
Abstract
c-Jun N-terminal kinase (JNK) is an important stress-responsive kinase that is activated by various forms of brain insults. In this study, we have examined the role of JNK activation in neuronal cell death in a murine model of focal ischemia and reperfusion; furthermore, we investigated the mechanism of JNK in apoptosis signaling, focusing on the mitochondrial-signaling pathway. We show here that JNK activity was induced in the brain 0.5 to 24 h after ischemia. Systemic administration of SP600125, a small molecule JNK-specific inhibitor, diminished JNK activity after ischemia and dose-dependently reduced infarct volume. c-Jun N-terminal kinase inhibition also attenuated ischemia-induced expression of Bim, Hrk/DP5, and Fas, but not the expression of Bcl-2 or FasL. In strong support of a role for JNK in promoting the mitochondrial apoptosis-signaling pathway, JNK inhibition prevented ischemia-induced mitochondrial translocation of Bax and Bim, release of cytochrome c and Smac, and activation of caspase-9 and caspase-3. The potential mechanism by which JNK promoted Bax translocation after ischemia was further studied using coimmunoprecipitation, and the results revealed that JNK activation caused serine phosphorylation of 14-3-3, a cytoplasmic sequestration protein of Bax, leading to Bax disassociation from 14-3-3 and subsequent translocation to mitochondria. These results confirm the role of JNK as a critical cell death mediator in ischemic brain injury, and suggest that one of the mechanisms by which JNK triggers the mitochondrial apoptosis-signaling pathway is via promoting Bax and Bim translocation.
Introduction
Emerging evidence has suggested that mitochondrial release of apoptogenic proteins play a critical role in neuronal cell death after cerebral ischemia (Fujimura et al, 1999; Sugawara et al, 1999; Graham and Chen, 2001). While the precise mechanism regulating this pathway is complex and remains only partially understood, Bcl-2 family proteins likely play an essential role (Graham et al, 2000). The antiapoptotic proteins such as Bcl-2 and Bcl-xL enhance cell survival after ischemia by maintaining mitochondrial membrane integrity (Lawrence et al, 1996; Martinou et al, 1994; Parsadanian et al, 1998; Zhao et al, 2003). In contrast, the proapoptotic proteins Bax, Bid, and Bim promote ischemic cell death by inducing the release of apoptogenic factors (Cao et al, 2001; Plesnila et al, 2001; Yin et al, 2002b). Most of these so-called BH3 domain-containing proapoptotic proteins normally reside within the cytoplasm in an inactive form by binding to their respective sequestration proteins (Guo et al, 2003; Lei and Davis, 2003; Nomura et al, 2003; Samuel et al, 2001). However, after ischemia or other stress stimuli, they become activated, and redistribute from the cytoplasm to mitochondria (Cao et al, 2001; Chen and Yin, 2000; Shibata et al, 2002). Thus far, the upstream signaling mechanism responsible for the translocation of Bax or Bim in ischemic neurons remains unknown, but has been an object of active investigation.
Among the stress-responsive signaling molecules that may promote the mitochondrial apoptosis-signaling pathway is the c-Jun N-terminal kinase (JNK) (Tournier et al, 2000), a member of the mitogen-activated protein kinase (MAPK) group. c-Jun N-terminal kinase activation plays a central role in stress-induced apoptosis (Harris et al, 2002; Irving and Bamford, 2002; Liou et al, 2003; Tournier et al, 2000). Activated JNK phosphorylates serine on a variety of cellular targets and, consequently, influences cell survival via transcriptional and posttranslational regulation of proteins involved in apoptosis (for a review, see Manning and Davis, 2003). After stress-induced activation, JNK permits phosphorylation of several transcription factors, particularly the c-Jun component of the transcription factor complex AP1 (Herdegen and Waetzig, 2001; Hibi et al, 1993), thus triggering the expression of a number of apoptosis-regulatory proteins (Sanchez and Yuan, 2001; Shaulian and Karin, 2002). Through protein phosphorylation, JNK also directly enhances the proapoptotic activity of p53, but reverses the antiapoptotic function of Bcl-2 and Bcl-xL (Fan et al, 2000; Mielke and Herdegen, 2000). More recently, it has been found that activated JNK can directly trigger the translocation of Bax and Bim to mitochondria by disrupting their association with cytoplasm sequestration proteins (Lei and Davis, 2003; Tsuruta et al, 2004). This latter mechanism provides a novel link between JNK signaling and Bax/Bak-dependent mitochondrial apoptosis.
The JNK signaling pathway is activated in the brain after cerebral ischemia (Ferrer et al, 1997, 2003; Herdegen et al, 1998; Ozawa et al, 1999; Wu et al, 2000). However, until recently there has been uncertainty as to whether JNK signaling is a causative factor in ischemic cell death (Irving and Bamford, 2002). The study by Kuan et al (2003), based on gene-knockout approaches, convincingly demonstrated the critical role of JNK3, the neural-specific JNK, in ischemia-induced apoptosis in the brain. The recent development of JNK-specific inhibitors has greatly accelerated our understanding of this signaling pathway in ischemic neurodegeneration. One such inhibitor is the small molecule organic compound SP600125 (Bennett et al, 2001; Han et al, 2001), which shows 300-fold selectivity of inhibition of JNK over the extracellular signal-regulated kinases (ERKs) and p38 MAPKs, the closest kinase relatives of JNK. Another class of newly developed JNK-specific inhibitors is peptide-based, such as the 21-amino-acid peptide derived from the JNK-binding domain (JBD) of JNK-interacting protein-1 (Barr et al, 2002), which recently has been tested successfully in models of cerebral ischemia (Borsello et al, 2003).
In this study, our first aim was to determine whether JNK activation is critical in ischemia-induced neuronal cell death in a murine model of focal ischemia/reperfusion. We used the SP600125 compound and the JBD peptide inhibitor to show that cerebral damage is causatively coupled to JNK activation. Our second aim was to test the hypothesis that JNK activation plays an essential role in triggering the mitochondrial apoptosis-signaling pathway in ischemic neurons.
Materials and experimental procedures
Murine Model of Transient Focal Ischemia
Animal surgery
Focal cerebral ischemia was produced by intraluminal occlusion of the left middle cerebral artery (MCA) as originally described (Kondo et al, 1997; Yang et al, 1994) with slight modifications (Cao et al, 2002). Male 10- to 12-week-old C57/B6 mice (25 to 30 g each, Jackson's Laboratory) were anesthetized with 1.5% isoflurane in a 30% O2/68.5% N2O mixture under spontaneous breathing. Rectal temperature was controlled at 37.0°C±0.5°C during and after surgery via a temperature-regulated heating pad. Mean arterial blood pressure was monitored during MCA occlusion through a tail cuff, and arterial blood gas was analyzed at 15 mins after the onset of ischemia. The animals underwent left MCA occlusion for 60 mins and then reperfusion for the indicated duration. Changes in regional cerebral blood flow (rCBF) before, during and after MCA occlusion were evaluated in animals using laser-Doppler flowmetry (Cao et al, 2002). After recovering from anesthesia, the animals were maintained in an air-conditioned room at 20°C.
Behavioral tests
Beginning 24 h after ischemia, two types of behavioral functional tests were performed in animals by an observer who was masked to the experimental conditions. Neurological deficits were scored on a 0 to 5 scale (Murakami et al, 1998): no neurological deficit (0); failure to extend the right forepaw fully (1); circling to the right (2); falling to the right (3); unable to walk spontaneously (4); dead (5). The second test, the corner test, which is a sensorimotor functional assessment, was performed in ischemic animals as described previously (Zhang et al, 2002). In this test, the ischemic mouse turns preferentially toward the nonimpaired (left) side. The turns in one versus the other direction were recorded from 10 trials for each test.
Determination of infarct volume
At 48 h after MCA occlusion, brains were removed and the forebrain was sliced into 8 coronal sections 1 mm thick. Sections were stained with 2,3,5-triphenyltetrazolium (3%). Infarct volume was determined using the MCID image analysis system as previously described (Chen et al, 1996). Animals that showed a massive hematoma in the brain or no infarction in the brain were omitted from further neurological or histological analysis.
In vivo administration of c-Jun N-Terminal Kinase inhibitors
Two types of JNK inhibitors were tested in the present study. For the test of the anthrapyrazolone JNK inhibitor SP600125, the compound was first dissolved in 100% dimethyl sulfoxide (DMSO) to generate a stock solution of 20 mmol/L, which was further diluted in PPCES vehicle (30% PEG–400/20% polypropylene glycol/15% cremophor EL/5% ethanol/30% saline) as described previously (Bennett et al, 2001). The vehicle in the volume of 5 mL/kg or SP600125 at the doses of 0.3, 1, 3, or 10 mg/kg was injected into the animals intravenously via the tail vein 15 mins before MCA occlusion. This was followed by another intravenous injection of the solution at the same doses at 3 h after MCA occlusion, based on the estimation that the half-life of SP600125 is between 3 and 4 h in animals (Bennett et al, 2001; Han et al, 2001). The animals that received administration of SP600125 or vehicle were assigned randomly to experimental groups consisting of nine animals each. To determine the time window of efficacy of SP600125 in the MCAO model, the optimal dose of the compound was administered as described above, beginning at 0, 0.5, 1, or 2 h after the onset of reperfusion.
In separate experiments, JBDTAT (Calbiochem, San Diego, CA, USA), the small peptide JNK inhibitor containing the TAT protein transduction domain, was tested in the murine MCAO model. JBDTAT was designed based on amino acids 143 to 163 of the JBD of the JNK scaffolding protein, JIP-1, which has been proved previously to be a potent and selective JNK inhibitor (Barr et al, 2002). For the administration of JBDTAT, the peptide was dissolved in DMSO, further diluted in artificial CSF, and then infused for 10 mins at a dose of 3 μg/5 μL into the intracerebroventricles 15 mins before or 30 mins after MCA occlusion (n=9 per group). Control animals received vehicle or the same amounts of the scramble control peptide (ConTAT).
Western Blot Analysis and Immunoprecipitation
Western blotting was performed using the standard method (Chen et al, 1998). The following antibodies were used: rabbit polyclonal antibodies of anti-phospho-MKK4 (thr261), anti-phospho-JNK (thr183/tyr185), anti-phospho-c-Jun (Ser63), and anti-JNK1/2. They were purchased from Cell Signaling, Beverly, MA, USA; rabbit antibodies for anti-Bcl-2, anti-Bax, anti-Hrk/DP5, anti-Bcl-xL, anti-Fas, and anti-FasL, and the goat anti-cytochrome c antibody were from Santa Cruz Biotechnology, Santa Cruz, CA, USA; antibodies for the detection of specific isoforms of 14-3-3 were also from Santa Cruz; the rabbit anti-Bim antibody was from BD Biosciences, San Jose, CA, USA; the mouse monoclonal anti-cytochrome c oxidase IV antibody was from Molecular Probes, Eugene, OR, USA; the rabbit anti-phospho-14-3-3σ (Ser186) was made as instructed by Dr Gotoh (University of Tokyo); the rabbit anti-cleaved caspase-3 antibody was from Cell Signaling; the rabbit anti-cleaved caspase-9 antibody was as described previously (Cao et al, 2002).
Immunoprecipitation was performed to examine protein—protein interactions between Bax and 14-3-3 using standard procedures (Cao et al, 2001). Briefly, cytosolic protein was isolated from cerebral cortices using the RIPA A (150 mmol/L NaCl, 1% Nonidet P40, 0.5% deoxycholate, 0.1% SDS, and 50 mmol/L Tris-HCl, pH 8.0) buffer. Equal amounts of protein from each experimental condition (300 μg per sample) were subjected to immunoprecipitation using the anti-14-3-3 antibody (K19, for total 14-3-3, Santa Cruz) followed by immunoblotting with anti-14-3-3 and anti-Bax antibodies, respectively.
Immunohistochemistry
Animals were killed in a carbon dioxide chamber at the indicated time points after ischemia, and the brains were prepared for freshly frozen coronal sectioning (15 μm of thickness) as described previously (Chen et al, 1997). For immunohistochemical staining of phospho-c-Jun (p-c-Jun), the rabbit polyclonal anti-p-c-Jun (Ser63) antibody was used at a dilution of 1:200 (Cell Signaling). For double-label immunofluorescence staining, sections were first incubated with the anti-p-c-Jun antibody at 4°C for 48 h followed by incubation for 2 h at room temperature with goat anti-rabbit Cy3.18 immunoconjugate (Jackson ImmunoResearch, West Grove, PA, USA) at 1:2,500 dilutions. Sections were then subjected to incubation for 24 h in the mouse anti-NeuN antibody (dilution at 1:500, Chemicon, Temecula, CA, USA). This was followed by incubation with biotin-conjugated anti-mouse antibody (dilution 1:3,000) and then fluorescein-avidin D (Vector Laboratories, Burlingame, CA, USA) at 8 μg/mL. The sections were washed four times in PBS, mounted in gelvatol, and coverslipped. For the assessment of nonspecific staining, alternating sections from each experimental condition were incubated without the primary antibody.
For immunohistochemical staining of Bax, the monoclonal antibody against the active Bax was used at a dilution of 1:150 (clone 6A7, Pharmingen, San Diego, CA, USA). The secondary antibody was the goat anti-mouse Cy3.18 immunoconjugate (Jackson ImmunoResearch) at 1:3,000 dilutions.
Protein Kinase Assays
Cell lysates were prepared under nondenaturing conditions as described (Derijard et al, 1994), and 150 μg of protein was used for each kinase assay. To assay for total JNK activity (JNK 1–3), a capture JNK assay was performed using a nonradioactive kinase assay kit according to the manufacturer's instructions (Cell Signaling). Briefly, cell lysates were incubated with recombinant GST-c-Jun (1–79) bound to glutathione-coupled agarose beads, and the complex was washed extensively with lysis buffer. Kinase activity in the complex was assayed by addition of cold ATP and subsequent immunoblot analysis for c-Jun phosphorylation using the anti-phospho-c-Jun (Ser-63) antibody. To assay for the JNK1-specific kinase activity, the cell lysates were first subjected to immunoprecipitation with the specific anti-JNK1 antibody (clone F-3, Santa Cruz), and then the immunoprecipitates were assayed for the kinase activity as described above. c-Jun N-terminal kinase 3-specific kinase activity was assayed as originally described (Namgung and Xia, 2000; Yang et al, 1997). Briefly, cell lysates were immunoprecipitated with a mixture of a monoclonal antibody that recognizes JNK1/2 (clone G151-666, BD PharMingen, San Diego, CA, USA) and a monoclonal antibody that recognizes JNK1 (clone F-3, Santa Cruz) to remove both JNK1 and JNK2 from the lysates. The remaining kinase activity (JNK3) in the supernatant was assayed by the capture JNK assay as described above. To ensure that JNK1 and 2 were completely removed from the supernatant, the supernatant was also examined by immunoblot analysis using the JNK1/2 antibody.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assay (EMSA) was performed to determine AP-1 binding activity in cerebral nuclear extracts as described previously (An et al, 1993; Yin et al, 2002a). In brief, the AP-1 consensus oligonucleotide (5′-CGCTTGATGAGTCAGCCGGAA-3′) was end-labeled with γ-32P-ATP using T4 polynucleotide kinase. The DNA—protein-binding reaction was performed in a total volume of 30 μL containing the binding assay buffer (10 mmol/L Tris-HCl, 20 mmol/L NaCl, 1 mmol/L DTT, 1 mmol/L EDTA, 5% glycerol, pH 7.6), 0.0175 pmol of labeled probe (>10,000 cpm), 30 μg of nuclear protein, and 1 μg of poly(dI-dC). After incubation for 20 mins at room temperature, the reaction mixture was subjected to electrophoresis on a nondenaturing 6% polyacrylamide gel for 2 h under low ionic strength conditions. The gel was dried and subjected to autoradiography. The specificity of AP-1 DNA-binding activity was determined by immunodepletion of c-Jun of the nuclear extracts before EMSA. Additional control experiments were performed in the presence of a 100-fold molar excess of cold AP-1 oligonucleotide.
Measurement of Caspase-Like Protease Activities
Caspase-3-like and caspase-9-like protease activities were measured in cell extracts using the fluorogenic substrates Ac-DEVD-AFC and Ac-LEHD-AFC, respectively (Cao et al, 2002). One unit activity corresponds to the caspase-like activity that cleaves 1 pmol of AFC per minute at 37°C at saturating substrate concentrations. To detect nonspecific protease activity, in parallel experiments the protein extracts were incubated in the reaction buffer with 5 μmol/L of the caspase-3 inhibitor DEVD-CHO or the caspase-9 inhibitor LEHD-CHO at room temperature for 30 mins before the addition of assay substrates, and the values were subtracted from those obtained without the inhibitors.
Statistical Analysis
Results are reported as mean values±s.d. The significance of difference between means was assessed by Student's t-test (single comparisons) or by ANOVA and post hoc Scheffe's tests (for multiple comparisons), with P<0.05 considered statistically significant.
Results
c-Jun N-Terminal Kinase is Activated in the Brain After Focal Ischemia and Reperfusion
We first wanted to confirm that JNK activation occurs in mice after focal ischemia and reperfusion. Whole-cell protein extracts were prepared from the MCA territory (depicted in Figure 1B) in the brain at various time points after 60 mins of ischemia or after sham operation. Immunoblot analysis was performed using antibodies against phosphorylated (p-) forms of MKK-4, JNK and c-Jun, and nonphosphorylated JNK1/2 (Figure 1A). Distinct increases in immunoreactivity were seen at 0 and 0.5 h after reperfusion for p-MKK4 and p-JNK, respectively. Densitometric analysis revealed that the relative immunoreactivities for p-MKK4 and p-JNK were both significantly enhanced beginning at 0.5 h after reperfusion, returning to sham control levels by 24 h (Figures 1C and 1D). The level of p-c-Jun, a major downstream target of JNK, did not significantly increase until 1 h postreperfusion (Figure 1E), but remained significantly elevated at 24 h, although at this late time point the magnitude of the elevation was substantially reduced from its peaks. Ischemia produced by MCA occlusion therefore produces a robust and temporally regulated increase in the MKK4/JNK/c-Jun signaling pathway in murine brain.
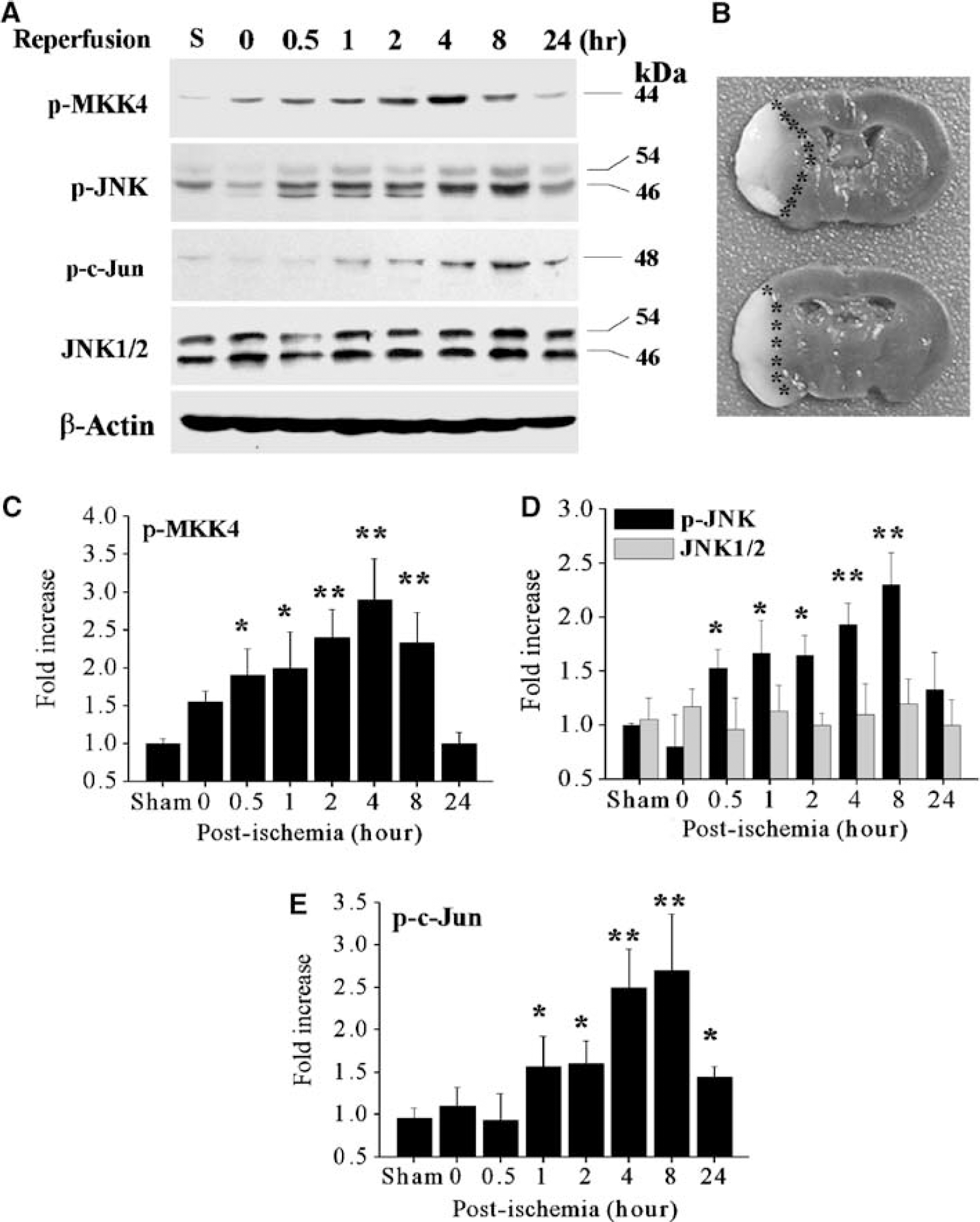
Temporal profiles of c-Jun N-terminal Kinase (JNK) activation after focal ischemia/reperfusion. (
c-Jun N-Terminal Kinase Activation is Inhibited After Ischemia by Specific Inhibitors
To test whether it was possible to modulate ischemia-induced brain JNK activity, two JNK inhibitors were administered 15 mins before MCA occlusion, respectively. The small molecule chemical inhibitor SP600125 was administered intravenoulsy at a dose of 1 or 3 mg/kg, followed by a second injection at the same dose 3 h after MCA occlusion. The peptide inhibitor JBDTAT (3 μg in 5 μL) was infused intracerebroventricularly. As shown (Figure 2A), Western blot analysis revealed that brain p-c-Jun levels were significantly attenuated at 4 and 8 h after reperfusion by either SP600125 or JBDTAT, whereas neither the SP600125 vehicle nor the control peptide (ConTAT) had any effect on p-c-Jun levels. c-Jun N-terminal kinase activity was next measured using a capture assay for total JNK. With the administration of either inhibitor, JNK activity in brain extracts was dramatically decreased (Figure 2B), indicating that either inhibitor was effective in blocking JNK activity in vivo. In parallel experiments, p38 kinase activity was also measured based on p38-immunoprecipitation and the p38 kinase substrate GST-ATF2, and the results revealed that neither inhibitor had a detectable effect on ischemia-induced p38 activation (Figure 2B). These results indicate that both SP600125 and JBDTAT provided specific JNK inhibition in the ischemic brain.
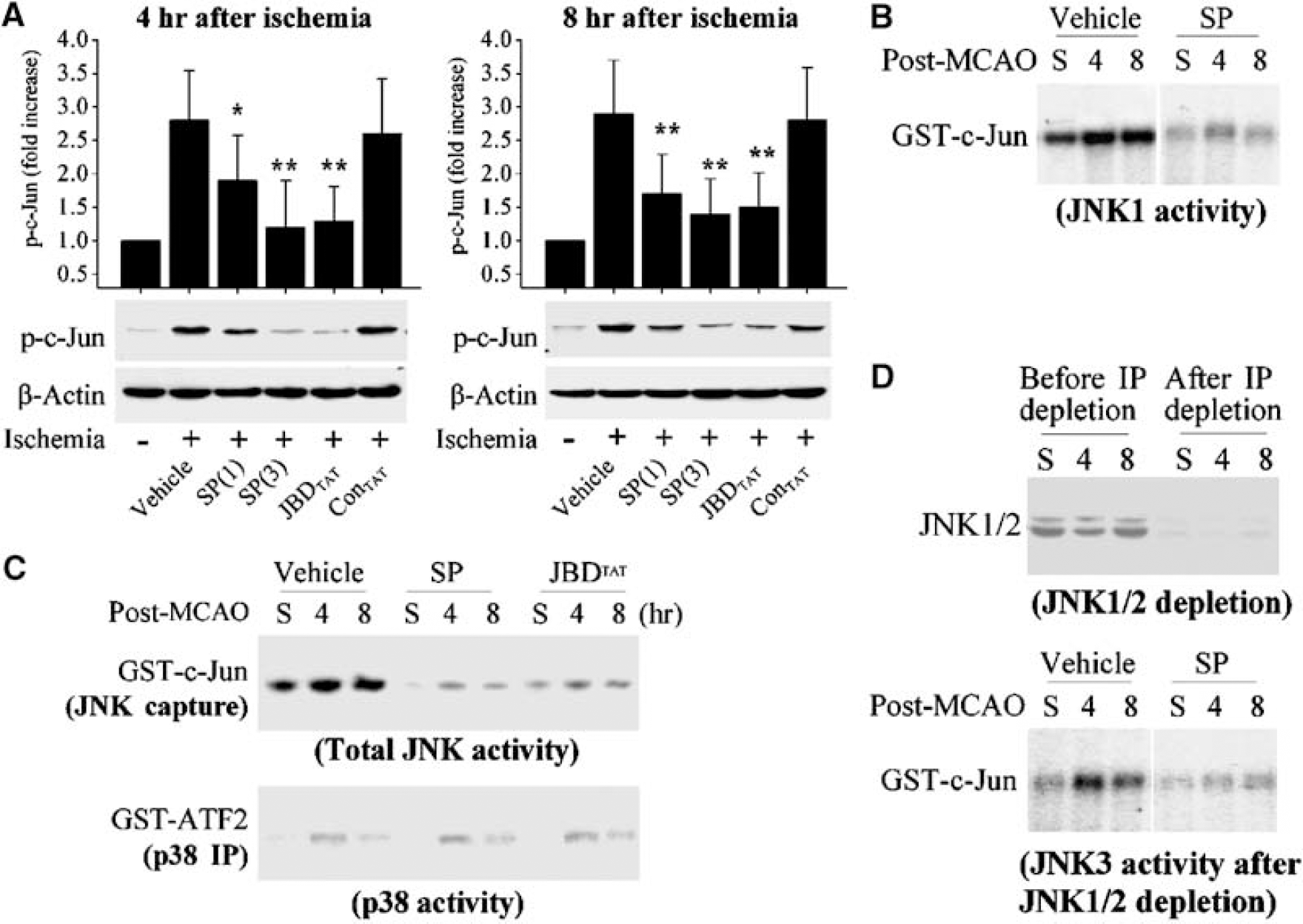
Pharmacological inhibition of c-Jun N-terminal Kinase (JNK) activity after focal ischemia/reperfusion. (
Next, the inhibitory effect of SP600125 on specific isoforms of JNK, JNK1 and JNK3, was examined after ischemia. A considerably high basal level of JNK1 activity was detected in the sham control brain, and it was increased approximately 2-fold at 4 and 8 h after ischemia. The administration of SP600125 (3 mg/kg × 2) remarkably attenuated JNK1 activity in ischemic brains and sham controls (Figure 2C). To determine JNK3 activity, JNK1 and JNK2 isoforms were first removed by immunodepletion (Figure 2D). Residual JNK activity could then be ascribed to the JNK3 isoform. JNK3 activity showed a very low basal level in the sham control brain, but the levels were increased ∼4-fold at 4 or 8 h after ischemia. The administration of SP600125 (3 mg/kg × 2) nearly completely eliminated the JNK3-specific activity (Figure 2D).
The recent study by Bain et al (2003) suggests that SP600125, as an ATP-competitive inhibitor of JNK, may also inhibit the activity of a number of other cellular proteins with similar or even greater potency. To address this issue in the brain, SP600125 (3 mg/kg) was administrated to additional sham-operated mice (n=3 per condition), and at 1 and 4 h after drug administration the brains were processed for Western blot analyses of several potential pharmacological targets for SP600125, including phospho-SGK (Ser78), phospho-p70 S6 Kinase (Ser371), and phospho-AMPK (Thr172) (all primary antibodies were from Cell Signaling). SP600125 treatment had no detectable effect on the levels of any of the above phosphorylated proteins (data not shown).
Activated JNK is a major contributor to the formation of the AP-1 transcription factor complex via phosphorylation of c-Jun. AP-1 DNA-binding activity can therefore be used as an index of downstream JNK efficacy. To verify that enhanced JNK activation after ischemia resulted in the activation of AP-1, AP-1 DNA-binding activity was measured in cortical nuclear extracts using an EMSA. In sham control cortex, low levels of AP-1 DNA binding were present (Figure 3A, leftmost lane). After 4 or 8 h postreperfusion, however, AP-1 activity was greatly enhanced (Figure 3A, center lanes). Brains receiving SP600125 treatment showed completely attenuated AP-1 activity after ischemia (Figure 3A, rightmost lanes). Specificity of the EMSA for AP-1 binding was tested by immunodepletion of c-Jun using anti-c-Jun antibody. Depletion of c-Jun eliminated nearly all AP-1 DNA binding (Figure 3B). The specificity of the AP-1 DNA-binding activity was also confirmed by adding 100-fold molar excess of cold AP-1 oligonucleotide to the assay, which eliminated the signals (not shown).
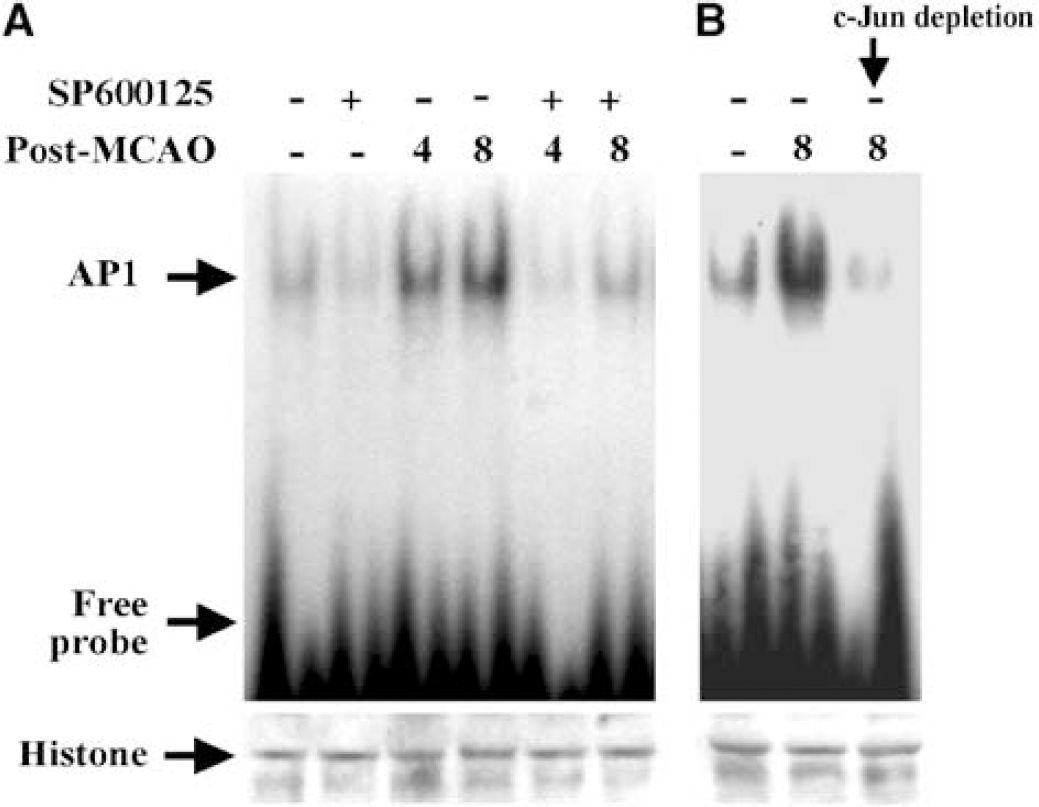
Pharmacological inhibition of c-Jun N-terminal Kinase (JNK) decreases AP-1 DNA-binding activity after ischemia. (
To determine whether the JNK inhibitory effects of SP600125 could be detected at the cellular level, p-c-Jun expression was examined using immunocytochemistry. Sections from sham control brains contained little or no p-c-Jun immunofluorescence (Figure 4A). Four hours after reperfusion, nuclear localization of p-c-Jun was found throughout the MCA cortical areas examined (Figure 4B). Twenty-four hours after reperfusion, the spatial distribution of nuclear p-c-Jun immunofluorescence was diminished, and mainly restricted to the inner border zone of the infarction (Figure 4C). Double staining for p-c-Jun and NeuN revealed that neurons were the principal cell type displaying nuclear p-c-Jun (Figures 4D–4F). In addition, TUNEL, a marker of fragmented DNA, indicative of cell death, was found to colocalize with many of the p-c-Jun-positive cells (Figures 4G–4I).
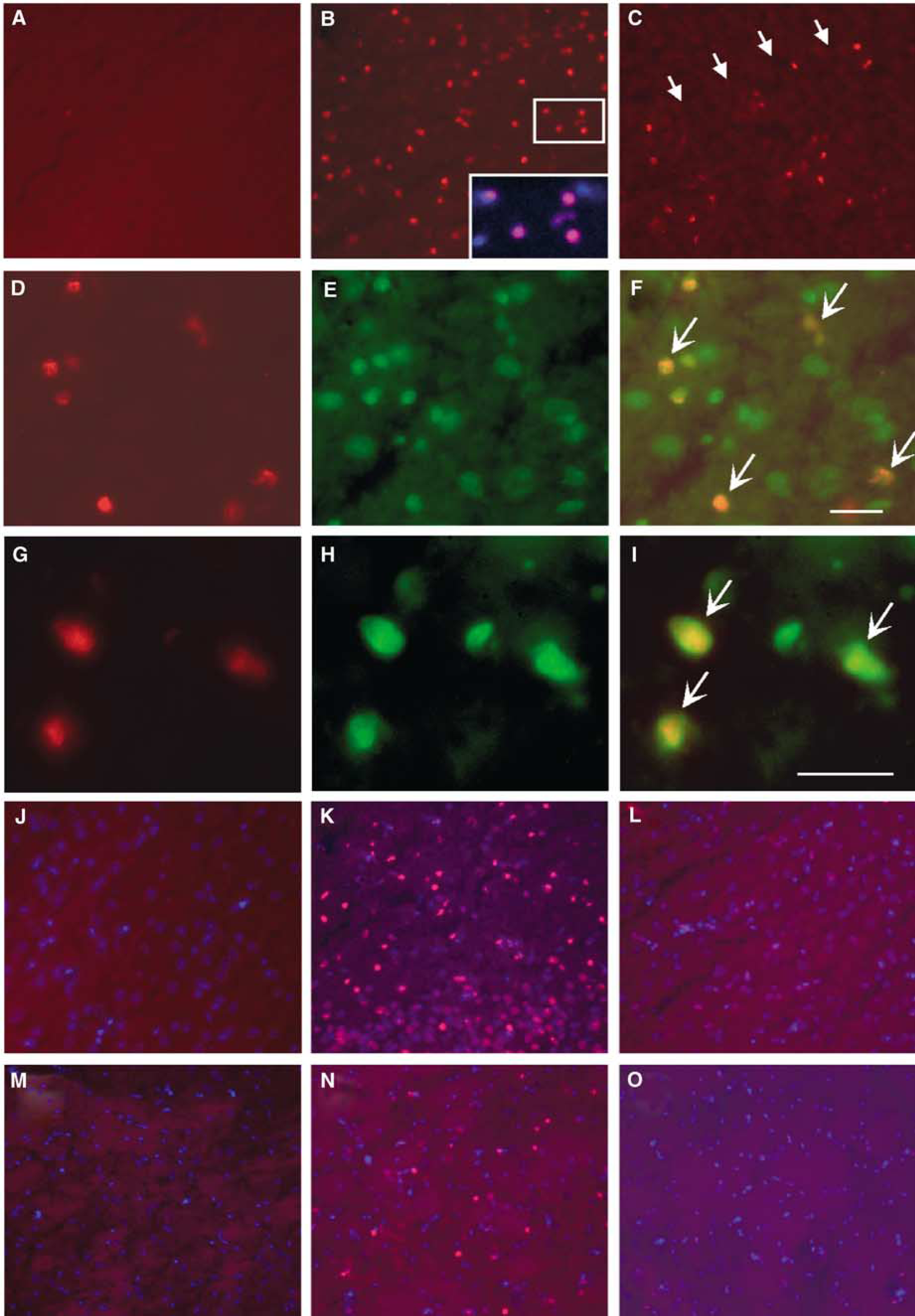
Immunofluorescent analysis of p-c-Jun after focal ischemia/reperfusion. In the non-ischemic control brain (
The effect of SP600125 treatment on ischemia-induced p-c-Jun induction was determined immunohistochemically at 4 h after reperfusion. Compared with vehicle-treated animals, the amounts of p-c-Jun positive cells were markedly reduced in both cortex (Figures 4J–4L) and striatum (Figures 4M–4O) in SP600125-treated animals. Thus, the results confirmed that inhibition of brain JNK activity resulted in diminished JNK-dependent downstream signaling after cerebral ischemia.
c-Jun N-Terminal Kinase Inhibition in the Brain Results in Reduced Ischemic Infarct Size
We aimed to determine whether JNK inhibition could mitigate ischemic brain damage. Infarct volume was measured based on the regional loss of TTC staining in brain sections 48 h postreperfusion after 60 mins of ischemia. MCA occlusion produced ipsilateral cerebral infarction averaging ∼42 mm3 in volume. Administration of SP600125 15 mins before MCA occlusion (second injection was administered 3 h after ischemia) reduced the infarct volume in a dose-dependent manner (Figure 5A). A significant reduction averaging ∼33% and ∼55% was reached at 1 (× 2 times) and 3 (× 2 times) mg/kg (P<0.05 versus vehicle controls), respectively, but an additional increase in the dose to 10 mg/kg (× 2 times) did not result in an additional reduction in infarct volume. Cerebral salvage occurred primarily in the periphery of the ischemic tissue, leading to enhanced survival of penumbral cortical tissue. Reduction of infarct size by SP600125 was not accompanied by significant alterations in arterial blood pressure and blood gases compared with animals that received vehicle alone (data not shown). Delayed treatment with SP600125 (3 mg/kg × 2 times) continued to significantly reduce infarct volume when the treatment was initiated at 0, 0.5, or 1 h after the completion of ischemia (Figure 5B), but the effect was lost when the compound administration was delayed by 2 h.
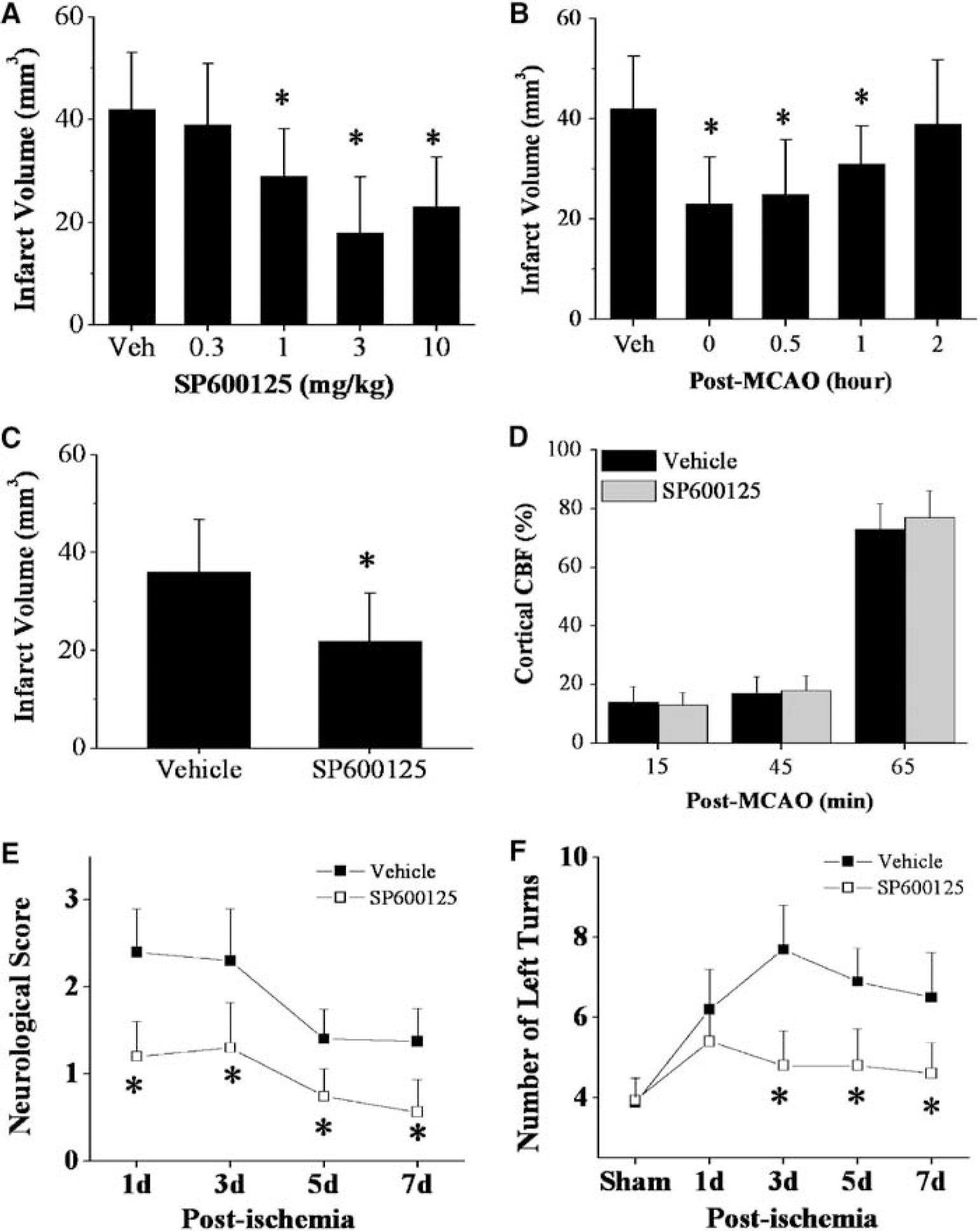
Pharmacological inhibition of c-Jun N-terminal Kinase (JNK) decreases infarct size after focal ischemia/reperfusion. (
To establish that SP600125-mediated protection was indeed sparing brain tissue and not simply delaying tissue loss, infarct volume was measured at 7 days after ischemia in two additional groups that received SP600125 (3 mg/kg × 2) or vehicle, respectively. Significant protection was still detected after 7 days (Figure 5C). To rule out the possibility that SP600125 might alter cerebral blood flow (CBF), laser Doppler flowmetry (LDF) was used to monitor cortical CBF in both vehicle- and SP600125-treated animals. No difference was found in CBF between animals that received SP600125 or vehicle during or after MCA occlusion (Figure 5D). To determine the effect of SP600125 treatment on functional outcomes after ischemia, both neurological deficits and sensorimotor functions were examined at 1, 3, 5, and 7 days after ischemia. The results revealed that the animals treated with SP600125 showed significantly improved functional outcomes than the vehicle-treated animals (Figures 5E and 5F).
The potential protective effect of the peptide JNK inhibitor, JBDTAT, was also assessed in the focal ischemia model. Intracerebroventricular injection of JBDTAT (3 μg) 15 mins before or 30 mins after MCA occlusion significantly decreased infarct volume as compared with vehicle-treated animals (Pretreatment: JBDTAT versus vehicle, 25.2±9.1 versus 45.3±11.4 mm3, P<0.05; posttreatment: 27.8±8.6 versus 44.2±10.6 mm3, P<0.05, n=9 per group). Injection of the control peptide did not show any effect on infarct volume (pretreatment, 43.5±10.4 mm3; posttreatment, 43.9±11.1 mm3).
c-Jun N-Terminal Kinase Inhibition Attenuates the Mitochondrial Apoptosis-Signaling Pathway after Ischemia
To show the cell death-suppressing effect of JNK inhibition by SP600125 at cellular levels, TUNEL was performed using coronal brain sections obtained 24 h after ischemia. In vehicle-treated mice, TUNEL-positive cells were robustly detectable in the MCA territory (Figure 6A). Two types of TUNEL-positive cells could be detected in this model as described previously (Li et al, 1995; Yin et al, 2002b). One type of cell had apoptotic morphology with condensed or fragmented nuclei (Figure 6A, panel c), and these were mainly detected in the inner border zone of the infarction, the region where apoptotic death most often occurs in this model (Kondo et al, 1997). The other type of TUNEL-positive cells had a diffusive cytoplasm-like staining (Figure 6A, panel e), which was suggestive of nonapoptotic cell death and could be detected in more than 80% of cells in the infarct core. In SP600125-treated ischemic brains, the numbers of total TUNEL-positive cells and apoptotic TUNEL-positive cells were both significantly reduced (Figures 6B and 6C). These effects of SP600125 were particularly evident in the infarct border zone, and, to a lesser extent, in the ischemic core region where cells die mainly via necrosis in this model. Taken together, the results show that JNK inhibition by SP600125 markedly suppressed the induction of apoptosis in the ischemic brain; however, the protective effect of SP600125 may involve partial inhibition of necrosis as well.
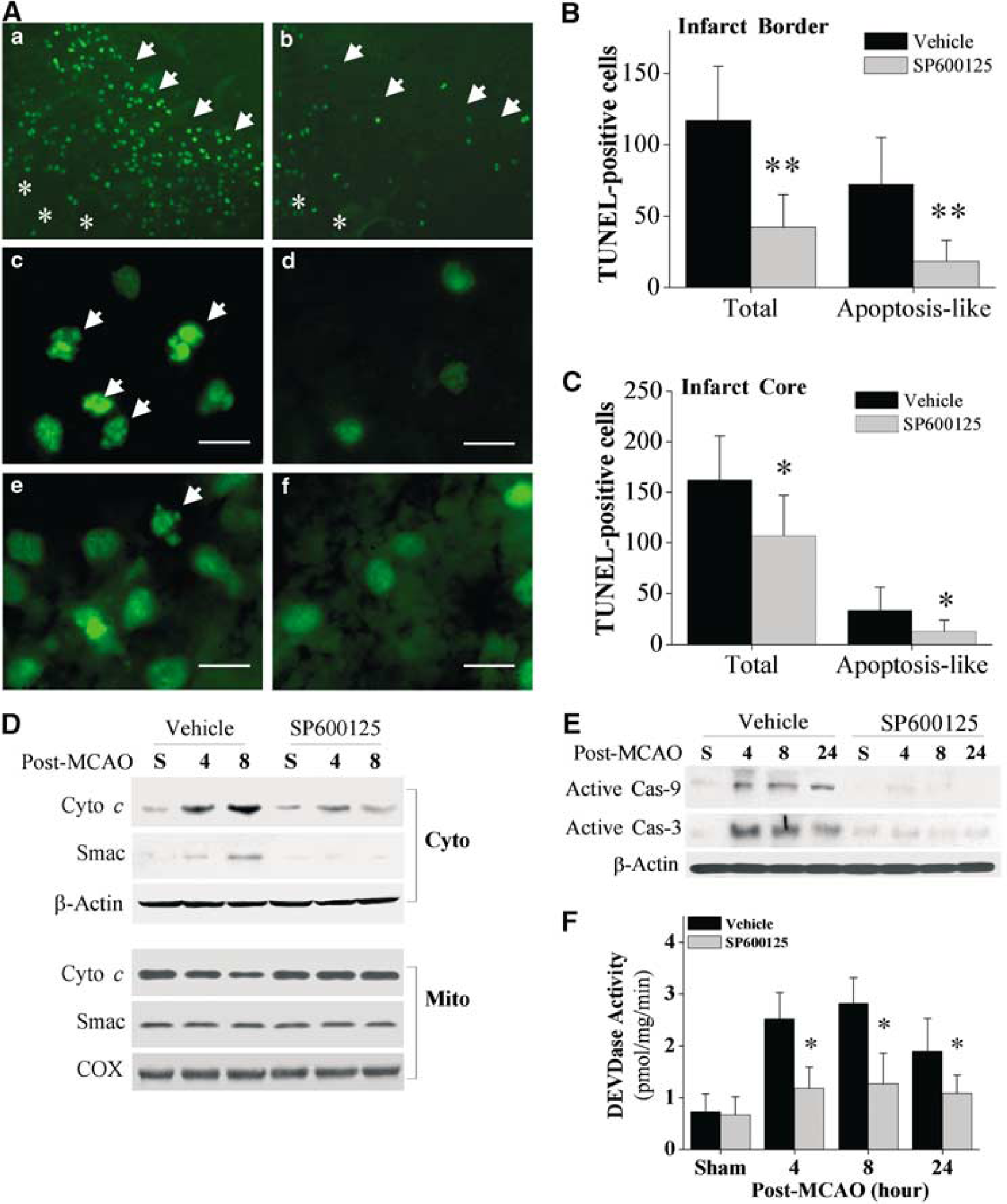
Pharmacological inhibition of c-Jun N-terminal Kinase (JNK) attenuates the mitochondrial apoptosis-signaling pathway. (
Activation of the mitochondrial apoptosis-signaling pathway plays a critical role in ischemic neuronal cell death and has been well characterized in rodent models of cerebral ischemia (Cao et al, 2001, 2002; Fujimura et al, 1999; Sugawara et al, 1999). In this study, we wanted to determine whether JNK activity contributes to mitochondrial damage and to the activation of the downstream apoptosis cascade after ischemia. First, the release of cytochrome c and Smac into the cytosolic fraction, an indicator of compromised mitochondrial integrity, was assessed using immunoblot analysis. Ischemia increased cytosolic cytochrome c and Smac staining at both 4 and 8 h after reperfusion, whereas obvious inhibition of cytochrome c and Smac release occurred in brains treated with SP600125 (Figure 6D). In line with its demonstrated inhibitory effect on the mitochondrial signaling pathway, SP600125 treatment diminished the activation of both caspase-9 and caspase-3 after ischemia (Figures 6E and 6F). The activation of caspases was confirmed at 4, 8, and 24 h after ischemia using the substrate-based protease assay and immunoblot analysis with antibodies recognizing the active fragments for caspase-9 and caspase-3. These results suggest that JNK activity is required for the mitochondrial damage-mediated activation of cell death executioners, including caspase-9 and caspase-3.
c-Jun N-Terminal Kinase Inhibition Attenuates the Expression of Proteins Involved in Apoptosis Signaling
It has been suggested that a number of proteins involved in apoptosis signaling are transcriptionally activated through the JNK pathway (Puthalakath and Strasser, 2002). Several proapoptotic proteins, such as Bim and Fas, have been identified as specific transcriptional targets for neuronal JNK3 signaling after cerebral ischemia (Kuan et al, 2003). In the present study, we have examined the effect of JNK inhibition on the expression of proteins that could potentially contribute to mitochondrial apoptosis signaling after ischemia. Whole-cell cortical extracts from sham control animals, or at 8 and 24 h after ischemia, were immunoblotted with specific antibodies against Bim, Bax, Hrk/DP5, Fas, Bcl-2, FasL, and Bcl-xL (Figure 7A). Relative quantities of proteins were measured using densitometric analysis and expressed as fold increases over sham control animals (Figure 7B). Ischemia significantly upregulated the expression of Bim (mainly BimEL), Hrk/DP5, Fas, and Bcl-2, but had no effect on Bax or FasL expression. Inhibition of JNK activity using SP600125 (3 mg/kg × 2 times) attenuated ischemia-induced Bim, Hrk/DP5, and Fas protein expression in the brain. Administration of SP600125 did not affect elevated Bcl-2 expression, nor did it alter Bax or FasL levels at any time point studied. In addition, the level of Bcl-xL was significantly decreased in the brain 24 h after ischemia, whereas SP600125 treatment retained the expression of Bcl-xL. The results suggest that JNK inhibition by SP600125 in the ischemic brain results in a shift of the balance between pro- and anti-apoptotic proteins in favor of promoting cell survival.
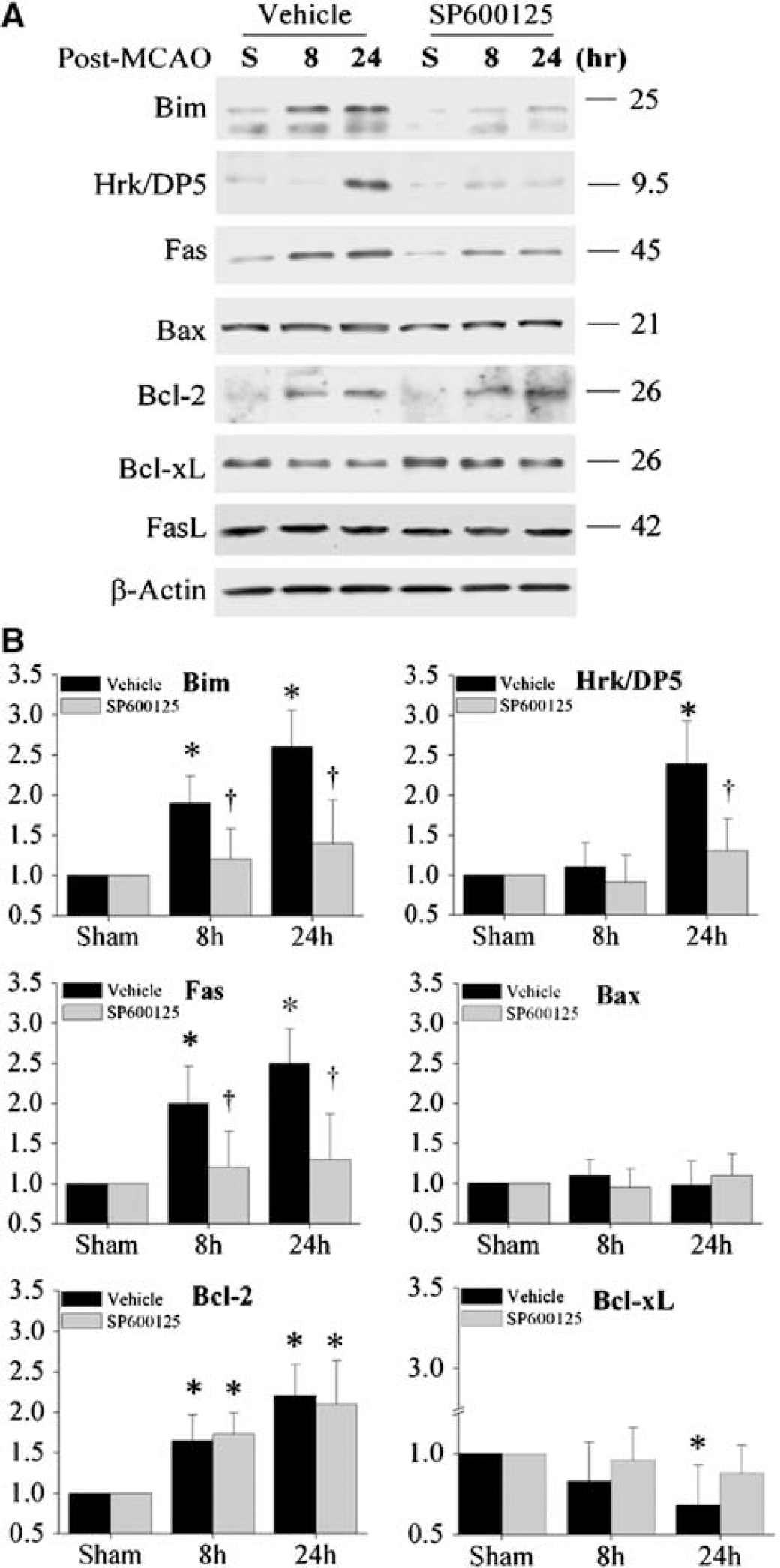
Effects of c-Jun N-terminal Kinase (JNK) inhibition on the expression of proteins that could contribute to mitochondrial apoptosis signaling after ischemia. (
c-Jun N-Terminal Kinase Inhibition Prevents Ischemia-Induced Translocation of Bax and Bim to Mitochondria
In addition to gene expression regulation, posttranslational regulation of BH3-containing proteins is an important mechanism by which JNK promotes apoptotic cell death via the mitochondrial pathway (Puthalakath and Strasser, 2002). Recent studies have shown that JNK can remarkably promote the proapoptotic activity of the proapoptotic proteins Bim and Bax, leading to their translocation to the mitochondria and subsequent activation of the mitochondrial apoptosis-signaling pathway (Lei and Davis, 2003; Tsuruta et al, 2004). Since Bim and Bax translocation appears to be important for neuronal cell death after ischemia (Chen and Yin, 2000; Cao et al, 2001; Shibata et al, 2002), in the present study we have investigated the effect of JNK on ischemia-induced Bim and Bax translocation.
The translocation of Bim and Bax from cytosol to mitochondria after ischemia was first examined using subcellular fractionation and immunoblot analysis. In agreement with our previous observations in the rat model of transient focal ischemia (Cao et al, 2001), the results presented in Figure 8A show that 4 and 8 h after MCA occlusion there was a marked accumulation of Bax in the mitochondrial fraction; a corresponding decrease in Bax was detected in the cytosolic fraction. Inhibition of JNK activity using SP600125 (3 mg/kg × 2 times) diminished ischemia-induced Bax translocation at both time points studied (Figure 8A). The ischemia-induced Bax translocation was then demonstrated by immunofluorescent staining of coronal brain sections using an antibody recognizing the active form of Bax. Consistent with previous observations (Cao et al, 2001), a punctate pattern of Bax immunofluorescence was seen in ischemic brains at 4 and 8 h after ischemia (Figure 8B, panels b, c), and this pattern was not detected in brains treated with SP600125 (Figure 8B, panel d).
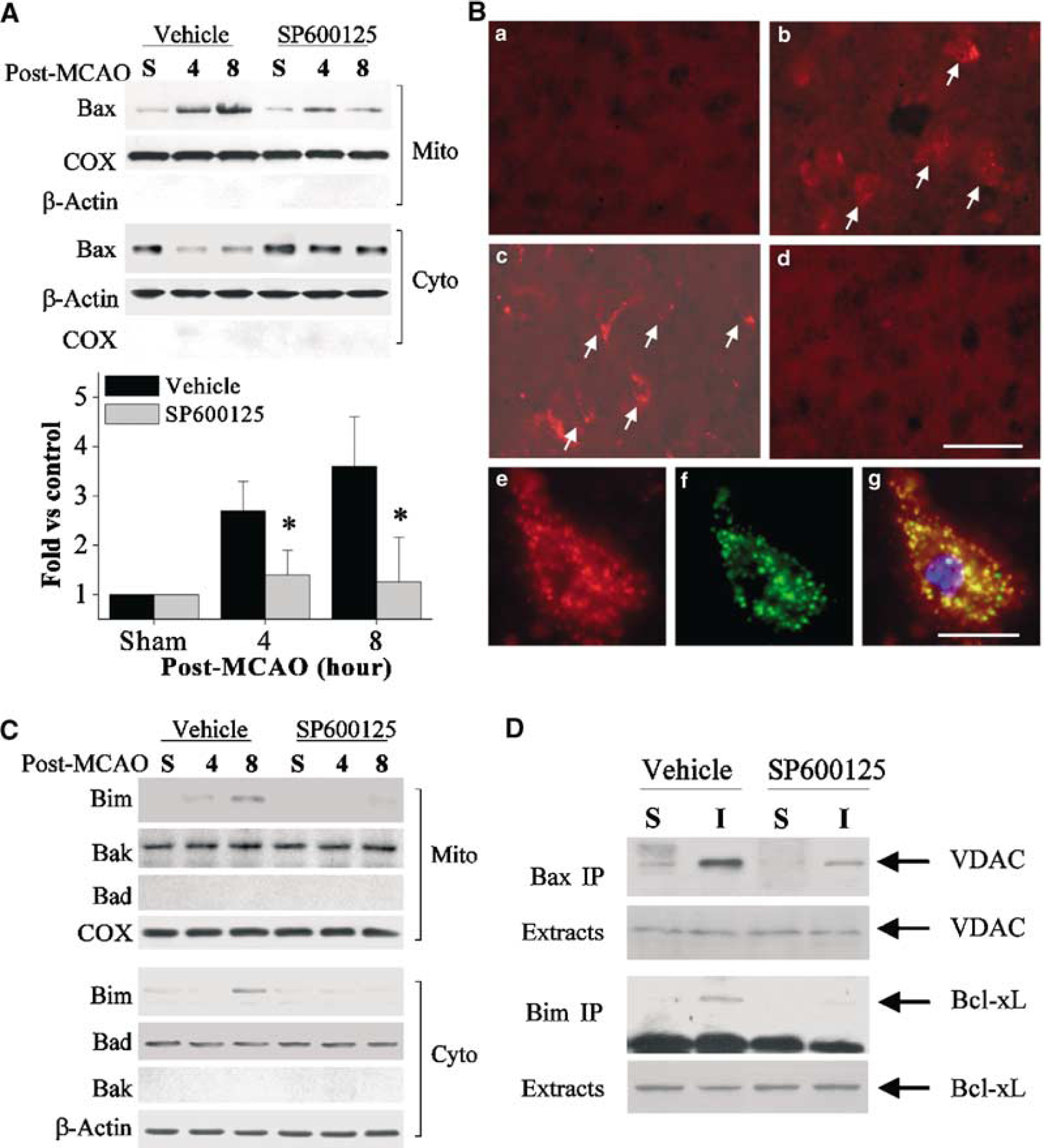
Effect of c-Jun N-terminal Kinase (JNK) inhibition on ischemia-induced Bax and Bim translocation. (
The levels of Bim in the mitochondrial fraction were moderately increased after ischemia (Figure 8C), and this was more readily detectable at the 8 h time point. In support of a role of JNK in Bim activation, this Bim translocation was reduced in brains treated with SP600125. Moreover, the levels of Bak and Bad, additional proapoptotic proteins involved in the mitochondrial apoptosis-signaling pathway, were also examined in subcellular fractions after ischemia. Bak and Bad, which in normal brain reside exclusively in the mitochondria and cytoplasm, respectively, did not show detectable changes after ischemia (Figure 8C).
To further confirm that Bax and Bim interact with mitochondrial proteins on their translocation, immunoprecipitation was performed to examine the protein—protein interactions between Bax and VDAC (Cao et al, 2001) and between Bim and Bcl-xL (Sanchez and Yuan, 2001). The 8 h-post-ischemia time point was chosen because it was the time at which translocation of both Bax and Bim was readily detectable. The results revealed a markedly increased formation of the Bax—VDAC complex and moderately increased formation of the Bim—Bcl-xL complex after ischemia, whereas the amounts of both complexes were decreased in brains treated with SP600125 (Figure 8D).
c-Jun N-Terminal Kinase Promotes Bax Translocation after Ischemia through Phosphorylation of 14-3-3 Proteins
Bax translocation to mitochondria is a robust phenomenon after ischemia, and it likely contributes to the mitochondrial apoptosis-signaling pathway (Cao et al, 2001; Ferrer et al, 2003). Thus far, we have found that JNK inhibition by SP600125 nearly completely blocked Bax translocation at 4 and 8 h after ischemia (Figure 8A), strongly suggesting that JNK activity is required for this translocation. Thus, we have further investigated the mechanism by which JNK mediates Bax translocation after ischemia, focusing on the interactions between Bax and 14-3-3 proteins, the recently identified cytoplasmic anchor of Bax (Putcha et al, 2003; Tsuruta et al, 2004). Initially, immunoprecipitation followed by Western blot analysis was performed to establish the binding partnership between 14-3-3 proteins and Bax in normal brain tissues. The anti-14-3-3β antibody (Clone K-19, Santa Cruz) that recognizes all 14-3-3 isoforms was used for immunoprecipitation of cytosolic extracts, and the immunoprecipitates were then subjected to immunoblot analysis for Bax. As shown (Figure 9A), Bax was detectable in the 14-3-3 immunoprecipitates, but not in the postimmunoprecipitation supernatant, suggesting that Bax resides in the cytosol exclusively as a 14-3-3/Bax complex. To determine which particular 14-3-3 isoform binds to Bax in the normal brain, immunoprecipitation was performed using the anti-Bax antibody, and the immunoblot was then probed with antibodies against 14-3-3β, 14-3-3γ, 14-3-3ɛ, 14-3-3η, 14-3-3ζ, and 14-3-3σ. Bax was found to be associated mainly with 14-3-3σ and, to a lesser extent, with 14-3-3η and 14-3-3ɛ (Figure 9B), but not with other 14-3-3 isoforms (not shown) in the cytosolic extracts from the cerebral cortices.
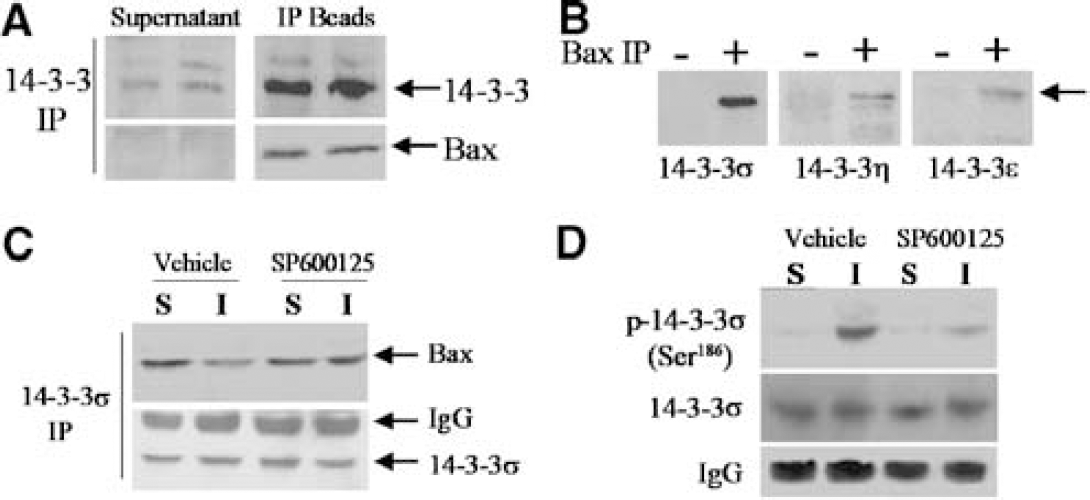
c-Jun N-terminal Kinase (JNK)-mediated alterations in Bax/14-3-3 interaction after ischemia. (
Next, we wanted to determine how ischemia/reperfusion alters the interactions between Bax and 14-3-3σ, and whether JNK activation contributes to this alteration. Cytosolic protein was extracted from cerebral cortices at 4 h after ischemia with or without SP600125 treatment, or after sham operation, and subjected to immunoprecipitation using the anti-14-3-3σ antibody. This was followed by immunoblot analysis using the anti-Bax antibody. While the amount of 14-3-3σ was not changed, the content of Bax was decreased in the immunoprecipitates after ischemia (Figure 9C), suggesting that cytosolic Bax was partially disassociated with 14-3-3σ. In contrast, this reduced association between Bax and 14-3-3σ was restored in brains treated with SP600125 (Figure 9C).
Finally, to determine whether ischemia/reperfusion induced JNK-attributable phosphorylation of 14-3-3, the 14-3-3σ immunoprecipitates described above were subjected to immunoblot analysis using the 14-3-3σ phosphoserine-specific antibody (Tsuruta et al, 2004). Phosphorylated 14-3-3σ (p-Ser186), barely detectable in normal brain extracts, was increased after ischemia; however, this ischemia-induced serine phosphorylation on 14-3-3σ was diminished in brains treated with SP600125 (Figure 9D). In contrast, in separate experiments, cytosolic dynein, the anchor protein for Bim, was immunoprecipitated from brain extracts 4 and 8 h after ischemia and then probed with the anti-phosphoserine antibody (Novus Biologicals, Littleton, CO, USA), but no evidence was found for an increased serine phosphorylation on dynein after ischemia (data not shown).
Discussion
Emerging evidence suggests that activation of JNK signaling may play an important role in ischemia-induced neuronal apoptosis (Irving and Bamford, 2002). Confirmative demonstration of a causative contribution of JNK to ischemic neuronal cell death is scant, however (Manning and Davis, 2003). In the present study, we provide new proof that specific JNK inhibition can reduce multiple components of the JNK signaling pathway and, consequently, attenuate neuronal cell death in a model of focal ischemia and reperfusion. Furthermore, we identify a novel molecular link between JNK and the mitochondrial apoptosis-signaling pathway, which is activated at least in part via JNK-mediated translocation of the proapoptotic proteins Bax and Bim.
Our study found that several components of the JNK signaling pathway were markedly activated after focal ischemia and reperfusion in the murine model. An increase in the phosphorylated p-MKK4, an upstream kinase for JNK, was detectable in the brain immediately after the completion of ischemia. This was followed by significant increases in the levels of p-JNK and p-c-Jun. The increases in p-MKK4 and p-JNK subsided to baseline levels at 24 h after ischemia, whereas the levels of p-c-Jun remained elevated for at least 24 h. This pattern of JNK activation was similar to that reported by others (Kuan et al, 2003; Wu et al, 2000). A number of previous studies have investigated the activation of JNK in several different rodent models of cerebral ischemia (Irving and Bamford, 2002). After transient global ischemia, the phosphorylation of JNK and c-Jun was increased in hippocampal neurons; however, the temporospatial distribution of this activation appears to correlate with selective vulnerability of neurons to global ischemia in some studies (Gillardon et al, 1999; Hu et al, 2000), but not in others (Ferrer et al, 1997; Sugino et al, 2000; Tsuji et al, 2000). In contrast, the results from studies involving focal ischemia appeared to be more akin to each other. Several studies showed rapid and sustained JNK activation within the ischemic lesion after MCA occlusion (Hayashi et al, 2000; Herdegen et al, 1998; Wu et al, 2000), and the increased c-Jun activity was found to colocalize with cell death markers (Herdegen et al, 1998). However, direct evidence for a causative effect of JNK activation on ischemic cell death was not available until two recent studies demonstrated diminished ischemic cerebral damage in mice deficient in the neuronal JNK3 gene (Kuan et al, 2003) or in mice treated with a peptide JNK inhibitor (Borsello et al, 2003). Nevertheless, the mechanism by which JNK activation ultimately leads to neuronal cell death after ischemia is poorly understood thus far.
In the present study, our first goal was to determine whether specific JNK inhibitors could reduce JNK signaling and attenuate ischemic brain injury. Two different types of JNK inhibitors were tested: the small molecule chemical inhibitor SP600125 and the peptide inhibitor JBDTAT. The specificity of these inhibitors has been well characterized previously (Barr et al, 2002; Bennett et al, 2001). As determined using both JNK activity assay and immunoblot/immunohistochemical analysis for p-c-Jun, administration of either inhibitor beginning 15 mins before MCA occlusion significantly ameliorated JNK activity and nuclear accumulation of p-c-Jun at 4 and 8 h after ischemia, and these effects by JNK inhibitors were without the accompanying effect on the activation of p38 MAP kinase, a close relative of JNK. Importantly, a clearly demonstrated consequence of specific JNK inhibition in this study was the dose-dependent and prolonged reduction in infarct volume by SP600125 administration, which, together with the antiinfarction effect of JBDTAT, confirms a critical role of JNK in mediating ischemic tissue loss. The protective effect of SP600125 or JBDTAT shown here is likely clinically relevant, as significant neuroprotection was achieved when JBDTAT or SP600125 was administered either before or after the onset of reperfusion. Our data also suggest that JNK inhibition may have a limited time window of efficacy; in particular, SP600125 showed effectiveness when it was administered up to 1 h after reperfusion. This time-dependent protection by JNK inhibition is consistent with the rapid-onset nature of JNK activation after cerebral ischemia. However, the recent study by Borsello et al (2003) showed that intraventricular administration of a TAT-linked, protease-resistant peptide JNK inhibitor, D-JNKI-1, had a therapeutic time window of 6 h in the murine model of MCA occlusion. The mechanism responsible for this extended therapeutic time window for D-JNKI-1 is unknown, but could be related to its ability to rapidly penetrate cell membranes and maintain prolonged kinetics in cells. More recently, Carboni et al (2004) reported that another small molecule JNK inhibitor AS601245 showed marked neuroprotective effect in a rat model of MCA occlusion. Taken together, these results suggest that development of new derivatives of JNK inhibitors with a prolonged therapeutic time window and those allowing systemic administration may have great clinical implications for the treatment of stroke.
The results presented here confirm that JNK contributes to ischemic neuronal cell death and also provide some new insights into the mechanism by which JNK triggers cell death execution pathways. Our second goal in this study was to understand whether and how JNK might link to the mitochondrial apoptosis pathway, a well-established mechanism for neuronal death in ischemic injury (Cao et al, 2001, 2002; Fujimura et al, 1999; Sugawara et al, 1999). Our results strongly suggest that JNK activity is required for the activation of the mitochondrial apoptosis-signaling pathway. The supporting evidence is the clearly demonstrated attenuation of mitochondrial cytochrome c and Smac release and caspase-9/caspase-3 activation after ischemia by JNK inhibition. To further elucidate the underlying mechanisms, we have investigated the effects of JNK on the expression and posttranslational regulation of several apoptosis-regulatory proteins. c-Jun N-terminal kinase is known to promote cell death via both transcriptional and posttranslational mechanisms (Shaulian and Karin, 2002). Through c-Jun/AP-1-mediated transcriptional regulation, JNK activity can enhance the expression of a number of gene products, such as Bim, Hrk/DP5, and Fas, which could ultimately contribute to mitochondrial apoptosis (Bozyczko-Coyne et al, 2001; Harris and Johnson, 2001; Ivanov et al, 2002; Whitfield et al, 2001). We found that AP-1 activation after ischemia was sensitive to JNK inhibition by SP600125, indicating that JNK activity contributed to AP-1-mediated gene regulation after ischemia. In fact, the expression of Bim, Hrk/DP5, and Fas proteins was increased up to 2.5-fold after ischemia, and this upregulation was eliminated in brains treated with SP600125. These results confirm the observations by Kuan et al (2003) showing the amelioration of Bim and Fas mRNA expression in JNK3 knockout mice after hypoxic-ischemia. Our results suggest that the net sum of the effects by JNK activation, which enhanced the expression of Bim, Hrk/DP5, and Fas, and decreased the expression of Bcl-xL, shifted the ratio of pro- and anti-apoptotic molecules to one favoring cell death.
Importantly, AP-1-mediated gene expression may not be the predominant and certainly not the only mechanism by which JNK promotes neuronal cell death after ischemia. Recent studies based on assays using isolated mitochondria have suggested that activated JNK may directly interact with mitochondria and results in the release of cytochrome c and Smac from mitochondria (Schroeter et al, 2003). The underlying mechanism for this direct effect of JNK is unclear, but it may involve JNK-mediated phosphorylation of certain mitochondrial proteins, such as Bcl-2 and Bcl-xl (Schroeter et al, 2003). Another area of increasing importance that has not been adequately explored is the posttranslational regulation of proapoptotic proteins by JNK. Several studies have identified Bax and Bim as important regulatory targets for JNK (Lei and Davis, 2003; Tsuruta et al, 2004). Given the potential importance of Bax and Bim in neuronal apoptosis and ischemic cell death (Putcha et al, 2001; Whitfield et al, 2001; Chen and Yin, 2000; Yuan and Yankner, 2000), we have studied the effect of JNK activation on the functional status of Bax and Bim after ischemia. Consistent with previous findings (Cao et al, 2001; Ferrer et al, 2003; Shibata et al, 2002), in the current study both Bax and Bim were subjected to mitochondrial translocation after ischemia. JNK inhibition by SP600125 prevented the translocation of Bax and Bim, indicating that JNK activity was required for their posttranslational activation. The mechanisms responsible for the respective effects of JNK on Bax and Bim translocation are different from each other, however. As shown by immunoprecipitation experiments, Bax and Bim use 14-3-3 protein and dynein, respectively, as the cytoplasm anchor, and, on liberation from the anchor proteins, they move to mitochondria as unbound, active forms. Using phosphoserine-specific antibodies, we detected JNK-dependent phosphorylation on the 14-3-3σ (serine186), but not on dynein, after ischemia. These results are consistent with the results from nonneural systems in that JNK triggers Bax translocation via phosphorylation of 14-3-3 (Tsuruta et al, 2004). Recently, the 14-3-3 proteins have merged as a critical cytosolic anchor for several prodeath members of the Bcl-2 family, including Bax and pBad. Similar to Bax, the disassociation of pBad from 14-3-3 may be required for its activation and translocations in ischemic neurons (Saito et al, 2003). What remain to be determined is if Bax and pBad bind to the same 14-3-3 isoform(s) in neurons and whether JNK activity contributes to the activation of both molecules after ischemia. In contrast to Bax activation, JNK activates Bim translocation by directly phosphorylating Bim rather than its anchor protein (Lei and Davis, 2003). The phosphorylation status of Bim was not confirmed in the present study, but should be a future objective on the availability of appropriate antibodies. Collectively, our results suggest that Bax and Bim translocation is a novel molecular link between JNK activation and the mitochondrial apoptosis-signaling pathway in ischemic neurons.
It should be noted that although JNK signaling is critical in neuronal cell death, other members of the MAPK family may also contribute to ischemic brain injury. The involvement of ERK1/2 and p38 MAPK in cerebral ischemia has been reported. Both ERK1/2 and p38 MAPK are activated in the brain after ischemia (Alessandrini et al, 1999; Walton et al, 1998), and inhibition of either kinase results in marked reduction of cerebral infarct volume (Alessandrini et al, 1999; Namura et al, 2001; Simi et al, 2000). Given the fact that JNK, ERK1/2, and p38 are distinctly activated and presumably function in parallel with each other, it is indeed an intriguing question why inhibition of one of them alone could provide significant protection after cerebral ischemia. A potential explanation for this apparent crossreaction of MAPK inhibition is the existence of considerable crosstalk between MAP kinases after ischemia; as a consequence, targeting one pathway may affect other pathways as well. Such crosstalk between different MAPKs has not been defined, but may involve certain downstream kinase substrates that may be critical for the regulation of cell death (Bogoyevitch et al, 1996; Gupta et al, 1995). Future studies are warranted to elucidate the converging points for various MAPK members, which certainly would help identify novel therapeutic targets.
In summary, the present study confirms the critical role of JNK signaling in neuronal cell death induced by transient cerebral ischemia. The study also identifies Bax and Bim mitochondrial translocation as a novel mechanism through which JNK may activate the mitochondrial apoptosis-signaling pathway in ischemic neurons. The results presented here reinforce the concept that the JNK signaling pathway may be a legitimate therapeutic target for the treatment of ischemic stroke and other neurological diseases.
Footnotes
Acknowledgments
The first two authors contributed equally to the work. The authors thank Liping Sheng, Shuping Wang, and Cristine O’Horo for their excellent technical assistance, Carol Culver for editorial assistance, and Pat Strickler for secretarial support.