
Abstract
Postresuscitation cerebral hypothermia is consistently neuroprotective in experimental preparations; however, its effects on white matter injury are poorly understood. Using a model of reversible cerebral ischemia in unanesthetized near-term fetal sheep, we examined the effects of cerebral hypothermia (fetal extradural temperature reduced from 39.4±0.1°C to between 30 and 33°C), induced at different times after reperfusion and continued for 72 hours after ischemia, on injury in the parasagittal white matter 5 days after ischemia. Cooling started within 90 minutes of reperfusion was associated with a significant increase in bioactive oligodendrocytes in the intragyral white matter compared with sham cooling (41±20 vs 18±11 per field, P < 0.05), increased myelin basic protein density and reduced expression of activated caspase-3 (14±12 vs 91±51, P < 0.05). Reactive microglia were profoundly suppressed compared with sham cooling (4±6 vs 38±18 per field, P < 0.05) with no effect on numbers of astrocytes. When cooling was delayed until 5.5 hours after reperfusion there was no significant effect on loss of oligodendrocytes (24±12 per field). In conclusion, hypothermia can effectively protect white matter after ischemia, but only if initiated early after the insult. Protection was closely associated with reduced expression of both activated caspase-3 and of reactive microglia.
Although white matter injury is well known to be the dominant cause of neural handicap in premature infants (Inder et al., 1999), there is increasing evidence from magnetic resonance imaging studies that it is also a significant contributor to neurodevelopmental problems at term (Barkovich et al., 1995; Mercuri et al., 1999; Okumura et al., 1997). Experimentally, mature, myelinating oligodendrocytes are at least as vulnerable to ischemic injury as neurons (Cao et al., 2003; Guan et al., 2000; Ikeda et al., 1998; Loeliger et al., 2003; Pantoni et al., 1996; Petersson et al., 2002). Indeed, after asphyxia in the near-term fetal sheep, the mildest pathologic findings were vacuolation and loss of myelin in white matter, rather than neuronal death (Ikeda et al., 1998).
There is now strong clinical and experimental evidence that a prolonged period of moderate cerebral hypothermia initiated within a few hours after severe hypoxia-ischemia can markedly reduce subsequent neuronal loss and improve behavioral recovery (Bernard et al., 2002; Colbourne and Corbett, 1995; Gunn et al., 1997, 1998b; The Hypothermia after Cardiac Arrest Study Group, 2002; Tooley et al., 2003). Although there are few data on perinatal white matter damage, early initiation of hypothermia can ameliorate posttraumatic spinal cord injury (Inamasu et al., 2003), and there is increasing evidence that many of the effects of cooling are relevant to the proposed mechanisms of hypoxic-ischemic oligodendrocyte loss.
Recent studies have shown that the pathogenesis of postischemic demyelination includes a substantial element of delayed or secondary loss (Cao et al., 2003), likely mediated by apoptosis and microglia activation (Hagberg et al., 2002; Shibata et al., 2000). Apoptosis is a significant component of postasphyxial cell death in the developing brain (Edwards et al., 1997; Scott and Hegyi, 1997). Although multiple pathways are likely to be involved in such postischemic apoptosis, caspase-3, one of the family of cysteine proteases, is reported to play a crucial role in the immature brain (Hu et al., 2000; Johnson et al., 1999; Zhu et al., 2003), and specifically in loss of oligodendrocytes (Cao et al., 2003; Shibata et al., 2000). Several reports suggest that hypothermia can specifically suppress the development of apoptotic injury (Edwards et al., 1995; Xu et al., 1998).
In addition, there is increasing evidence that cytokine induction and the associated inflammatory reaction are major contributors to perinatal brain injury (Hagberg et al., 2002). In particular, microglia activated by interferon-gamma induce contact-dependent oligodendroglial death (Nicholas et al., 2003). Hypothermia potently reduces cytokine release and microglial activation both in vivo and in vitro (Gibbons et al., 2003; Inamasu et al., 2000; Wang et al., 2002).
The effects of cooling on neuronal loss are known to be highly dependent on the timing of initiation of cooling, with rapid loss of effect as delay is increased (Gunn and Bennet, 2002). This relationship has not been established for perinatal white matter injury. Therefore, in the present study we examined the effect of time of initiation of moderate cerebral hypothermia for 3 days after ischemia on white matter damage after transient cerebral ischemia in near-term (0.85 gestation) fetal sheep. In terms of cerebral maturity and myelination, this stage is comparable to the human brain at term (McIntosh et al., 1979). The effects of hypothermia on postischemic white matter apoptosis and the inflammatory reaction after 5 days recovery from ischemia were assessed using proteolipid protein (PLP) mRNA to identify bioactive oligodendrocytes and immunohistochemistry to identify other glial cell types.
MATERIALS AND METHODS
Experimental preparation
All procedures were approved by the Animal Ethics Committee of the University of Auckland. Singleton Romney/Suffolk fetal sheep, while under general anesthesia (2% halothane in O2), were instrumented at 117 to 124 days of gestation (term = 147 days) using sterile techniques. Ewes were given 5 mL of Streptopen (Stockguard Labs Ltd., Hamilton, New Zealand) intramuscularly before the start of surgery. Polyvinyl catheters were placed in the brachial arteries, a brachial vein, and the amniotic sac. Electrocardiogram (ECG) electrodes were sewn across the chest to record the fetal electrocardiogram. A thermistor was placed in the fetal esophagus at the level of the right atrium to measure fetal core body temperature (Gunn et al., 1997). The vertebral-occipital anastomoses were ligated bilaterally to restrict vertebral blood supply to the carotid arteries. A double-ballooned inflatable occluder was placed around each carotid artery (Gunn et al., 1997). Two pairs of electroencephalographic electrodes (AS633-5SSF, Cooner Wire Co., Chats-worth, CA, U.S.A.) were placed on the dura over the parasagittal parietal cortex (5 mm and 15 mm anterior to bregma and 10 mm lateral), with a reference electrode sewn over the occiput. Cortical impedance was measured using a third pair of electrodes (AS633-3SSF, Cooner Wire Co.) placed over the dura, 5 mm lateral to the electroencephalographic electrodes. A second thermistor (Incu-temp-1; Mallinckrodt Medical, St. Louis, MO, U.S.A.) was placed over the parasagittal dura 20 mm anterior to bregma and the burr holes were sealed and the skin over the fetal skull was secured with cyanoacrylate glue.
A cooling coil made from silicone tubing (external diameter, 7.9 mm; internal diameter, 4.8 mm; Silclear, Degania Silicone, Degania Bet, Israel) was attached over the dorsal surface of the scalp and extended over the lateral surface of the cranium down to the level of the external auditory meatus. The fetus was returned to the uterus and 80 mg of gentamicin was administered into the amniotic sac before closure of the uterus. All fetal leads were exteriorized through the maternal flank. The maternal long saphenous vein was catheterized for administration of antibiotics and for euthanasia. A thermistor was inserted with the catheter to permit measurement of maternal temperature.
After surgery, sheep were housed together in separate metabolic cages with access to water and food ad libitum. They were kept in a temperature-controlled room (16±1°C, humidity 50%±10%), in a 12-hour light/dark cycle. A period of 3 to 5 days' postoperative recovery was allowed, during which the ewe was injected with daily intravenous (i.v.) gentamicin (80 mg gentamicin, Pharmacia & Upjohn, Perth, Australia). Fetal arterial blood was taken daily from the brachial artery, for blood gas analysis. Catheters were maintained patent by continuous infusion of heparinized saline (40 U.mL−1 at 0.2 mL.h−1).
Experimental design and recordings
Fetuses were randomly assigned to one of four groups: sham control, ischemia plus sham cooling, or ischemia plus cooling, starting either 2 hours (2-hour cooling group) or 6 hours (6-hour cooling group) after the start of ischemia. Fetal mean arterial pressure, corrected for maternal movement by subtraction of amniotic fluid pressure, fetal heart rate, carotid artery blood flow, fetal electroencephalogram, spectral edge, and impedance were recorded continuously from 12 hours before the experiment until 5 days afterwards. Data were stored at 1-minute intervals to disk by custom software for off-line analysis (Labview for Windows, National Instruments Ltd, Austin, TX, U.S.A.).
Cerebral ischemia was induced by rapid inflation of the carotid artery occluders bilaterally for 30 minutes with sterile saline. Successful occlusion was confirmed by observation of a rapid, sustained flattening of the electroencephalogram. Cooling was started either 2 or 6 hours after the onset of the ischemic period (i.e., 90 minutes vs. 5.5 hours after the initiation of reperfusion) and was continued until 72 hours after occlusion. Fetal arterial blood was taken 60 minutes before carotid occlusion, and then at 35 minutes after the start of occlusion (i.e., 5 minutes after the end of occlusion), and 2, 4, 6, 12, 24, 48, 72, 96, and 120 hours and analyzed for pH and blood gas determination (Ciba-Corning Diagnostics 845 blood gas analyzer and co-oximeter, East Walpole, MA, U.S.A.) and for glucose and lactate measurements (YSI model 2300, Yellow Springs, OH, U.S.A.).
On completion of the experiment at 120 hours, the ewe and fetus were killed by an overdose of pentobarbitone sodium given IV to the ewe. The fetal brain was perfusion fixed with saline followed by 10% neutral buffered formalin, stored overnight for fixation in 10% neutral buffered formalin, then embedded in paraffin using a standard paraffin tissue preparation.
The pH, blood gas data, and glucose and lactate measurements, carotid blood flow and electrophysiologic changes, and neuronal loss scores have been published in a subset of these animals (Gunn et al., 1997, 1998b).
Immunohistochemistry
Immunohistochemical staining was performed on coronal sections (6 μm), at the level of the parietal cortex, cut and mounted on chrome alum—coated slides. Before specific staining, all the slides were deparaffinized in xylene, twice for 15 minutes, dehydrated in a series of ethanol (100%, 100%, 95%, 95%, 70%, for 2 minutes each), and incubated in 0.1 mol/L phosphate buffered saline (PBS) twice for 5 minutes. The sections were pretreated with 1% H2O2 in 50% methanol for 20 minutes, washed in PBS (3×5 minutes) and incubated with the primary antibody for 2 days at 4°C. The following primary antibodies and dilutions were used: mouse anti-myelin basic protein (MBP, Roche, Mannheim, Germany; 1:200); isolectin B-4 (Sigma, St. Louis, MO, U.S.A.; 1:100); mouse anti-glial fibrillary acidic protein (Sigma; 1:200); mouse-proliferating cell nuclear antigen PC10 (PCNA; DAKO, Denmark, diluted 1:100); rabbit anti-Caspase-3 Asp175 (cleaved Caspase-3 Antibody, detects endogenous levels of the large fragment of activated caspase-3 (17–20kDa), Cell Signaling Technology, Beverly, MA, U.S.A., diluted 1:1,000). The antibodies were diluted in PBS containing normal horse serum/goat serum. The primary antibodies were washed off with PBS (3 × 10 minutes) and then incubated with anti-mouse/anti-goat biotinylated immunoglobulin G (Vector Laboratories, Burlingame, CA, U.S.A., diluted 1:200) overnight at 4°C. The sections were washed, incubated in avidin-biotin complex (ABC, Vector Laboratories, Peterborough, England, diluted 1:100) in the ratio 1:500 for 3 hours at room temperature, washed again in PBS, and then reacted in diaminobenzidine tetrahydrochloride (DAB, Sigma). Sections were then dehydrated in a series of alcohol to xylene washes and coverslipped with mounting medium. Control sections were processed in the same way except that the primary antibody was omitted from the incubation solution.
In situ hybridization
In situ hybridization, using a sheep PLP RNA probe, was used to identify bioactive oligodendrocytes, as previously described (Guan et al., 2001).
Analysis and statistics
Scoring of oligodendrocytes, astrocytes, microglia, and density of myelin was performed on coronal sections by light microscopy at ×200 magnification. Within these sections, cells were counted using a fixed grid (0.42 × 0.21 mm), in three areas in the intragyral white matter of the parasagittal cortex, and one in the corona radiata, of both hemispheres (Fig. 1). The density of MBP from the same areas and their background was measured using image analysis (Sigmascan, SPSS, Chicago, IL, U.S.A.). The difference between the MBP density and the background reading from adjacent gray matter was calculated and used for data analysis. The number of cells positive for glial fibrillary acidic protein, IB-4, PLP, and PCNA and the MBP density were averaged from the three selected intragyral areas. Because of the relatively low density of caspase-3 (Asp 175) positively stained cells, the measurement represents the total number of caspase-3 (Asp 175)-positive cells present in the white matter tract of the first parasagittal gyrus.
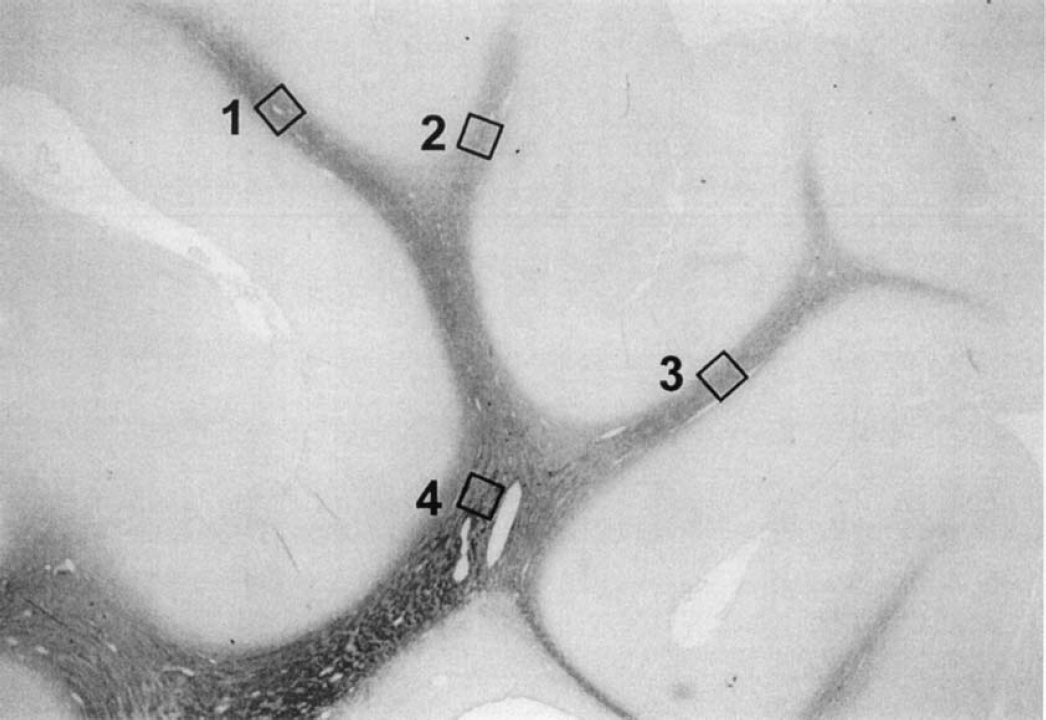
Photomicrograph showing the regions used to count different cell types and to assess the average density of myelin in the intragyral white matter and the corona radiata. Three regions in the intragyral white matter (squares 1, 2, and 3) and one area in the corona radiata (square 4), in both hemispheres, were used for assessment.
The sham cooling (ischemia) group showed marked tissue swelling in the intragyral regions, with a significant increase in intragyral white matter areas compared with sham controls. To correct for decreased cell and staining density because of tissue swelling, cell counts and MBP staining density measurements in the sham cooling group were adjusted by the relative increase in white matter area (the ratio of intragyral white matter area in sham cooled fetuses to the sham control group) before statistical analysis. There was no significant tissue swelling in the two hypothermia groups; therefore the counts and staining density were not adjusted. Treatment effects were evaluated by analysis of variance (SPSS v10, SPSS Inc.), followed by the protected least-significant difference post-hoc test when a significant overall effect was found. Significance was accepted at P < 0.05. Data are mean ±SD.
RESULTS
Myelin Basic Protein
Myelin basic protein immunohistochemistry showed specific staining of myelin sheaths in the intragyral and corona radiata without obvious staining of cell bodies (Figs. 2a and 2e). Cerebral ischemia was associated with a severe loss of MBP density compared with sham controls (Figs. 2b and 2f, and Fig. 3, P < 0.05), which was greatest in the parasagittal intragyral white matter (P < 0.05). Morphologically, the normally parallel microstructure of MBP was disorganized and fragmented. The intragyral white matter tracts were markedly swollen (Figs. 2b and 2f), with a significant increase of the intragyral white matter area compared with sham controls (12.70±2.25 vs 7.11±1.63 mm2, P < 0.05).
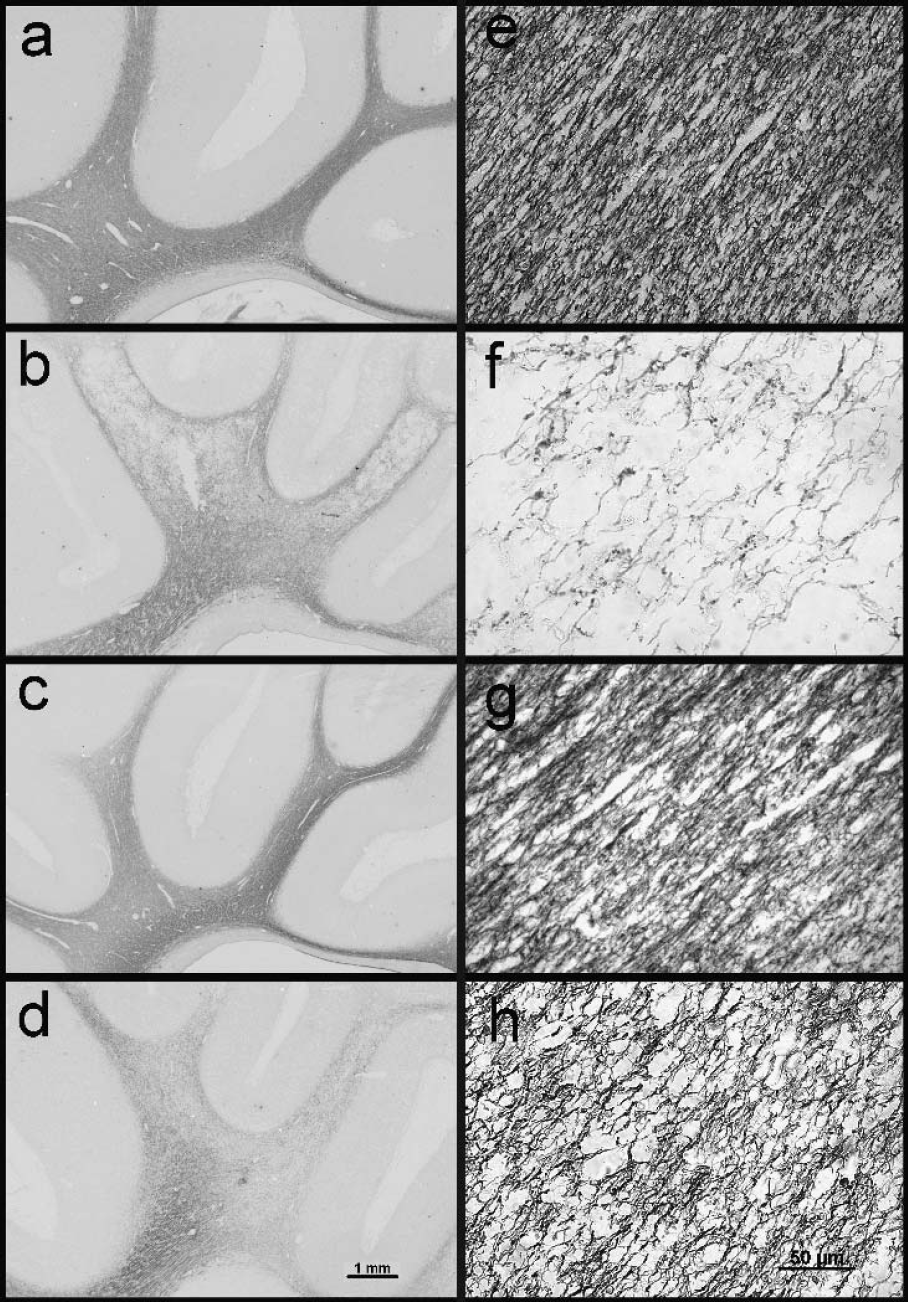
Photomicrographs showing the distribution (
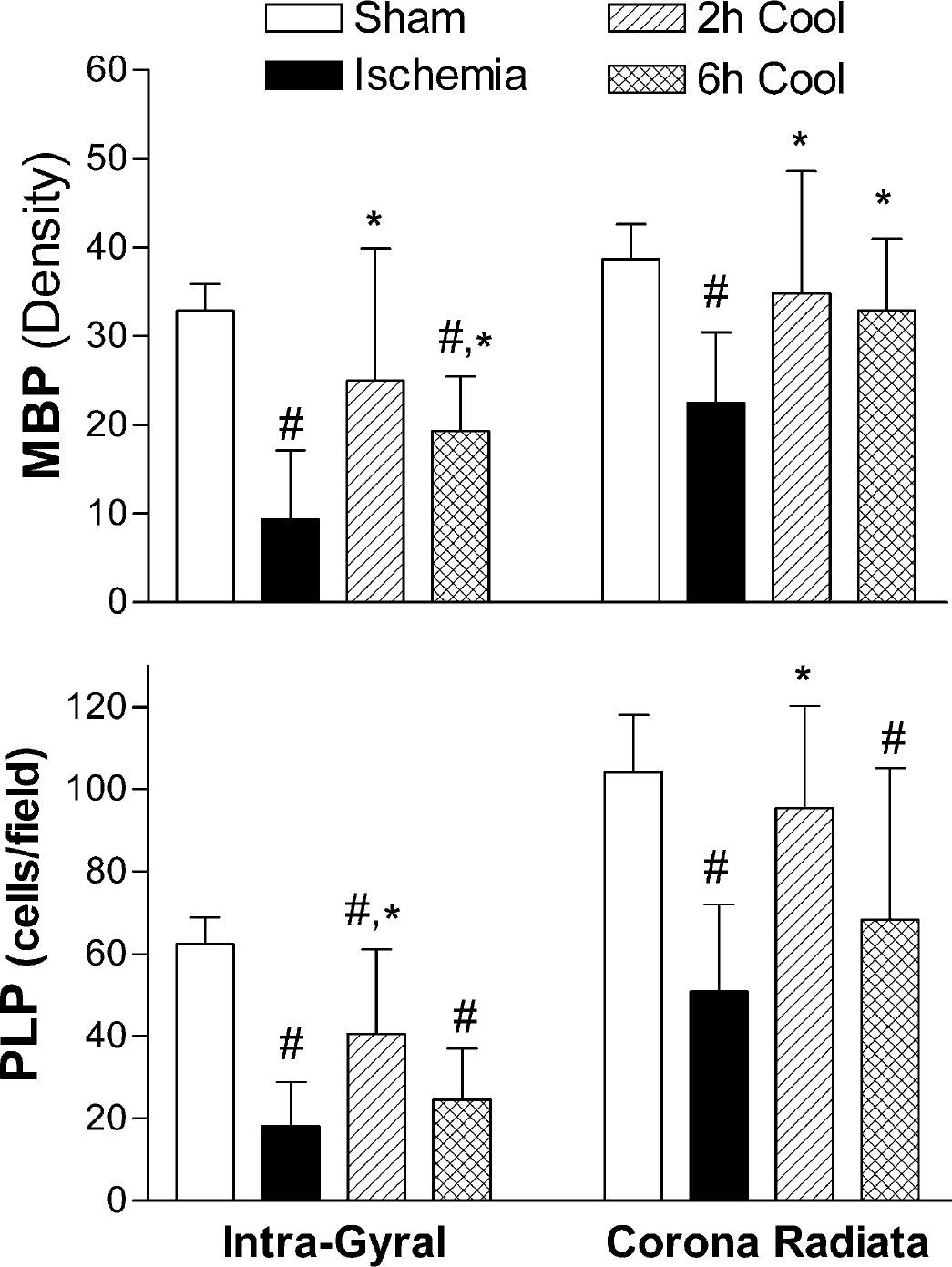
Density of myelin basic protein (MBP, top) immunostaining and numbers of proteolipid protein (PLP, bottom) mRNA-positive cells in parasagittal white matter. Ischemia resulted in a severe loss of MBP density (top) after 5 days recovery compared to the sham control group. Both the 2-hour and the 6-hour cooling groups showed a significant increase in average MBP density after ischemia compared to sham cooled (ischemia) fetuses, both in the intragyral white matter and the corona radiata. There was a significant loss of PLP mRNA-containing cells (bottom) in the sham cooling (ischemia) group compared to sham controls both in the intragyral white matter and the corona radiata. The 2-hour but not the 6-hour cooling group showed a significant increase in PLP mRNA-positive cells compared to the sham cooling (ischemia) group. #P < 0.05 vs sham controls, *P < 0.05 vs ischemia with sham cooling. Data are mean±SD.
Both cooling groups showed reduced loss of MBP staining density in the intragyral white matter and corona radiata after 5 days recovery compared with the sham-cooled ischemia group, which was greatest in the 2-hour group (Figs. 2c, 2d, 2g, 2h, and Fig. 3, P < 0.05). On visual assessment, MBP staining was denser, more compact and less fragmented than in the sham-cooled (ischemia) group (Figs. 2g and 2h). Consistent with this, the area of the intragyral white matter area was significantly reduced in both the 2- and 6-hour cooling groups compared to ischemia alone (P < 0.05, 8.30±243 and 9.02±3.29 mm2, respectively) and not significantly different from sham controls.
Proteolipid protein mRNA expression
In the sham control group, PLP mRNA staining was densely distributed from the corona radiata area toward the intragyral regions of the parasagittal white matter tracts. Proteolipid protein staining was localized in the periphery of the cytoplasm of oligodendrocytes in a bipolar distribution, as previously described (Guan et al., 2001). Ischemia was associated with a significant reduction in the number of the PLP staining oligodendrocytes compared to sham controls (P < 0.05; bottom panel, Fig. 3); the loss was greater in intragyral white matter tracts compared with the corona radiata (P < 0.05). The 2-hour, but not the 6-hour, cooling group showed a significantly greater number of PLP mRNA–positive cells than in the sham cooling (ischemia) group in both white matter regions (P < 0.05). Numbers of PLP mRNA cells in the 2-hour cooling group were still less than in sham controls in the intragyral white matter (P < 0.05) but similar to sham controls in the corona radiata.
Astrocytes and microglia
Glial fibrillary acidic protein–positive astrocytes were localized throughout the fetal white matter tracts of sham control animals (Fig. 4a). Double labeling of glial fibrillary acidic protein with PLP mRNA showed a close association between astrocytes and oligodendrocytes in the white matter (data not shown). Ischemia was associated with an apparent reduction in numbers of astrocytes in the intragyral white matter (Fig. 4b) but not the corona radiata (Fig. 5). However, this was not statistically significant after correction for tissue swelling in this group (Fig. 5). There was no significant effect of cooling (Figs. 4c, 4d, and Fig. 5).
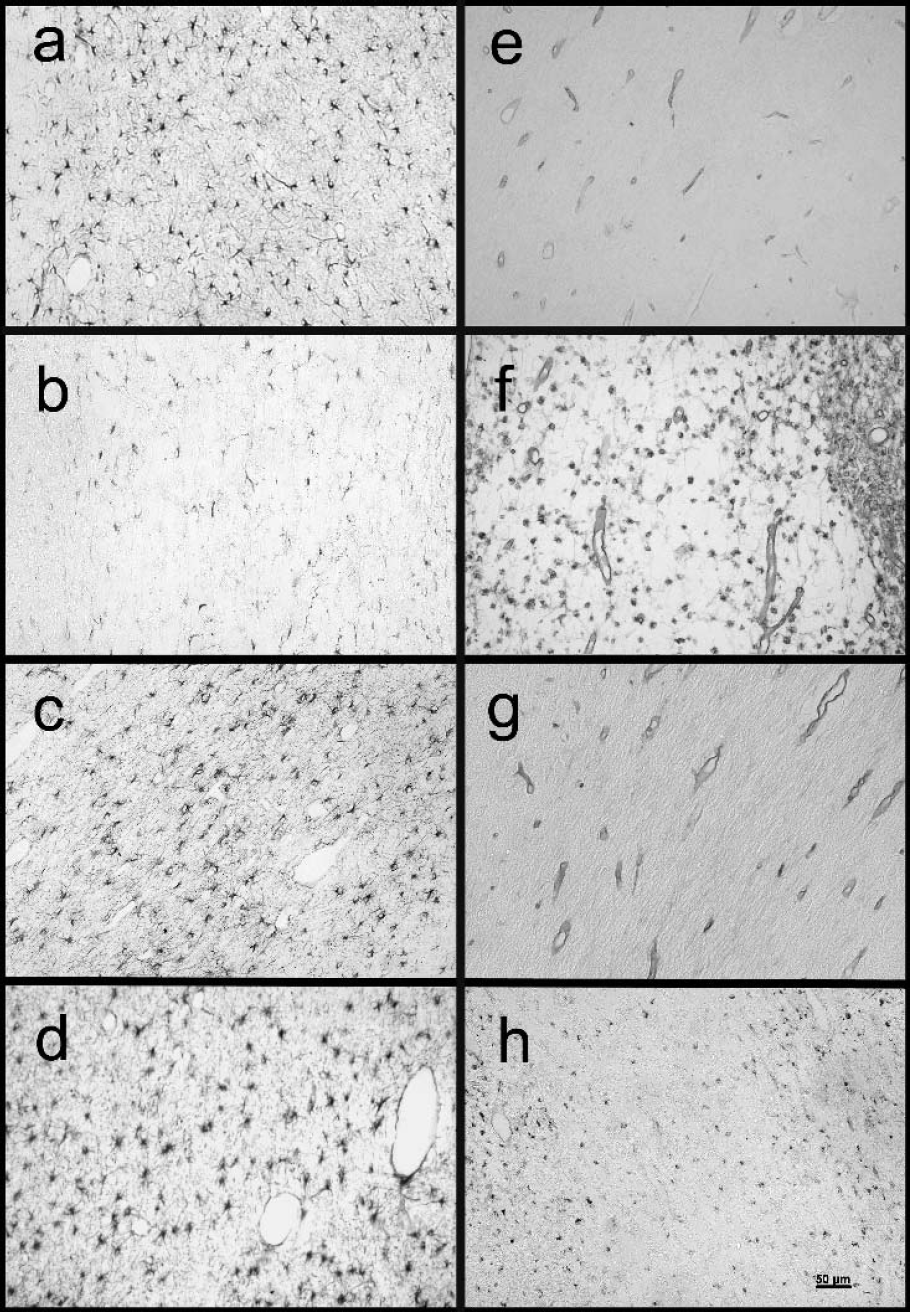
Photomicrographs showing glial fibrillary acidic protein immunostaining (
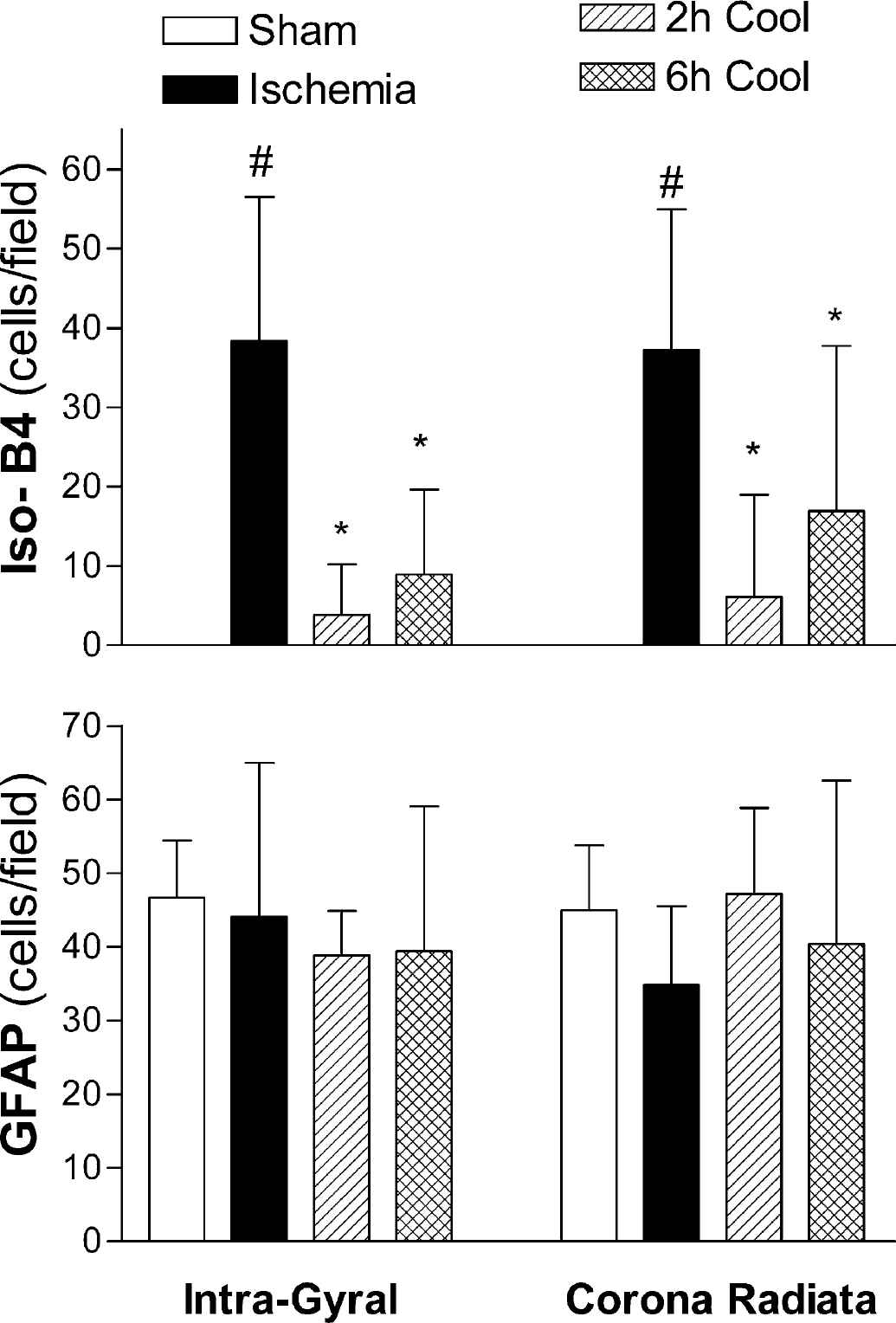
Numbers of isolectin B-4 (Iso-B4, top) and glial fibrillary acidic protein (GFAP, bottom) immunopositive cells in predefined areas of the intragyral cerebral white matter and parasagittal corona radiata. The number of Iso-B4-positive cells was significantly increased after ischemic injury compared to the sham controls. Treatment with either early (2-hour group) or late (6-hour group) initiation of hypothermia significantly suppressed isolectin B-4 positive cells compared to the sham cooled group, although the degree of suppression was less with late cooling. Corrected for the area of the intragyral white matter tracts, there was no effect of ischemia on numbers of GFAP cells (bottom), and no effect of hypothermia. #P < 0.05 vs sham controls, *P < 0.05 vs ischemia with sham cooling. Data are mean±SD.
Sham control animals showed virtually no isolectin B-4 staining in the intragyral regions or corona radiata (Fig. 4e). Ischemia was associated with a significant increase of isolectin B-4 staining activated microglia in the intragyral and the corona radiata regions (Fig. 4f and Fig. 5). Both the 2-hour and 6-hour cooling groups showed a significant reduction in numbers of microglia compared with the sham cooled (ischemia) group (Figs. 4 g and 4h, respectively, P < 0.05) in both white matter regions, which was relatively greater in the 2-hour cooling group (Fig. 5).
Activated caspase-3 expression
Only occasional cleaved 17-kD caspase-3 positive cells were seen in a few sham control animals. After ischemia in the sham-cooled (ischemia) and both cooling groups, there was a significant increase in activated caspase-3 expression in the parasagittal cerebral white matter (Fig. 6). The density of caspase-3 staining varied between cells because of the timing of cleavage of caspase-3. Cells were counted that had evidence of newly cleaved caspase-3, as shown by morphologically dense caspase-3 (Asp175) staining evenly distributed within the ectoplasm, with coarse granular fragments around the cells (Fig. 7). Thionine counterstaining showed condensed chromatin fragments within the caspase-3 (Asp175)-positive cells. Both cooling groups showed significantly reduced numbers of cells expressing activated caspase-3 compared to sham cooling; the reduction in the 2-hour cooling group was greater than in the 6-hour cooling group (Fig. 6, P < 0.05).
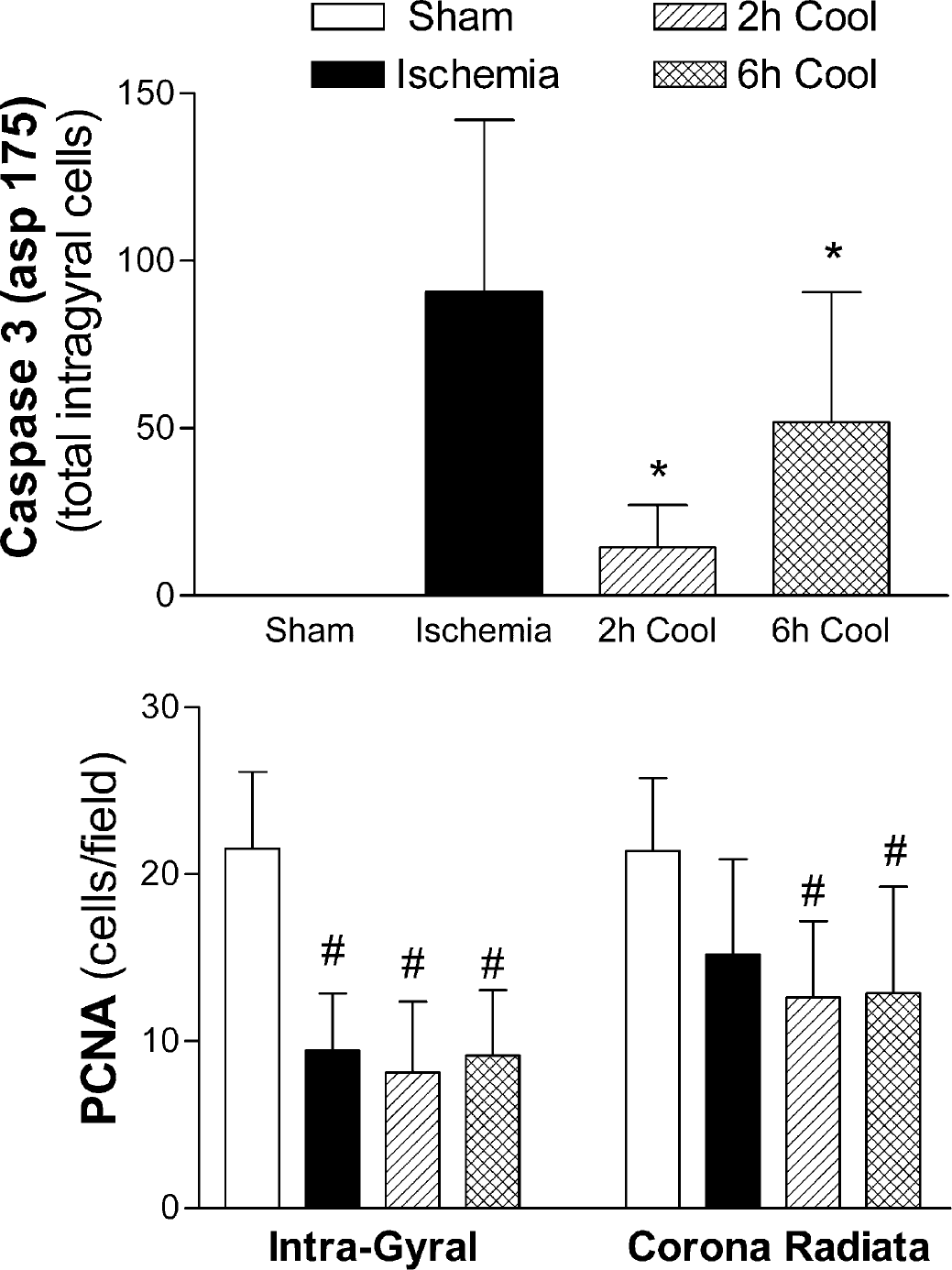
The total number of caspase-3 (Asp175)-positive cells in the parasagittal cerebral white matter tract (top) was significantly increased after 5 days' recovery from cerebral ischemia compared to the sham control group. The 2-hour cooling group showed near-complete suppression of caspase-3 activation compared to the sham cooled group, with a much smaller effect in the 6-hour cooling group. Ischemia reduced the number of proliferating cell nuclear antigen (PCNA, bottom)–positive cells in the intragyral white matter tract. There was no significant additional effect of hypothermia. #P < 0.05 vs sham controls, *P < 0.05 vs ischemia with sham cooling. Data are mean±SD.
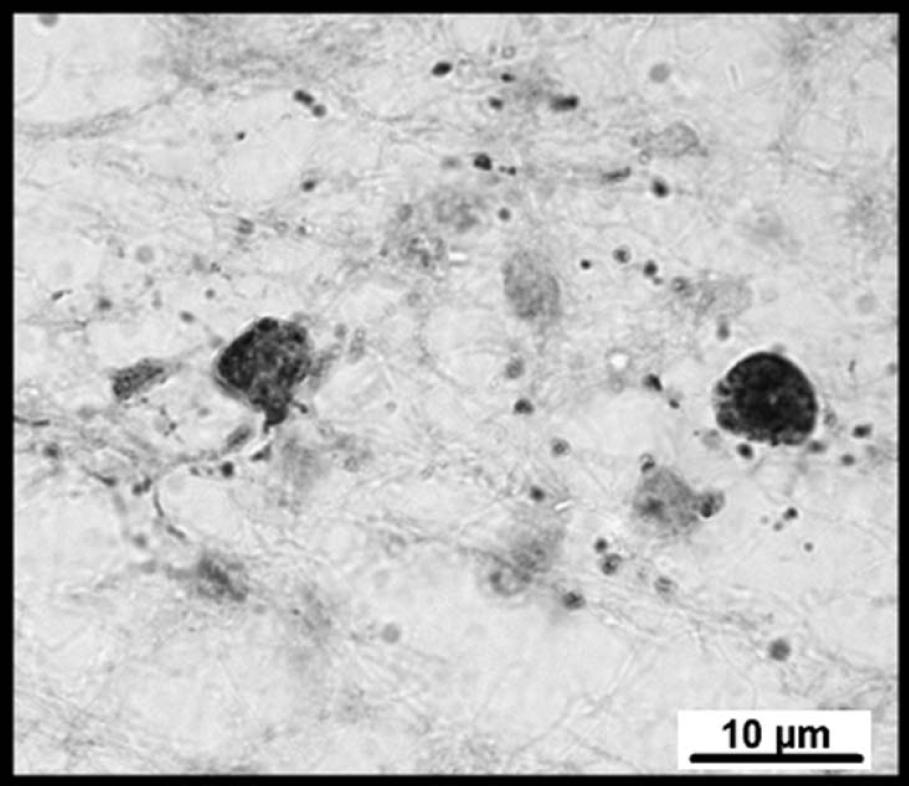
Photomicrograph showing an example of caspase-3-positive staining (brown) in the intragyral white matter tracts after cerebral ischemia in the near-term fetal sheep. Note the nuclear condensation within the caspase-3 (Asp175)-positive cells and coarse granular fragments of caspase-positive material around the cells.
Proliferating cell nuclear antigen expression
Figure 6 shows changes in cell proliferation in the white matter tracts assessed by PCNA immunohistochemistry. There were abundant PCNA labeling cells in sham control animals. Ischemia was associated with a significant reduction in the number of PCNA labeling cells compared to sham control (P < 0.05). The number of PCNA labeled cells was not significantly different between sham cooling (ischemia) and the two cooling groups.
DISCUSSION
The present study demonstrates that moderate, delayed cerebral hypothermia is associated with potent, delay-dependent suppression of postischemic cerebral demyelination in the near-term fetal sheep. Moderate hypothermia was begun 90 minutes after reperfusion (2-hour cooling group) and continued until 3 days after insult, and was associated with greater oligodendrocyte survival and myelin density and with reduced tissue edema in the parasagittal white matter tracts when evaluated 5 days after the ischemic insult. Oligodendrocyte protection was lost when initiation of cooling was delayed until 5.5 hours after reperfusion. The effects of hypothermia were strongly associated with suppression of both activated caspase-3 expression and the microglial reaction.
Proteolipid protein (PLP) gene expression has been used as a sensitive marker to detect bioactive oligodendrocytes at the translation stage of myelination (Guan et al., 2001; Mandai et al., 1997). Expression of PLP in the central nervous system is classically reported only in differentiated, myelinating oligodendrocytes, although trace amounts may be found earlier in development (Schindler et al., 1990). In vitro and in vivo evidence now clearly demonstrate that both mature and immature oligodendrocytes are exquisitely vulnerable to hypoxic-ischemic injury (Back et al., 2001; Pantoni et al., 1996; Skoff et al., 2001). Although in vitro myelinating oligodendrocytes are reported to be relatively resistant to excitotoxicity compared to immature oligodendrocytes (Rosenberg et al., 2003), there is increasing evidence that white matter injury is also a significant contributor to neurodevelopmental disability after hypoxic-ischemic encephalopathy at term (Mercuri et al., 1999; Okumura et al., 1997; Rutherford et al., 1998). Because cooling is already being tested clinically as a potential neuroprotective treatment for newborn infants exposed to perinatal asphyxia (Gunn et al., 1998a; Shankaran et al., 2002), it is important to ascertain whether cooling is effective in reducing white matter loss at term.
We have previously reported, in the same experimental model as the present study, that improved neuronal loss and electrophysiologic recovery with cerebral hypothermia are dependent on the timing of initiation of cooling after reperfusion. There is dramatic improvement if moderate cooling is initiated within a few hours of severe ischemia (Gunn et al., 1997), but only partial protection if cooling is delayed until 6 hours from the start of ischemia (5.5 hours after reperfusion) (Gunn et al., 1998b). Clinically, the development of seizures in the recovery period after asphyxia is one of the major prognostic factors associated with an adverse outcome (Caravale et al., 2003), and most clinicians would prefer to treat only infants who demonstrate seizures. However, experimentally, when cooling was delayed until after seizures were established (8.5 hours after reperfusion), there was no electrophysiologic or overall histologic improvement (Gunn et al., 1999).
These studies demonstrated the importance of starting cooling during the latent phase of recovery (approximately the first 6 hours in this paradigm), before the phase of secondary deterioration with seizures and cytotoxic edema (Gunn et al., 1999). This finding supports the hypothesis that there are events during early recovery, well after resolution of the insult itself, which contribute significantly to the final severity of injury. The present data show that hypothermia is associated with an even more acute time dependence for improvement in oligodendrocyte loss than for neuronal loss, such that cooling delayed by 5.5 hours after reperfusion did not improve numbers of oligodendrocytes after 5 days recovery. Thus, the current study further highlights the considerable importance of early initiation of cooling for cerebral protection.
The mechanisms of this rapid loss of protection of both oligodendrocytes and neurons with hypothermia with increasing delay are unclear. One potential mechanism is the timing of induction of caspase-mediated cell death. In vitro, cytoplasmic activation of caspases occurs in the latent phase, before the active or “execution” phase of programmed cell death involving intranuclear mechanisms (Samejima et al., 1998). Apoptotic pathways are suggested to have a relatively more important role in ischemic brain damage in the neonate compared to the adult (Hu et al., 2000), particularly in mediating posthypoxic loss of oligodendrocytes (Shibata et al., 2000). Similarly, we have reported that ischemia in the near-term fetal sheep leads to a marked increase in cells expressing activated caspase-3, and that this colocalized exclusively with PLP mRNA expression (Cao et al., 2003).
Several studies have suggested that hypothermia acts to reduce morphologic apoptotic cell loss in the neonatal brain (Edwards et al., 1995; Xu et al., 1998). Supporting this concept, mild hypothermia can reduce mitochondrial cytochrome C translocation, a critical step in the intrinsic pathway of apoptosis (Xu et al., 2002; Yenari et al., 2002), and has been shown to suppress expression of caspase-3 both after hypoxia-ischemia in vivo (Fukuda et al., 2001; Tomimatsu et al., 2001) and serum deprivation in vitro (Xu et al., 2002). Consistent with these results, the present study confirms a significant time of initiation-dependent suppression by hypothermia of postischemic activated caspase-3 in the cerebral white matter.
However, quantitatively, caspase-3 expression after 5 days' recovery can only account for a minority of oligodendrocyte cell loss, with a relatively low density of caspase-positive cells observed relative to the extent of loss of PLP-labeled cells. Speculatively, this discrepancy is likely related to the timing of histologic evaluation, with the preponderance of activation of caspases occurring much earlier after ischemia (Northington et al., 2001). A recent study has demonstrated that in the early stages of the development of cerebral necrosis, neurons display a number of features of early apoptosis, including cytoplasmic and nuclear condensation and specific caspase activation (Benchoua et al., 2001). Furthermore, in addition to caspase-mediated mechanisms, caspase-independent apoptotic pathways, such as apoptosis-inducing factor, contribute to delayed cell death and to necrotic processes (Leist and Jaattela, 2001; Sanchez-Gomez et al., 2003). Often, more than one of these pathways seem to be activated simultaneously, and cell fate is then determined by the relative speed of each process (Leist and Jaattela, 2001).
Alternatively, suppression of secondary inflammatory processes after hypoxic-ischemic insults may be an important mechanism of protection with hypothermia. There is considerable evidence that such processes, including induction of activated microglia with subsequent release of proinflammatory cytokines, contribute to both evolving neuronal and white matter injury (Silverstein et al., 1997). Cytokines exacerbate brain injury through various pathways including direct neurotoxicity and induction of apoptosis or by promoting stimulation of capillary endothelial cell proinflammatory responses and leukocyte adhesion and infiltration into the ischemic brain (Mallard et al., 2003; Silverstein et al., 1997).
However, the role of microglia in the brain is complex. Under physiologic conditions, microglia may actually help promote oligodendrocyte survival (Nicholas et al., 2003; Rabchevsky and Streit, 1997), but once activated, microglia induce contact-dependent oligodendroglial death in vitro (Nicholas et al., 2003). Thus, the present finding that early postischemic hypothermia profoundly reduced microglial induction suggests that this may be a significant contributor to improved oligodendrocyte survival. The effect on microglia was specific, because there was no effect of hypothermia on numbers of astrocytes. Clearly this effect could be either primary, or secondary to reduced cell loss and local tissue damage, leading to a reduced stimulus to further microglial induction. The markedly reduced effect of late cooling on microglia closely paralleled the loss of effect on oligodendrocyte numbers, despite an identical period of recovery after rewarming to the early cooling group. This time dependence strongly suggests a significant secondary element.
Nevertheless, there is good evidence that postischemic cooling directly suppresses the inflammatory reaction. In vitro, hypothermia potently inhibits proliferation and superoxide and nitric oxide (NO) production by cultured microglia (Si et al., 1997), with a greater effect on inducible NO production than on other sources (Gibbons et al., 2003). Similarly, to the present study, postischemic hypothermia delays microglial activation after transient focal ischemia in the adult rat (Inamasu et al., 2000). Thus, a direct effect of early cooling on microglia cannot be ruled out. It is striking that in the present study, late cooling in the 6-hour group was associated with reduced tissue swelling and with substantial preservation of myelin, despite no significant effect on numbers of oligodendrocytes. This raises the possibility that a major effect of suppression of microglial induction by hypothermia may not have been on cell survival per se, but rather to reduce local effects of microglia on the extracellular matrix and degradation of myelin.
The present findings that early initiation of cerebral cooling improved oligodendrocyte survival with reduced microglial activation and reduced total proliferation provide an interesting contrast with our previous report that early IGF-1 treatment also improves oligodendrocyte survival, but with significantly increased glial induction and overall proliferation (Cao et al., 2003). This difference is consistent with in vitro data showing that hypothermia acts to broadly suppress the cell death program (Bossenmeyer-Pourie et al., 2000), whereas IGF-1-mediated receptor activity actively initiates cellular programs including anti-apoptotic pathways and proliferation (Mason et al., 2000; McMorris et al., 1986). The changes in proliferation, as measured by PCNA immunohistochemistry, are consistent with this interpretation. In the control group there was abundant PCNA expression in the white matter tracts, which was markedly reduced after ischemia. Given the severity of cell death, it is likely that the residual proliferation in the sham cooling (ischemia) group reflected almost entirely microglia. In contrast, the persistent reduction in PCNA expression after hypothermia despite increased oligodendrocyte survival must reflect general suppression of glial proliferation (Lee et al., 2002), not just microglia (Si et al., 1997).
Finally, evolving neuronal and white matter injury may extend for longer than the 5-day recovery from ischemia in the present study (Geddes et al., 2001). The continued presence of activated caspase-3 at this time also suggests ongoing injury, and thus it is possible that the observed white matter protection will reduce over time. Nevertheless, there was markedly reduced caspase-3 expression as well as greater oligodendrocyte survival in the 2-hour group 48 hours after rewarming, suggesting a reduction in ongoing damage. Consistent with this, in adult rodents persistent neuronal protection with hypothermia has been found 2 to 6 months after ischemia (Colbourne and Corbett, 1995; Corbett et al., 2000), although clearly further longer term outcome studies are necessary to confirm the present findings.
CONCLUSION
Prolonged moderate cerebral hypothermia can prevent postischemic white matter damage in the near-term fetal sheep, but there is rapid attenuation of this effect with increasing delay in the initiation of cooling. Furthermore, hypothermic white matter protection was closely associated with reduced activation of both caspase-3 and microglia. These data strongly support the critical importance of early initiation of cooling in clinical trials.