced by the increased expression of CD16/32 protein in Iba+ cells. Subsequently, an alternatively activated subset of microglia/macrophage (M2) occupied the leading position at day 5 after TBI. The present study as well as previous reports
16
further indicate that M2 microglia/macrophages are healthier cells with enhanced phagocytic activity and reduced production of inflammatory mediators. Therefore, the recruitment of M2 microglia/macrophages into the cortex, striatum, and CC may represent an endogenous effort to clean injured tissue and restrict brain damage. Previous studies also suggest that expression of M2 microglial factors promote CNS repair while limiting secondary inflammation-mediated injury after spinal cord injury.
18
Maintaining the M2 microglia/macrophage phenotype could therefore benefit the injured brain in multiple ways.
16
However, the present study reveals that the M2 phagocyte responses in both gray and white matter are transient and phased out within 7 days after injury. During these changes, M1 microglia/macrophages, which are characterized by reduced phagocytosis and increased secretion of proinflammatory mediators, begin to dominate the injured landscape. These M1 phagocytes may exacerbate nerve cell demise through proinflammatory cytokines (tumor necrosis factor-
Interestingly, the M1 microglial phenotype was also transiently activated at low levels in the contralateral cortex and CC area. Previous studies have revealed the presence of damage in contralateral brain areas after TBI.32, 33 The direct injury on the ipsilateral hemisphere may stress remote areas through transcallosal or transhemispheric diaschisis, 34 resulting in alterations in electrical activity, cerebral blood flow, and metabolites in the contralateral hemisphere. Inflammatory processes are also known to contribute to the contralateral responses after TBI. As part of the neuroimmune response, a cascade of pro- and anti-inflammatory cytokines are released and can be detected at the site of injury as well as contralateral regions after brain injury. 35 Our present study further suggests that the M1 microglia/macrophage phenotype is also related to the extent of WMI in the contralateral brain. Further studies of the effect of microglia/macrophage phenotype on white matter alterations in both hemispheres may guide us toward a more complete understanding of TBI and help us develop therapeutic interventions.
Our results indicate that microglia/macrophages respond dynamically to TBI, experiencing a transient M2 phenotype followed by a shift to the M1 phenotype in both white and gray matter. The current study focused on WMI after TBI because the severity of WMI, manifested by immunohistochemical staining for neurofilament SMI-32, was strongly correlated with the number of M1-like phagocytes. In contrast, there was poor correlation between microglia/macrophage dynamics and the number of surviving neurons, as revealed by NeuN staining, in this model (data not shown). Given the importance of WMI to the long-term deficiencies in motor and cognitive functions in TBI patients, our study not only identifies a novel mechanism of WMI after TBI, but also provides a potential target for TBI treatment.
The findings of the present study support the hypothesis that the M1 phenotype exacerbates WMI. First, we found a strong positive correlation between the extent of WMI and activation of M1 (but not M2) microglia on both the ipsilateral and contralateral sides. Second,
As mentioned earlier, neuroinflammation in the CNS predominantly involves microglia and macrophages, and is believed to elicit secondary expansion of injury after trauma.3, 9, 13 As a result, there is significant interest in developing novel anti-inflammatory agents to ameliorate or even prevent CNS inflammation.9, 36 However, classic anti-inflammatory agents such as steroids and nonsteroidal anti-inflammatory drugs have not had a major role in the management of CNS inflammatory conditions. 36 Among the potential factors that might have prevented their use in inflammatory conditions is that broad suppression of microglia/macrophages may deprive the brain of their normal phagocytic roles and cause the buildup of cellular debris. This notion is supported by observations that selective depletion of proliferative microglia exacerbate brain injuries24, 37 and, conversely, that injections of exogenous microglia into the brain ameliorate CNS injuries.38, 39 Many authors have concluded that acute inflammation serves several protective functions, 16 whereas chronic inflammation exacerbates injury.5, 6 In keeping with this traditional view of chronic inflammation, the results of our study demonstrate that microglia/macrophages experience a sustained rise in the destructive M1 phenotype within 1 week after TBI.
It is important to note here that mononuclear phagocytes exhibit extensive heterogeneity and plasticity in response to different physiologic and pathologic stimuli. Indeed, there are likely to be additional activation states, as well as transitional stages between M1 and M2.40, 41 Microglia/macrophages present during TBI are therefore not just comprising 1 or 2 discrete subpopulations but may include a spectrum of diverse activation states. The concept of M1 and M2 microglia/macrophage polarization is a simplified framework that represents two extremes along a continuum.
41
This framework is nevertheless useful to help understand the different functional status of phagocytes during injury progression and to explore therapeutic strategies targeting microglia/macrophage responses. One caveat to using signature markers, even in combination, to characterize phagocyte polarization is that many established M1 and M2 markers are not specific for phagocytes or for the polarization processes. To this end, further analyses of microglia/macrophage functions, including migration, phagocytosis, and protein production, are warranted to conclusively define the functional states of these cells in different disease stages. Furthermore, whether microglia and macrophages have similar activation patterns in humans is still under debate. For example, some
In conclusion, a comprehensive analysis of white matter changes in the mouse brain after TBI suggests that mechanical trauma induces both acute and chronic local inflammatory changes in microglia/macrophages. The transient M2 response is replaced by a chronic M1 response and the M1 response is correlated with the extent of WMI. Our results suggest that the goals of anti-inflammatory therapy for TBI should be shifted from blanketed microglia/macrophage suppression towards a more specific titration of the inflammatory response away from the destructive M1 phenotype. This more nuanced approach may avoid disruptions in beneficial phagocyte responses while reducing the secondary expansion of gray and WMI in trauma victims.
Footnotes
The authors declare no conflict of interest.